
Abstract
Sulforaphane (SFN) has been considered as an indirect antioxidant and potential inducer of the Nrf2-ARE pathway. This study was conducted to investigate the protective role of SFN against oxidative stress in bovine granulosa cells (GCs). GCs were collected from antral follicles (4–8 mm) and cultured according to the experimental design where group 1 = control, group 2 = treated with SFN, group 3 = treated with hydrogen peroxide (H2O2), group 4 = pretreated with SFN and then with H2O2 (protective) and group 5 = treated with H2O2 followed by SFN treatment (rescuing). Results showed that SFN pretreatment significantly increases cell viability and reduces cytotoxicity in GCs under oxidative stress. Following H2O2 exposure, expression of NRF2 was found to be significantly increased (p < 0.05) in SFN-pretreated cells, while no significant differences were observed between group 3 and group 5, although the expression was significantly increased compared to the control group. Moreover, the relative abundance of the NRF2 downstream target antioxidant genes (CAT, PRDX1, SOD1 and TXN1) were higher (fold change ranged from 7 to 14, p < 0.05) in sulforaphane pretreated GCs. Low level of ROS and lipid accumulation and higher mitochondrial activity were observed in GCs pretreated with SFN, whereas no such changes were observed in GCs treated with SFN after exposure to oxidative stress (group 5). Thus, we suggest that SFN pretreatment effectively protects GCs against oxidative damage through the activation of the NRF2-ARE pathway, whereas addition of SFN during oxidative insult failed to rescue GCs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oxidative stress mediated by oxygen-derived free radicals (also known as reactive oxygen species or ROS) is a common state affecting nearly all living organisms due to suboptimal environmental conditions. In homeostatic condition, there is a balance between the generation of ROS and ROS scavenging ability of cells through the cellular antioxidant system. However, when the production of ROS overwhelms the cellular antioxidant capability, it potentially leads to a condition known as oxidative stress (Agarwal et al. 2005). The ROS level can be raised endogenously during many physiological processes including ovulation (Agarwal et al. 2005). Moreover, the use of oxygen as a respiratory substrate was reported to develop oxidative stress during aerobic metabolism and energy production (Frisard and Ravussin 2006). In addition, other endogenous sources (mitochondria, inflammatory cell activations, plasma membrane NADPH oxidase, endoplasmic reticulum, lysosomes and peroxisomes) can influence the production of ROS in mammalian cells (Klaunig et al. 2010). Recently, showed that moderate to low levels of oxidative stress induced by hydrogen peroxide (H2O2) in GC could trigger the release of the RNA molecule as a defense mechanism into the extracellular space mediated by exosomes (Saeed-Zidane et al. 2017).
Despite its essential role in the ovulatory process, ROS-mediated oxidative stress was found to have a detrimental role in female reproduction, particularly on ovarian function and a number of reproductive diseases including endometriosis, polycystic ovarian syndrome and unexplained infertility (Agarwal et al. 2012). Ovarian GCs, the major cellular constituent in a follicle, have two pivotal roles in female reproduction: steroid production and protect the oocyte during ovulation (Yada et al. 1999; Sohel and Cinar 2015). At the end of follicular growth, GCs in the dominant follicle are differentiated into luteal cells by an ovulatory luteinizing hormone (LH) surge (Duffy and Stouffer 2003). This process is crucial for a successful ovulation and subsequent formation of corpus luteum to maintain pregnancy. On the other hand, during ovulation after the preovulatory surge of LH, inflammatory cells especially neutrophils and macrophages are massively recruited to produce ROS to facilitate follicular rupture and the release of the oocyte (Shkolnik et al. 2011), indicating exposure of GCs to some sort of oxidative stress during ovulation. In addition to an endogenous source, environmental sources of ROS could make the situation more complex.
During the evolutionary process, mammalian cells have developed incredible defense mechanisms against harmful environmental insults including oxidative stress. One of the major mechanisms is the production of different phase II antioxidant enzymes via activation of the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway, which is considered as an innate cellular defense mechanism against oxidative stress (Zhang et al. 2013). It has recently been shown that sulforaphane (SFN) triggers the induction of phase II antioxidant enzymes via activation of the Nrf2-ARE pathway in GCs (Sohel et al. 2017). Sulforaphane is a natural dietary isothiocyanate found in cruciferous vegetables such as cabbage, brussel and broccoli sprouts (Guerrero-Beltrán et al. 2012). By reacting with thiol groups of KEAP1, SFN forms thionoacyl adducts and promotes the disruption of NRF2-KEAP1 interaction leading to Nrf2-ARE pathway activation (Hong et al. 2005) and enhances the abundance of phase II enzymes. As a result, cells acquire an enhanced ability to handle oxidative insults and survive under suboptimal environmental conditions (Zhang et al. 2013). Recently, we showed that the survival ability of preimplantation embryos under oxidative stress is associated with the activation of the Nrf2-ARE oxidative stress response pathway (Amin et al. 2014). However, the effect of oxidative stress on bovine GCs and the use of SFN as a potent antioxidant in order to activate the Nrf2 pathway have not been investigated yet. The aims of this study were to investigate whether SFN supplementation is able to activate the NRF2-ARE antioxidant mechanism and protect and/or rescue bovine GCs under oxidative stress conditions. For this, we used the primary culture of GCs as a model and H2O2 as an inducer of oxidative stress to determine the protective and/or rescuing ability of SFN.
Materials and methods
Experimental groups
To determine the protective or rescuing effects of SFN supplementation on bovine GCs under oxidative stress condition, primary cultures of GCs were grown in five groups, where group 1: untreated control, group 2: treated only with 10 μM SFN (Sigma-Aldrich, S4441-5MG, St. Louis, MO), group 3: treated only with 500 μM H2O2 (Sigma-Aldrich, 216,763, Steinheim, Germany) for 40 min, group 4: first pretreated with 10 μM SFN and then treated with 500 μM H2O2 40 min and group 5: no pretreatment but 10 μM SFN added after 500-μM H2O2 exposure. Before classified into the different treatment groups, recovered cells were grown until they reached up to 40–50% confluency.
Collection of ovaries and GCs
GCs were collected and cultured according to the procedure described in our previous work (Sohel et al. 2013). In brief, a total of 120 ovaries were obtained from a local slaughterhouse and transported in 0.9% saline solution at 38 °C within 2 h of collection. GCs from a minimum of 10 ovaries were used in each replicate in order to obtain a sufficient number of cells for different experiments. Following washing with new saline solution and 70% ethanol, follicular materials (follicular fluid containing GCs and cumulus-oocyte complex) were aspirated from antral follicles of 4–8 mm in diameter by an 18-gauge needle attached to a 5-mL syringe and placed in a 50-mL sterile falcon tube containing 10-mL Media 199 (Sigma-Aldrich, M5017, Steinheim, Germany). After collection, tubes were left for 15 min at 37 °C to allow the oocyte cumulus complex and cellular debris to settle at the bottom. The upper liquid containing GCs was then collected in a 15-mL tube and centrifuged at 500×g for 5 min to obtain the GCs in the form of pellets. GC pellets were washed once in calcium-magnesium free PBS (CMF) followed by centrifugation at 500×g for 5 min and resuspended in 1 mL of red blood cell (RBC) lysis buffer (8.26 g/L NH4Cl) for 1 min to remove erythrocytes. Isotonicity was restored by adding 5 mL of DMEM/F-12 medium supplemented with 10% FBS (Pan-Biotech GmbH, P30–8500, Aidenbach, Germany). The purity of isolated GCs was determined by the presence of FSHR (GC-specific marker) and the absence of CYP17A1 (Theca cell-specific marker), and the results are presented in Electronic Supplementary Material Fig. S1.
Culture and treatment of GCs
Approximately 50,000, 1.5 × 105 and 6 × 105 viable cells were seeded in a 8-well chamber slide, 24-well and 6-well culture plate (Corning Incorporated, Kennebunk, ME, USA), respectively and cultured in DMEM/F-12 medium (Sigma-Aldrich, D6046, Steinheim, Germany) supplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (100 μg/mL) (Sigma-Aldrich, P4333, Steinheim, Germany) at 37 °C in a humidified atmosphere of 5% CO2 until 40–50% confluency. To investigate the effects of SFN during oxidative stress, primary cultures of GCs were exposed to 10 μM SFN and 500 μM H2O2 and culture according to the experimental design. The concentrations of SFN and H2O2 were chosen based on our previous experiences (Sohel et al. 2016, 2017). A time-course experiment was performed in order to find out the appropriate exposure time of 10 μM SFN against GCs. Based on the time-course result, we decided to expose the GCs to SFN for 24 h which is presented in Electronic Supplementary Material Fig. S2. A simplified experimental design is presented in Fig. 1. Following the treatments, cells were either investigated for different morphological characterization or harvested using trypsin EDTA (Biological Industries, catalog no. 03–052-1B, Kibbutz Beit Haemek, Israel) and stored at − 80 °C for further genetic analysis.

Simplified experimental groups. Cells in all groups were grown to 40–50% confluence and continued different treatments under optimum culture conditions. Group 1: no treatment, cells were grown with complete medium; group 2: cells were treated with 10 μM SFN; group 3: cells were continued with complete medium up to 40–50% confluence followed by an incubation with 500 μM H2O2 for 40 min and continued with complete medium; group 4: cells were treated with 10 μM SFN for 24 h followed by 40-min incubation with 500 μM H2O2; group 5: cells were continued with complete medium followed by an incubation with 10 μM SFN + 500 μM H2O2 for 40 min and continued with 10 μM SFN
Cell morphology and viability
After each treatment, GCs from different treatment groups were observed using inverted microscopy in a Nikon Eclipse TS100 microscope (Nikon, Tokyo, Japan) for confluency and changes in morphology. The viability of cells was determined using the trypan blue exclusion test as previously described (Strober 2001) with some modifications. Briefly, after the treatment, both adherent and floating cells from each treatment group were collected and resuspended in 1 mL of complete medium. Following that, 100 μL of cell suspension and 100 μL of 0.4% trypan blue were mixed into a micro-centrifuge tube and allowed to incubate for 1–2 min at room temperature. Ten microliters of cell mixture/trypan blue was applied to the hemocytometer and placed under a microscope for counting live and dead cells. GC viability was calculated as a percentage of viable cells from the total cell count.
Cytotoxicity assay
Damaging effects of H2O2 or protective/rescuing ability of SFN on preventing cytotoxicity were determined using a colorimetric assay (WST-1-based) for nonradioactive quantification of cell proliferation, cell viability and cytotoxicity kit (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer’s protocol. Briefly, 1 × 104 viable GCs were plated in each well of the 96-well microplate (Clear, Flat Bottom TC-Treated, Corning Incorporated) containing 100 μL/well culture media until 40–50% confluency in a humidified atmosphere (37 °C and 5% CO2). Cells were treated according to the experimental plan (Fig. 1). Following the culture period, 10 μL WST-1 reagent was added to each well of the microplate and incubated for 5 h at 37 °C in the presence of 5% CO2. Finally, the microplate was shaken for 1 min and absorbance was measured using a microplate (ELISA) reader (Glomax Multi Detection System, Promega BioSystems Sunnyvale, CA, USA) with a wavelength of 460 nm.
RNA isolation and complementary DNA synthesis
Total RNA was isolated from GCs using the miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to manufacturer’s instruction. An on-column DNase digestion step using RNase-Free DNase set (catalog no. 79254, Qiagen, Hilden, Germany) was performed during total RNA isolation in order to remove any additional DNA contamination. Total RNA concentration and purity were determined using a NanoDrop ND-1000 spectrophotometer. Approximately, 1 μg of total RNA was then reverse transcribed to complementary DNA (cDNA) using oligo (dT)-25 and random primers (Promega, Madison, WI) and Superscript II reverse transcriptase (Invitrogen, Karlsruhe, Germany). The cDNA synthesis reaction contains 4-μL 5× first-strand buffer (375 mM KCl, 15 mM MgCl2, 250 mM Tris–HCl, pH 8.3), 2 μL dithiothreitol (Promega), 1 μL dNTP, 0.7 μL Superscript II reverse transcriptase and 0.3-μL RNase inhibitor (Promega) and was incubated at 42 °C for 90 min and then denatured at 70 °C for 15 min.
Real-time quantitative PCR
An ABI PRISM® 7000 sequence detection system (Applied Biosystems, Foster City, CA) instrument was used to quantify the transcript abundance of NRF2-ARE pathway genes including NRF2 and KEAP1; antioxidant genes, namely, superoxide dismutase-1 (SOD1), peroxiredoxin-1 (PRDX1), thioredoxin-1 (TXN1) and catalase (CAT); and the endogenous control gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primer sequences of genes used in this experiment were designed via Primer3 online software v.0.4.0 (http://frodo.wi.mit.edu/primer3/) and listed in Supplementary Table S1. Before performing the quantitative RT-PCR reaction, the suitability of the endogenous control was confirmed by investigating the consistency of GAPDH expression among the experimental samples. The qRT-PCR reactions were set up in 20 μL using 2-μL first-strand cDNA template, 7.4 μL deionized H2O, 0.3 μM of forward and reverse gene specific primers and 10 μL 1× Power SYBR Green I master mix with ROX as a reference dye (Bio-Rad Laboratories, 10032041, CA, USA). The thermal cycling conditions were 3 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Relative expressions of each messenger RNA (mRNA) were analyzed using a comparative CT (2ΔΔCT) method.
Immunocytochemical analysis of NRF2 protein
Cells in each group were processed for immunocytochemistry according to the protocol described previously (Amin et al. 2014) with some modifications. Briefly, cells from each category were washed three times with warm DPBS and fixed in 4% paraformaldehyde at 4 °C overnight. Following that, cells were washed three times with DPBS and permeabilized using 0.2% Triton X-100 (Sigma-Aldrich) in PBS at room temperature for 1 h. After blocking with 3% normal donkey serum in DPBS for 1 h at room temperature, cells were incubated with specific primary antibody against NRF2 (1:100 dilution, SC-722; Santa Cruz Biotechnology, Dallas, TX) overnight at 4 °C. The next day, cells were washed twice with 0.05% Tween 20 (P9416, Sigma) in DPBS and incubated further with FITC conjugated donkey anti-rabbit secondary antibody (1:200 dilution, SC-2090, Santa Cruz Biotechnology, Dallas, TX) for 1 h at 37 °C in the dark. After washing twice with DPBS, cells were incubated with 5 ng/mL 4′,6′-diamidino-2-phenylindole (DAPI; Vector Laboratories) in DPBS to stain the nucleus for 5 min in the dark and fluorescence microscopy was performed with a Nikon Eclipse Ti-S microscope (Nikon Instruments Inc., Tokyo, Japan) using a green and blue-fluorescence filter and images were acquired by NIS Elements software.
Intracellular ROS detection
Intracellular ROS accumulation in different treated and control groups was assessed by 6-carboxy-2′,7′-dichlorodihydrofluorescin diacetate (H2DCFDA; Sigma-Aldrich) according to the protocol described elsewhere (Sohel et al. 2017). Briefly, following the treatments according to Fig. 1, GCs from each group were incubated with 400 μL of 15 μM H2DCFDA for 20 min in the dark at 37 °C. Cells were then washed twice with PBS and images were immediately captured with a Nikon Eclipse Ti-S microscope (Nikon Instruments Inc., Tokyo, Japan) using a green-fluorescence filter and images were acquired by NIS Elements software. For quantitative analysis, the mean fluorescence intensity of five nonoverlapping fields in each well was measured using ImageJ software. Data are presented as mean ± SD.
Assessment of lipid accumulation
Accumulation of lipid droplets in GCs of different groups was assessed using Oil Red O stain (Sigma-Aldrich, O1391, St. Louis, MO) using previously described protocol (Prastowo et al. 2016). Briefly, approximately 1 × 105 viable GCs were added to each well of a 24-well culture plate and treated according to the experimental design (Fig. 1). Following the incubation period, media was completely removed and GCs were washed twice with PBS and fixed in 10% formalin for 30 min at room temperature. Immediately prior to the staining, Oil Red working solution was prepared and then incubated with cells for 40 min at room temperature. Following staining, cells were washed three to four times with distilled water and visualized by a Leica DMIL LED inverted microscope (Leica Microsystem Ltd., Germany).
Mitochondrial activity
Mitochondrial activity in bovine GCs was determined using MitoTracker1 Red CMXRos (M7512, Invitrogen) according to the previous protocol (Prastowo et al. 2016) with small modifications. GCs from each group were incubated with 200-nM MitoTracker red dye for 45 min, followed by two washings with PBS and were then fixed overnight at 4 °C with 4% formaldehyde. The next day, fixed cells were mounted with Vectashield (H-1200) containing DAPI (Vector Laboratories). The mitochondrial activity of GC samples was visualized under a laser scanning confocal microscope (LSM 710; Carl Zeiss) using specific excitation lasers (579–599 nm). A constant level of laser gain (master gain = 700), pinhole (1 μm) and pixel size (1024 × 1024) were applied during image acquisition aim to allow image fluorescence signal comparison. Resulted images were then processed using ZEN 2011 software (Carl Zeiss). For quantitative analysis, the mean fluorescence intensity of five nonoverlapping fields in each well was measured using ImageJ software. Data are presented as mean ± SD.
Statistical analysis
Minimum of three biological replicates were used in each experiment. Normal distribution of data was analyzed and results are presented in Electronic Supplementary Material Fig. S3. Statistical differences between treatment group means (quantitative variables) were analyzed by one-way ANOVA, followed by multiple pairwise comparisons (Tukey test, α = 0.05). For statistical analyses and graphs, Microsoft Excel 2016 was used. Data are expressed as mean ± SD of three biological replicates. Differences were considered as significant at p < 0.05.
Results
Pretreatment with SFN increased cell viability and reduced cytotoxicity during oxidative stress
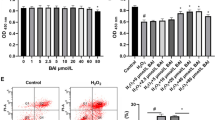
GCs from both treated and control groups were examined using the inverted microscope for their morphological changes after each treatment. Based on microscopic observation, there were no visible differences between the cells of group 1 and group 2 in terms of growth and confluence (Fig. 2a–e). It clearly showed that 10 μM of SFN was not detrimental to cells in terms of growth and proliferation (Fig. 2b). However, treatment with 500 μM H2O2 caused oxidative damage to the cells, which was characterized by a higher number of dead cells in group 3 (Fig. 2c). Preincubation with SFN clearly protects the cells from oxidative stress, which is evidenced by apparently healthy cells and presence of a lower number of dead cells in group 4 (Fig. 2d). On the other hand, upon exposure to H2O2, administration of SFN did not appear to be beneficial to the protection of GCs from oxidative damage in group 5 (Fig. 2e).

Effects of different treatments on cellular morphology, viability and cytotoxicity. a–e Representative micrographs of different treatments (n = 3), scale bar 200 μm. f Cells were subjected to trypan blue dye exclusion test and were counted using a hemocytometer for cell viability. g Cytotoxicity of different treatments was determined using WST-1 kit using an ELISA reader. Data are presented as mean ± SEM (n = 3) of three independent experiments. Different superscript letters (a, b) denote a significant difference between groups, such that groups not sharing a similar letter are significantly different from each other (p < 0.001)
Analysis of cell viability using the trypan blue dye exclusion test revealed that treatment of cells with 10 μM SFN had no significant effects on the viability of GCs in group 2, whereas oxidative stress induced by H2O2 caused a significant reduction of viability (< 40%) of GCs in group 3. However, more than 80% cell viability was observed in GCs when pretreated with SFN in group 4 and an almost similar viability as group 3 was observed when SFN was added along with H2O2 in group 5 (Fig. 2f). Therefore, these results indicated that pretreatment of GCs with SFN protects them from H2O2 induced cytotoxicity.
Moreover, the protecting and/or rescuing ability of SFN from H2O2 induced cytotoxicity on GCs was determined using WST-1 reagent and results are presented in Fig. 2g. Accordingly, results revealed that 10 μM SFN was not toxic to GCs, whereas 500 μM H2O2 appeared to be extremely cytotoxic. While SFN pretreatment effectively decreases H2O2 induced cytotoxicity in group 4, co-incubation of SFN could not reduce H2O2 induced cytotoxicity in group 5. Both the viability and cytotoxicity tests suggest that SFN pretreatment effectively protects GCs from H2O2 induced oxidative stress; however, SFN cannot rescue GCs from already existing oxidative stress.
Expression of NRF2 and KEAP1 in relation to SFN and H2O2 treatment
To understand whether the protective effect of SFN against H2O2 induced oxidative insult is associated with activation of the Nrf2-ARE antioxidant pathway, we first investigated the expression levels of NRF2 and its negative regulator KEAP1 at the mRNA level under different treatment conditions. Analysis of qRT-PCR data revealed that, compared to untreated control, expression of NRF2 was significantly increased in the cells of group 2 or group 3 (Fig. 3a). The highest expression of NRF2 was observed in the cells when they were pretreated with SFN and subjected to oxidative stress (group 4). Surprisingly, no differences were observed in the expression of NRF2 mRNA levels between group 3 and group 5 (Fig. 3a).

(a-f) Expression of NRF2, KEAP1 and candidate genes downstream to NRF2 activation. Expression of genes was quantified using qRT-PCR and relative abundance was analyzed using comparative CT (2ΔΔCT) method. Data are presented as mean ± SD of three biological replicates. Different superscript letters (a, b, c, d) denote a significant difference between groups (p < 0.05) as determined by Student’s t test
As expected, the expression of KEAP1 significantly decreased in all treatment groups compared to untreated control, i.e., group 1 (Fig. 3b). However, no significant difference in KEAP1 expression was found within SFN, H2O2 and SFN-resc groups. Moreover, significantly lower KEAP1 expression was observed in the SFN-pretreated group compared to any other groups (Fig. 3b).
Expression of antioxidant genes
A significant increase in the expression and nuclear translocation of NRF2 strongly suggests the activation of the Nrf2-ARE pathway including the activation of antioxidant enzymes. Therefore, we investigated the expression of selected antioxidant genes, namely, PRDX1, CAT, TXN1 and SOD1 as the downstream genes of NRF2 activation. Interestingly, almost similar expression patterns were observed for all candidate genes (Fig. 3c–f). Significantly higher expression of all candidate genes was observed in group 4 compared to any other groups (Fig. 3c–f). Although the expression of candidate genes was found to be significantly increased in groups 3 and 5 in comparison to group 1, no expression differences were observed between groups 3 and 5 (Fig. 3c–f). These results also demonstrated that the protective effects of SFN against oxidative stress are firmly associated with the activation of the Nrf2-ARE pathway.
Nuclear translocation of Nrf2 during SFN and H2O2 treatment
Immunofluorescence staining and fluorescence microscopy revealed that under the basal condition, in group 1, NRF2 was predominantly localized in the cytoplasm (Fig. 4a, a′, a″). Interestingly, in group 2, NRF2 was found to be evenly localized in both the cytoplasm and nuclear fraction (Fig. 4b, b′, b″). In contrast, NRF2 was almost absent in the cytoplasm and translocated into the nucleus in the presence of H2O2 (Fig. 4c, c′, c″, e, e′, e″). Although, NRF2 was predominantly localized in the nucleus in group 4, plenty of NRF2 was localized in the cytoplasm as well (Fig. 4d, d′, d″). This is perhaps due to the fact that pretreatment with SFN helped cells to promote their NRF2 activity against oxidative stress and hence, they recovered quicker than the cells of other groups (group 3 and group 5) within a given period of time.

Nuclear translocation of NRF2. Following different treatments, GCs were subjected to immunocytochemistry to localize NRF2 proteins. Images were acquired with a fluorescence microscope using a green fluorescence filter. Representative images showing the location of NRF2 proteins in green (a–e), while nuclear staining with DAPI (a′–e′) and merged image of NRF2 protein and DAPI (a″–e″). Scale bar 100 μm. White arrows indicate NRF2 in the cytoplasm, while red arrows represent the translocated NRF2 in the nucleus
Preincubation with SFN decreases ROS and lipid accumulation in GCs
Here, we next investigated the accumulation of ROS in the GCs of different treatment groups (Fig. 5a–e). A low level of ROS was detected in the cells of group 1 (Fig. 5a). When GCs were treated with 10 μM SFN, it slightly increased the accumulation of ROS in group 2. However, exposure to 500 μM H2O2 significantly increased ROS accumulation in the GCs of group 3, which is evidenced by a higher intensity of the fluorescence signal (Fig. 5c) and mean fluorescence signal quantified by ImageJ (Fig. 5k). Compared to group 3, accumulation of ROS was significantly decreased in group 4 (Fig. 5d), but no such decrease was observed in group 5 (Fig. 5e).

ROS and lipid droplet accumulation in GCs after different treatments according to Fig. 1. GCs were (a–e) washed and loaded with H2DCFDA (15 μM for 20 min) and visualized under a fluorescent microscope, scale bar 100 μm. f–j Following treatments, GCs were loaded with Oil Red O stain working solution (for 40 min), washed several times and visualized with an inverted microscope (n = 3). Scale bar 100 μm. k Mean fluorescence intensity was quantified by ImageJ software from five nonoverlapping fields per well; experiments were performed in triplicate, ***p < 0.001. Data are presented as mean ± SD
Accumulation of lipid droplets is the hallmark of cellular stress. Considering the results of ROS accumulation, we next investigated whether pretreatment of GCs with SFN can inhibit the formation of lipid droplets upon oxidative stress. In agreement with the ROS accumulation results, considerable differences were found in lipid accumulation when the cells were pretreated with SFN (Fig. 5f–j). The lowest level of lipid droplets was observed in cells of groups 1 and 2. Exposure to H2O2 considerably enhanced the lipid droplet formation in group 3 (Fig. 5h), while pretreatment with SFN reduced the formation of lipid droplet accumulation in group 4 (Fig. 5i). However, administration of SFN at the time of H2O2 exposure could not reduce the level of lipid droplet accumulation in group 5 (Fig. 5j).
Pretreatment of GCs with SFN enhances mitochondrial activity upon exposure to H2O2
While the mitochondrial electron transport chain is the major source of cellular ROS, mitochondria are one of the major targets of ROS-induced oxidative damage. Therefore, we investigated the activity of mitochondria in GCs of different groups and results are shown in Fig. 6a–e. High mitochondrial activity was observed in groups 1 and 2 as evidenced by higher fluorescence signal intensity detected by confocal microscopy (Fig. 6a, b) and mean fluorescence signals quantified by ImageJ (Fig. 6f) compared to other groups. Cells exposed to H2O2 exhibited lower mitochondrial activity in group 3, while pretreatment of GCs with SFN increased the activity of mitochondria in group 4 (Fig. 6c, d). However, SFN had no effects on enhancing mitochondrial activity when administrated along with H2O2 in group 5 (Fig. 6e).

Mitochondrial activity in GCs of different treatment groups. GCs were treated according to the experimental plan and 100 nM Mito Tracker red dye was added and incubated for 45 min at 37 °C. Images were acquired with a fluorescence microscope using a red filter. a–e show active mitochondria in red, while a′–e′ show nuclear staining with DAPI in blue and a″–e″ show merged image of active mitochondria and nucleus staining. Scale bar 50 μm, n = 3
Discussion
In a follicular microenvironment, GCs are the major cellular component and perform several important functions during follicular growth and ovulation (Yada et al. 1999). Shortly before ovulation, after the LH surge, massive recruitment of inflammatory cells (endothelial cells, macrophages and neutrophils) at the site of follicular rupture results in overproduction of ROS and acute inflammation, which facilitate follicular rupture, indicating that GCs are also exposed to a higher concentration of ROS and face oxidative stress during the ovulatory process. On the other hand, overproduction of ROS is the major cause of GC apoptosis in follicular atresia (Agarwal et al. 2012). It appears that there is an existence of a complex relationship between GCs and oxidative stress. Antioxidants, both enzymatic and nonenzymatic, play a crucial role in survival and maintaining function and integrity of GCs under oxidative stress. Several chemical compounds including SFN and melatonin have been found to be effective in resulting the induction of phase II antioxidant enzymes to exert protective effects against oxidative insult in different cell types (Guerrero-Beltrán et al. 2012). Among them, SNF has been studied intensively not only because of its extraordinary ability to induce phase II antioxidant responsive element (ARE) genes but also because it shows anticarcinogenic properties. Numerous studies have suggested that a certain concentration of SFN is cytotoxic to cancer cells (Chaudhuri et al. 2007; Chuang et al. 2007) but not in normal cells (Nair et al. 2007). Most importantly, SFN exerts protective effects in several normal cells against different stressors including oxidative stress-induced damages (Liu et al. 2013; He et al. 2014; Li et al. 2015).
In the present study, we investigated the protective effects of SFN pretreatment in GCs before induction of oxidative stress induced by H2O2. To the best of our knowledge, this is the first study to report SFN modulation of the Nrf2-ARE pathway under oxidative stress in ovarian GCs. Our results indicated that a specific concentration of SFN (10 μM) is noncytotoxic to GCs; however, 500 μM of H2O2 was extremely cytotoxic and causes oxidative stress-induced cell death. Cytotoxic effects due to oxidative stress induced by H2O2 in different cell types were found to be dose and cell type dependent. Furthermore, the working concentration of both chemicals largely depends on the experimental setup and the objectives of the study. For instance, a wide range of concentrations, starting from 1 up to 50 μM, of SFN has been used in different cell types to induce the protective effects in vitro (Gamet-Payrastre et al. 2000; Kim et al. 2016; Juengel et al. 2016; Sohel et al. 2017; Visalli et al. 2017). We used 10 μM SFN to induce protective effects against oxidative stress in vitro. The protective effects of SFN against vestibular Schwannoma have been shown in both in vitro and in vivo experiments (Kim et al. 2016), in which 10 μM SFN was used in in vitro and intraperitoneal administration of 25 mg/kg SFN in mice. It is important to note that the in vivo concentration of SFN primarily depends on the experimental setup and model organisms. Existing literature suggests that it may range from 5 to 500 mg/kg (Fuentes et al. 2015). It has been shown that a standard serving of broccoli sprout contains 7.5 mg of SFN, which is sufficient to modulate inflammatory and vascular prostanoid responses in healthy humans (Medina et al. 2015).
We found that 10 μM SFN can effectively protect the GCs from oxidative stress induced by H2O2 as supported by the results of different genetic, phenotypic and stress marker parameters. It is important to note that SFN exerts positive effects against oxidative stress only when GCs were pretreated with it before exertion of oxidative stress. However, administration of SFN at the time of oxidative stress cannot rescue GCs from oxidative damage. This protective effect seems to be mediated via activation of the Nrf2-ARE oxidative stress response pathway, which is evident by a significant higher expression of both NRF2 mRNA and its downstream antioxidant genes in SFN-treated cells compared to control. In addition, translocation of NRF2 into the nucleus after SFN treatment strongly suggests the activation of the Nrf2-ARE antioxidant pathway.
During evolution, the cell has developed an inherent defense mechanism against different stress or stimuli, i.e., activation of the Nrf2-ARE signaling pathway to produce a wide range of antioxidant enzymes (Poljsak et al. 2013), where NRF2 orchestrates the whole process. In our study, expression of NRF2 was significantly increased (5-fold) in cells treated with SFN compared to control. However, a sharp increase in the expression of NRF2 (15–20-fold) and loss of expression of KEAP1 was observed in cells exposed to H2O2 and notably, the highest expression of NRF2 was observed when the cells were pretreated with SFN and then exposed to H2O2 (Fig. 3a). The dramatic shift in the abundance of NRF2 and KEAP1 transcripts upon H2O2 exposure strongly suggests the disassociation of Nrf2-Keap1 interaction and release of NRF2 due to the existence of oxidative stress. It has been proposed that SFN seems to have both direct and indirect actions on the expression of NRF2 at the transcriptome level. An indirect way of NRF2 upregulation by SFN could be the mechanism of positive autoregulation of NRF2 (NRF2 protein upregulates its own transcript) (Kwak et al. 2002). A direct action of SFN on NRF2 was suggested by Su et al. (2014), where increased NRF2 mRNA expression was evident by decreasing the methylation ratio of the NRF2 gene promoter. SFN-mediated upregulation of NRF2 is consistent with our results, where we showed that SFN treatment significantly increases the expression of NRF2 in GCs, although this increase was significantly lower when cells were in a stressed condition, i.e., exposed to H2O2.
The endogenous antioxidant defense mechanism comprises a wide range of phase II detoxification enzymes such as glutathione transferase, superoxide dismutase (SOD), catalase (CAT) and heme oxygenase (HO-1). Expression of these antioxidant enzymes is regulated by a promoter sequence known as ARE, which is modulated by Nrf2 transcription factor (Petri et al. 2012; Ma 2013). The antioxidant potential of SFN is related to its free radicals scavenging abilities through increasing the levels and activities of these cellular noncatalytic antioxidant proteins. Thus, we examined gene expression of several major antioxidant/detoxificant enzymes, including PRDX1, CAT, TXN and SOD1, involved in cytoprotection against oxidative stress. Among the phase II antioxidant enzymes, SOD and CAT are considered as first-line cellular antioxidant defense enzymes. Under oxidative stress condition, SOD first reacts with highly reactive superoxide anion (O2˙¯) to generate either ordinary molecule O2 or H2O2, which is later catalyzed to H2O by CAT (Hemachandra et al. 2016). We found the expression of all phase II antioxidant genes that we investigated were higher in SFN-pretreated cells, whereas co-incubation of SFN with H2O2 could not enhance the expression of these genes when there was no SFN pretreatment.
In order to start the transcription of phase II antioxidant enzymes, NRF2 has to be translocated into the nucleus and interact with small MAF proteins to form a bonding with the ARE (Itoh et al. 1997). A hallmark of oxidative stress is the translocation of cytosolic NRF2 into the nucleus (Ma 2013). In addition, accumulation and translocation of NRF2 would also indicate the activation of NRF2 and induction of the Nrf2-ARE pathway induced by SFN (Liu et al. 2013). It is therefore of interest to examine the pattern of distribution of NRF2 in cells in response to SFN and H2O2 treatment. Our data confirm that nuclear translocation of NRF2 does occur in GCs; however, it showed an interesting pattern of NRF2 distribution and translocation into the nucleus in response to SFN and H2O2 exposure. For instance, when the cells were treated with 10 μM SFN, NRF2 was evenly distributed in both the cytoplasm and nucleus, which indicates that nuclear translocation occurred to a limited extent and activation of the Nrf2-ARE pathway as well. On the other hand, complete accumulation and translocation of NRF2 into the nucleus were observed when the cells were exposed to H2O2 (Fig. 4c, c′, c″), which is the indicative parameter of oxidative stress in different cell types (Buckley et al. 2003; Peng et al. 2015; Dou et al. 2016; Jin et al. 2016). However, oxidative stress induced by H2O2 in SFN-pretreated cells showed an interesting NRF2 distribution pattern; i.e., most of the NRF2 localized into the nucleus, while others started to relocalize into the cytoplasm, which may indicate that these cells are well equipped to handle oxidative stress and did not face similar stress like other groups (groups 3 and 5) did. It is perhaps due to the protective effects mediated via partial activation of the Nrf2-ARE pathway that has been reported in many cell types (Liu et al. 2013; Li et al. 2015; Dong et al. 2016; Qin et al. 2016). In contrast, when there was no SFN pretreatment (group 5), administration of SFN at the time of H2O2-induced oxidative stress could not create any difference compared to H2O2 in terms of localization of the NRF2 protein. These results clearly indicate that the degree of nuclear translocation of NRF2 is closely related to the extent of cellular stress faced by GCs during H2O2 exposure and the ability to activate survival pathways to handle oxidative stress.
Excessive production of intracellular ROS is considered as one of the major stress markers in oxidative stress. Indeed, the modulation of intracellular ROS is crucial to maintaining cellular homeostasis, as different ROS levels can initiate a different biological response (Gorrini et al. 2013). At the low or moderate level, ROS may act as a signaling molecule that modulates cell differentiation and proliferation and can activate stress-responsive survival pathways (Janssen-Heininger et al. 2008; Saeed-Zidane et al. 2017). However, excessive production of ROS damages major cellular components including DNA, protein and lipid, which ultimately cause cell death (Gorrini et al. 2013). In our study, we observed a slightly higher accumulation of ROS in SFN-treated cells than the cells in the control group. This slight alteration of redox status may induce the activation of the Nrf2-ARE pathway in GCs that eventually started the transcription of phase II antioxidant enzymes and subsequently, cells acquired an enhanced ability to maintain redox status and finally protect the cells against H2O2-induced oxidative stress in the SFN-pretreated group. On the other hand, a significantly higher accumulation of ROS was observed in cells of only H2O2- and SFN-post-treated groups, which a perhaps the main reason for higher cell death in these groups. Elevated ROS level can induce lipogenesis by activating SREBPs in cells under culture condition resulting in a higher accumulation of lipid droplets (Lee et al. 2015). Similar results were reported previously by our group, where we showed that excess generation of ROS, due to serum addition, resulted in a higher lipid accumulation in bovine embryos (Prastowo et al. 2016). Therefore, it is not surprising that we found an elevated accumulation of lipid droplets in H2O2- and SFN-post-treated groups. However, in this study, we did not check the underlying mechanism of lipid accumulation in GCs in response to SFN and H2O2 treatment, which warrants further experimental attention. In addition, higher mitochondrial activity in SFN-pretreated cells and comparatively lower mitochondrial activity in SFN-post-treated cells were observed. In an active mitochondrion, mitochondrial antioxidant defense enzymes such as glutathione peroxidase and manganese SOD scavenge ROS produced during ATP synthesis (Dröse and Brandt 2012). However, under stress conditions, excessive generation of ROS can impair the capacity of this defense system, resulting in mitochondrial damage. Importantly, damaged mitochondria can produce even more ROS due to an impaired electron transportation chain (Dröse and Brandt 2012). Subsequently, excessive ROS can decrease the mitochondrial membrane potential and facilitate the release of cytochrome C and apoptosis inducing factors that eventually activate the pro-apoptotic cascade resulting in cell death through apoptosis. It seems that SFN pretreatment effectively protects the integrity of mitochondrial membrane potential in GCs through scavenging of excessive ROS by inducing the production of antioxidant enzymes during oxidative stress induced by H2O2.
In summary, pretreatment of GCs with a low concentration of SFN maintains the cellular redox homeostasis during oxidative stress via modulating the antioxidant defense system by activating the Nrf2-ARE pathway. Hence, GCs acquired an enhanced ability to handle oxidative stress during ovulation, which ultimately maintains the integrity and function of GCs. By contrast, the addition of SFN at the time of oxidative stress failed to maintain redox homeostasis in GCs and proved to be useless. Therefore, it is speculated that SFN preincubation effectively protects GCs from oxidative insult but cannot rescue when added at the time of H2O2-induced oxidative stress.
Change history
06 November 2018
There is an error in the original publication of this paper. Figures 1-6 were shown in the wrong version, thus corrected figures provided were in this article.
References
Agarwal A, Gupta S, Sharma RK (2005) Role of oxidative stress in female reproduction. Reprod Biol Endocrinol 3(28). https://doi.org/10.1186/1477-7827-3-28
Agarwal A, Aponte-Mellado A, Premkumar BJ et al (2012) The effects of oxidative stress on female reproduction: a review. Reprod Biol Endocrinol 10:49. https://doi.org/10.1186/1477-7827-10-49
Amin A, Gad A, Salilew-Wondim D et al (2014) Bovine embryo survival under oxidative-stress conditions is associated with activity of the NRF2-mediated oxidative-stress-response pathway. Mol Reprod Dev 81:497–513
Buckley BJ, Marshall ZM, Whorton A (2003) Nitric oxide stimulates Nrf2 nuclear translocation in vascular endothelium. Biochem Biophys Res Commun 307:973–979. https://doi.org/10.1016/S0006-291X(03)01308-1
Chaudhuri D, Orsulic S, Ashok BT (2007) Antiproliferative activity of sulforaphane in Akt-overexpressing ovarian cancer cells. Mol Cancer Ther 6:334–345. https://doi.org/10.1158/1535-7163.MCT-06-0404
Chuang LT, Moqattash ST, Gretz HF et al (2007) Sulforaphane induces growth arrest and apoptosis in human ovarian cancer cells. Acta Obstet Gynecol Scand 86:1263–1268. https://doi.org/10.1080/00016340701552459
Dong Z, Shang H, Chen YQ et al (2016) Sulforaphane protects pancreatic acinar cell injury by modulating Nrf2-mediated oxidative stress and NLRP3 inflammatory pathway. Oxidative Med Cell Longev 2016:1–12. https://doi.org/10.1155/2016/7864150
Dou T, Yan M, Wang X et al (2016) Nrf2/ARE pathway involved in oxidative stress induced by paraquat in human neural progenitor cells. Oxidative Med Cell Longev 2016:1–8. https://doi.org/10.1155/2016/8923860
Dröse S, Brandt U (2012) Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol 748:145–169. https://doi.org/10.1007/978-1-4614-3573-0_6
Duffy DM, Stouffer RL (2003) Luteinizing hormone acts directly at granulosa cells to stimulate periovulatory processes: modulation of luteinizing hormone effects by prostaglandins. Endocrine 22:249–256. https://doi.org/10.1385/ENDO:22:3:249
Frisard M, Ravussin E (2006) Energy metabolism and oxidative stress: impact on the metabolic syndrome and the aging process. Endocrine 29:27–32. https://doi.org/10.1385/ENDO:29:1:27
Fuentes F, Paredes-Gonzalez X, Kong A-NT (2015) Dietary glucosinolates sulforaphane, phenethyl isothiocyanate, indole-3-carbinol/3,3′-diindolylmethane: anti-oxidative stress/inflammation, Nrf2, epigenetics/epigenomics and in vivo cancer chemopreventive efficacy. Curr Pharmacol Rep 1:179–196. https://doi.org/10.1007/s40495-015-0017-y
Gamet-Payrastre L, Li P, Lumeau S et al (2000) Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res 60:1426–1433. https://doi.org/10.1158/0008-5472.can-06-0139
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12:931–947. https://doi.org/10.1038/nrd4002
Guerrero-Beltrán CE, Calderón-Oliver M, Pedraza-Chaverri J, Chirino YI (2012) Protective effect of sulforaphane against oxidative stress: recent advances. Exp Toxicol Pathol 64:503–508. https://doi.org/10.1016/j.etp.2010.11.005
He C, Li B, Song W et al (2014) Sulforaphane attenuates homocysteine-induced endoplasmic reticulum stress through Nrf-2-driven enzymes in immortalized human hepatocytes. J Agric Food Chem 62:7477–7485. https://doi.org/10.1021/jf501944u
Hemachandra LPMP, Chandrasekaran A, Melendez JA, Hempel N (2016) Regulation of the cellular redox environment by superoxide dismutases, catalase, and glutathione peroxidases during tumor metastasis. Springer International Publishing, New York, pp 51–79
Hong F, Freeman ML, Liebler DC (2005) Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol 18:1917–1926. https://doi.org/10.1021/tx0502138
Itoh K, Chiba T, Takahashi S et al (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322. https://doi.org/10.1006/bbrc.1997.6943
Janssen-Heininger YMW, Mossman BT, Heintz NH et al (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 45:1–17. https://doi.org/10.1016/j.freeradbiomed.2008.03.011
Jin X, Wang K, Liu H et al (2016) Protection of bovine mammary epithelial cells from hydrogen peroxide-induced oxidative cell damage by resveratrol. Oxidative Med Cell Longev 2016:1–15. https://doi.org/10.1155/2016/2572175
Juengel E, Maxeiner S, Rutz J et al (2016) Sulforaphane inhibits proliferation and invasive activity of everolimus-resistant kidney cancer cells in vitro. Oncotarget 7:85208–85219. https://doi.org/10.18632/oncotarget.13421
Kim BG, Fujita T, Stankovic KM et al (2016) Sulforaphane, a natural component of broccoli, inhibits vestibular schwannoma growth in vitro and in vivo. Sci Rep 6:36215. https://doi.org/10.1038/srep36215
Klaunig JE, Kamendulis LM, Hocevar BA (2010) Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol 38:96–109. https://doi.org/10.1177/0192623309356453
Kwak M-K, Itoh K, Yamamoto M, Kensler TW (2002) Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol 22:2883–2892. https://doi.org/10.1128/mcb.22.9.2883-2892.2002
Lee J, Homma T, Kurahashi T, et al (2015) Oxidative stress triggers lipid droplet accumulation in primary cultured hepatocytes by activating fatty acid synthesis. Biochem Biophys Res Commun 464:229–235. https://doi.org/10.1016/j.bbrc.2015.06.121
Li B, Kim DS, Yadav RK et al (2015) Sulforaphane prevents doxorubicin-induced oxidative stress and cell death in rat H9c2 cells. Int J Mol Med 36:53–64. https://doi.org/10.3892/ijmm.2015.2199
Liu H, Smith AJO, Lott MC et al (2013) Sulforaphane can protect Lens cells against oxidative stress: implications for cataract prevention. Invest Opthalmol Vis Sci 54:5236. https://doi.org/10.1167/iovs.13-11664
Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. https://doi.org/10.1146/annurev-pharmtox-011112-140320
Medina S, Domínguez-Perles R, Moreno DA et al (2015) The intake of broccoli sprouts modulates the inflammatory and vascular prostanoids but not the oxidative stress-related isoprostanes in healthy humans. Food Chem 173:1187–1194. https://doi.org/10.1016/J.FOODCHEM.2014.10.152
Nair S, Li W, Kong A-NT (2007) Natural dietary anti-cancer chemopreventive compounds: redox-mediated differential signaling mechanisms in cytoprotection of normal cells versus cytotoxicity in tumor cells. Acta Pharmacol Sin 28:459–472. https://doi.org/10.1111/j.1745-7254.2007.00549.x
Peng S, Yao J, Liu Y et al (2015) Activation of Nrf2 target enzymes conferring protection against oxidative stress in PC12 cells by ginger principal constituent 6-shogaol. Food Funct 6:2813–2823. https://doi.org/10.1039/C5FO00214A
Petri S, Korner S, Kiaei M (2012) Nrf2/ARE signaling pathway: key mediator in oxidative stress and potential therapeutic target in ALS. Neurol Res Int 2012:1–7. https://doi.org/10.1155/2012/878030
Poljsak B, Suput D, Milisav I (2013) Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxidative Med Cell Longev 2013:1–11. https://doi.org/10.1155/2013/956792
Prastowo S, Amin A, Rings F et al (2016) Fateful triad of reactive oxygen species, mitochondrial dysfunction and lipid accumulation is associated with expression outline of the AMP-activated protein kinase pathway in bovine blastocysts. Reprod Fertil Dev 49:193–202. https://doi.org/10.1071/RD15319
Qin W-S, Deng Y-H, Cui F-C (2016) Sulforaphane protects against acrolein-induced oxidative stress and inflammatory responses: modulation of Nrf-2 and COX-2 expression. Arch Med Sci 12:871–880. https://doi.org/10.5114/aoms.2016.59919
Saeed-Zidane M, Linden L, Salilew-Wondim D et al (2017) Cellular and exosome mediated molecular defense mechanism in bovine granulosa cells exposed to oxidative stress. PLoS One 12(11):e0187569. https://doi.org/10.1371/journal.pone.0187569
Shkolnik K, Tadmor A, Ben-Dor S et al (2011) Reactive oxygen species are indispensable in ovulation. Proc Natl Acad Sci U S A 108:1462–1467. https://doi.org/10.1073/pnas.1017213108
Sohel MMH, Cinar MU (2015) Advancement in molecular genetics to understand the molecular reproduction of livestock—follicular development. Res Agric Livest Fish 1:47–60. https://doi.org/10.3329/ralf.v1i1.22355
Sohel MMH, Hoelker M, Noferesti SS et al (2013) Exosomal and non-exosomal transport of extra-cellular microRNAs in follicular fluid: implications for bovine oocyte developmental competence. PLoS One. https://doi.org/10.1371/journal.pone.0078505
Sohel MH, Cinar MU, Kalibar M et al (2016) Appropriate concentration of hydrogen peroxide and sulforaphane for granulosa cells to study oxidative stress in vitro. J Biotechnol 231:S24. https://doi.org/10.1016/j.jbiotec.2016.05.104
Sohel MMH, Konca Y, Akyuz B et al (2017) Concentration dependent antioxidative and apoptotic effects of sulforaphane on bovine granulosa cells in vitro. Theriogenology 97:17–26. https://doi.org/10.1016/j.theriogenology.2017.04.015
Strober W (2001) Trypan blue exclusion test of cell viability. Curr Protoc Immunol Appendix 3:Appendix 3B. https://doi.org/10.1002/0471142735.ima03bs21
Visalli G, Facciolà A, Bertuccio MP et al (2017) In vitro assessment of the indirect antioxidant activity of Sulforaphane in redox imbalance vanadium-induced. Nat Prod Res 31:2612–2620. https://doi.org/10.1080/14786419.2017.1286485
Yada H, Hosokawa K, Tajima K et al (1999) Role of ovarian theca and granulosa cell interaction in hormone production and cell growth during the bovine follicular maturation process. Biol Reprod 61:1480–1486
Zhang M, An C, Gao Y et al (2013) Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol 100:30–47. https://doi.org/10.1016/j.pneurobio.2012.09.003
Acknowledgements
The authors would like to thank Mr. Heinz Biörnsen and Mr. Mahmut Kaliber for their assistance during sample collection.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Statement on the welfare of animals
This article does not contain any studies with live animals performed by any of the authors.
Additional information
The original version of this article was revised: There is an error in the Original Publication of this paper. Figures 1-6 were unupdated, thus corrected figures provided below:
Electronic supplementary material
Fig. S1
The purity of GCs. GCs were collected according to the protocol mentioned in the “Materials and methods” section. Specific primers targeting FSHR, CYP17A1 and GAPDH were designed using primer3web version 4.0.4 (http://bioinfo.ut.ee/primer3/). Primer sequences are available in Supplementary Table 1. The polymerase chain reaction (PCR) was set in a thermocycler with thermal cycle conditions of pre-incubation at 95°C for 5 min, 35 cycles of denaturation at 95°C for 30 s, annealing at 55°C FSHR, 56°C GAPDH and 57°C CYP17A1 for 30 s, extension at 72°C for 1 min and a final extension at 72°C for 10 min. The corresponding PCR products were mixed with loading buffer and loaded into 2% agarose gel stained with Ethidium bromide (EtBr) and visualized using Chemidoc XRS (Bio-Rad) instrument (BIO-RAD, München, Germany) to detect the presence or absence of gene-specific bands. The results showed that the GC specific marker gene FSHR was detected at a higher level as indicated by a strong band, while no expression was detected for the CYP17A1 gene in GCs. These results confirmed that the isolation procedure of GCs was completely free of contamination from thecal cells. (PDF 117 kb)
Fig. S2
Expression of NRF2, KEAP1 and CASP3 in response to 10 μM SFN at different time points. Approximately 6 × 105 viable pure GCs were seeded in each well of a 6-well plate and grown until 40-50% confluence. Cells were then treated with 10 μM SFN for different time points (6 h, 12 h, 24 and 48 h) to identify the appropriate exposure time of SFN. We checked three different genes; NRF2 (inducer of the NRF2 antioxidant pathway), KEAP1 (negative controller of NRF2) and CASP3 (an indicator of apoptosis induction). The results showed that the expression of NRF2 was significantly higher in all exposure time points compared to the 6 h time point, however, the highest expression was observed in the 12 h and 24 h time points. On the other hand, the expression of KEAP1 was more or less similar across the time points. In addition, the expression of CASP3 was similar to control at 12 h and 24 h, however, it significantly increased at the 48 h time point. The expression analysis of these genes revealed that both the 12 h and 24 h exposure times are suitable for activating the antioxidant defense system without creating any damage to GCs. As most of the mammalian cell cycle is completed within 20-24 h, we decided to expose the GCs against 10 μM SFN for 24 h. (PDF 154 kb)
Fig. S3
Normality test of qPCR data. Normality of the qPCR data (Ct value) was tested using Microsoft Excel 2016. For this, all Ct values were organized in ascending order (smallest to largest). Afterwards, the cumulative distribution factor (CDF) was calculated using the 1 / (2*total count) formula for the first number, CDF1+2 / (2*total count) for the second number, CDF2+2 / (2*total count) for the third number and so on. Expected value was calculated using the NORM.INV(CDF, mean, standard deviation) formula and Z-score was calculated using the NORM.S.INV(CDF) formula. Normal probability plot for the Ct values shows that the data appear to be very close to being normally distributed. The actual Ct values (red) match very closely with the normally distributed (blue) Ct values that clearly indicate the data are normally distributed. (PDF 161 kb)
Table S1
(PDF 91.4 kb)
Rights and permissions
About this article

Cite this article
Sohel, M.M.H., Amin, A., Prastowo, S. et al. Sulforaphane protects granulosa cells against oxidative stress via activation of NRF2-ARE pathway. Cell Tissue Res 374, 629–641 (2018). https://doi.org/10.1007/s00441-018-2877-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-018-2877-z