
Abstract
RNAscope® technology provided by Advanced Cell Diagnostics (ACD) allows the detection and evaluation of coinciding mRNA expression profiles in the same or adjacent cells in unprecedented quantitative detail using multicolor fluorescent in situ hybridization (FISH). While already extensively used in thinly sectioned material of various pathological tissues and, to a lesser extent, in some whole mounts, we provide here a detailed approach to use the fluorescent RNAscope method in the mouse inner ear and thick brain sections by modifying and adapting existing techniques of whole mount fluorescent in situ hybridization (WH-FISH). We show that RNAscope WH-FISH can be used to quantify local variation in overlaying mRNA expression intensity, such as neurotrophin receptors along the length of the mouse cochlea. We also show how RNAscope WH-FISH can be combined with immunofluorescence (IF) of some epitopes that remain after proteinase digestion and, to some extent, with fluorescent protein markers such as tdTomato. Our WH-FISH technique provides an approach to detect cell-specific quantitative differences in developing and mature adjacent cells, an emerging issue revealed by improved cellular expression profiling. Further, the presented technique may be useful in validating single-cell RNAseq data on expression profiles in a range of tissue known or suspected to have locally variable mRNA expression levels.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The inner ear presents one of the most complex cellular assemblies of the mammalian body, with two different types of hair cells and seven distinct types of supporting cells organized into a single hearing organ, the organ of Corti (Jahan et al. 2015a). In addition, at least two types of spiral ganglion cells innervate the organ of Corti (OC) with distinctly distributed afferent nerve fibers (Dabdoub et al. 2016). These different cell types are not only tightly spaced and distinct in radial direction but are also known to show longitudinal expression variation in several genes related to tonotopic organization (Fritzsch et al. 2016). These variations can be seen in locally distinct reactions to gene mutations (Jahan et al. 2015b; Nakano et al. 2012). Available data suggest that variable levels of expression at the cellular level of more than one gene need to be analyzed to reveal local cellular variability and its dynamic development changes (Imayoshi and Kageyama 2014; Isomura et al. 2017; Jahan et al. 2015b). Such information could help us understand how local variations of common cell types, such as length variation of hair cells from base to apex (Booth et al. 2018), develop in order to guide restoration of lost cell types. We present here initial data suggesting that such approaches are possible using RNAscope whole mount fluorescent in situ hybridization (here referred to as WH-FISH) alone or in combination with other techniques (Wang et al. 2012).
Since its introduction only 6 years ago (Wang et al. 2012), RNAscope in situ hybridization (ISH) methods have been cited in over 500 papers covering a wide range of applications, tissues and species but with thus far limited use in the brain and sensory organs. Given the design of multiple RNA binding sites, RNAscope surpasses traditional immunofluorescent approaches to detect mRNA in pathological tissue (Bingham et al. 2017). In addition, RNAscope ISH can reveal microRNA species (miRNA; Yin 2018) and possibly also long non-coding RNA (lncRNA; Soares et al. 2017), both of which have emerged as major regulators of early development (Alvarez-Dominguez and Lodish 2017; Booth et al. 2018; Kersigo et al. 2011; Weston et al. 2011). Fluorescent in situ hybridization with RNAscope multiplexing promises to evaluate distribution of several species of mRNA (Wang et al. 2015) that can go beyond enzymatically amplified multicolor fluorescent in situ hybridization approaches (Hauptmann 2015; Hauptmann et al. 2016) by adding quantification. Indeed, multiplexed WH-FISH could allow complete quantification of local mRNA within a given set of cells, if properly calibrated using quantitative real-time PCR (qPCR; Bustin et al. 2010).
Taking cues from existing RNAscope FISH protocols (Gross-Thebing et al. 2014; Morrison et al. 2017), we modified our well-established inner ear whole mount ISH protocol (Duncan et al. 2015; Fritzsch et al. 2016) to provide a simple whole mount FISH (WH-FISH) protocol that can be combined with immunofluorescence (IF). We show that this WH-FISH mouse inner ear protocol can be applied not only across stages of ear development but also to other tissues as well. We also show that RNAscope WH-FISH can be combined with immunofluorescence and endogenous fluorescent protein markers like tdTomato.
Materials and methods
Animal model(s)
Mice derived from a mixed C57BL/6J and BALB/cJ background collected at embryonic day 18.5 (E18.5) or postnatal day 2 (P2) were used for this study. The mice were wild type and exhibited no phenotype. In addition, we used knock in TrkC-tdTomato reporter line to analyze its possible combination with the RNAscope method (Orefice et al. 2016; Zheng et al. 2014). Specifically, the TrkC-tdTomato mouse was generated by inserting a myristolyation-signal-tagged tdTomato after the ATG start sequence in exon1 of the TrkC gene to visualize the expression of TrkC (Bai et al. 2015). The University of Iowa, Decibel Therapeutics, or Harvard University Institutional Animal Care and Use Committee reviewed and approved all procedures used here.
Tissue preparation
Transcardial perfusion/fixation
Mice were anesthetized with 1.25%, v/v 2,2,2-tribromoethyl alcohol (Avertin) at 0.025 ml/g body weight IP. After assessing the depth of anesthesia with toe/tail pinch and ocular reflex, the mice were secured in a supine position to a large dissecting dish using straight pins through the extremities. The skin was superficially incised on the midline from below the xiphoid process to the neck being sure to avoid major vasculature. The thoracic cavity was exposed by cutting the diaphragm laterally from the xiphoid process and then rostrally avoiding the heart and lungs. The heart was perfused with 4% paraformaldehyde (PFA) in 1× phosphate-buffered saline (PBS) through the left ventricle using a 30G ½″ needle and the Masterflex C/L peristaltic pump. Forceps were used to puncture the right atrium to allow drainage. The perfusion continued until the liver appeared blanched, the body was stiff and the drainage fluid ran clear.
The head was removed at the neck with blunt scissors and skin incised from caudal to rostral along the midline to expose the sagittal suture of the skull. Using forceps for stabilization, a razor blade was used to hemisect the head. Tissues of interest were then removed (brain and/or inner ear with the stapes footplate removed) in 0.4% PFA in 1× PBS and immersion fixed in 4% PFA with gentle agitation at room temperature for 1 h.
Dissection/sectioning
All microdissection and sectioning were done in 0.4% PFA in RNase-free 1× PBS to ensure RNase-free conditions. Using fine instruments, the cartilaginous capsule was removed from around the inner ear and cochlea exposed. The cochlea was removed and cut into three sections: apex, middle turn and base. For best results, the tectorial membrane was removed. The vestibular end organs were also dissected for optimal penetration and flat-mount ability.
Brain tissue was prepared after perfusion by cutting the cerebellum with a razor blade by hand along the sagittal plane and collected in a non-treated 24-well tissue culture plate. The thickness of the sections varied between 200 and 500 μm as assessed using the z-axis motor of the confocal microscope. Using brain tissue slabs of variable thicknesses allowed us to estimate penetration depth.
RNAscope assay
Pretreatment and probe hybridization
Dissected tissue was washed in 0.1% Tween 20 in RNase-free 1× phosphate-buffered saline (PBT) three times followed by dehydration in a graded methanol (MeOH)/PBT series (50%, 75% MeOH-PBT, 100% MeOH) with gentle agitation at room temperature for 5 min each. Tissue was safely stored in 100% MeOH before continuing if needed (up to 3 weeks at − 20 °C appeared not to affect the assay). The tissue was rehydrated in a reverse MeOH/PBT series and washed three times for 5 min each in PBT. The tissue was treated with Protease III solution (ACD 322337, 1×) according to type (7–17 min, see Table 1) with gentle agitation at room temperature. Probe solution (ACD; see chart in “Amplification and Signal Enhancement”) was pre-warmed (40 °C for 10 min), cooled (room temperature 10 min), gently mixed and briefly centrifuged to ensure that there was no precipitate in solution. Up to three different probes/channels were used (C1-C3). Probes were diluted per manufacturer's instructions (C1 is a 1× probe, C2 and C3 probes are 50× concentrations). To make a multiplex probe mixture at 1× concentration, the C2 and C3 probes were mixed 1:50 with the C1 probe. Only enough probe solution to cover the tissue was used. The tissue was incubated for 8–10 h (see Table 1) at 40 °C with light agitation (or periodic manual agitation) being sure that they remained free-floating in the tube.
Amplification and label probe hybridization
All incubation periods were carried out with gentle agitation (best practice) or periodic manual tube inversion. Probe solution was recovered in a new tube and stored at 4 °C for reuse up to 6 months. Tissue was washed in RNase-free 0.2× saline sodium citrate (SSC) three times at room temperature for 15 min each. The tissue was refixed in 4% PFA for 10 min to ensure tissue integrity and then rewashed with 0.2× SSC three times for 5 min each at room temperature. SSC solution was replaced with Amp1 (ACD, 1×) pre-amplifier hybridization solution and allowed to incubate for 35 min at 40 °C before washing as above with SSC. SSC was replaced by Amp2 (ACD, 1×) signal enhancement solution and incubated for 20 min at 40 °C before washing as above. SSC was replaced by Amp3 (ACD, 1×) amplifier hybridization solution and incubated for 35 min at 40 °C before washing as above. SSC was replaced by Amp4 (ACD, Alt A-C 1× varieties, see Table 2) label probe hybridization solution and incubated for 20 min before washing with SSC three times for 5 min each at room temperature. The tissue was protected from light from this point forward.
Immunofluorescence and nuclear stain
Following the RNAscope protocol, it is possible to add immunofluorescence (IF) and/or nuclear staining as previously described (Duncan et al. 2015). We only used antibodies previously shown to recognize their epitope after proteinase K digestion. Tissue was washed several times in 1× PBT and blocked with 5% normal goat serum in 1× PBS containing 0.25% Triton X-100 for 1 h at room temperature. The tissue was incubated in primary antibodies anti-acetylated tubulin (Millipore-Sigma T7451, 1:800) and/or anti-myosin-VIIa (Myo7a, Proteus Biosciences 25–6790, 1:200) for 48 h (ear) or 72 h (brain) at 4 °C. The tissue was washed for several hours and blocked again as above before incubating in secondary antibodies Alexa Fluor IgG (H + L) goat anti-mouse and/or goat anti-rabbit (Invitrogen, 1:500) and Hoechst nuclear stain (Polysciences Inc., 5 μg/ml) for 12 h (ear) or 24 h (brain) at 4 °C. Finally, the tissue was washed for several hours in 1× PBS before viewing.
Combination with endogenous fluorescent proteins
Fluorescent proteins of various colors are widely used to label specific proteins and their distribution within cells. In situ hybridization requires some proteinase digestion to allow penetration, particularly in thicker tissue. While our initial attempts using the digestion times recommended by previous RNAscope whole mount techniques resulted in near complete loss of such signals, shortening the period to 7 min allowed us to retain enough fluorescent protein to verify that the RNAscope in situ signal overlapped with the fluorescent protein signal in spiral ganglion neurons (TrkC-tdTomato in Fig. 3).
Imaging
Prolong Diamond Anti-fade Mountant (Invitrogen) was used to preserve all fluorescent signals while imaging per manufacturer’s instructions. Imaging was with a Leica SP5 or Zeiss 800 confocal microscope using appropriate filter settings (Table 3) for the various dyes used. Images were taken with 10× (0.5 NA), 20× (0.7 NA) and 40× (1.4 NA) lenses as single stacks with spacing varying between 1 and 4 μm between images. Image stacks were collapsed to generate single images.
Quantification of puncta
Images were analyzed in ImageJ software (v1.51p) using the threshold function. The settings for the lower and upper threshold levels were set at 70 and 255, respectively, to minimize risk of false positives but still capture faint puncta signal. After, the analyzed particle function was applied with pixel size (pixel2) set from 0-∞, circularity from 0 to 1.0 to include all puncta. For image presentation, the selected puncta are displayed from the resulting output. To mimic the MIQE qPCR approach (Bustin et al. 2010) that requires for each data point to have three biological replicas and three technical replicas, we recommend to quantify puncta in at least three equal-sized squares for a given area as indicated (Fig. 4) to generate distribution differences per area. We recommend using collapsed images of complete stacks through the tissue (Fig. 5) to avoid any depth segregation of signals due to variable penetration.
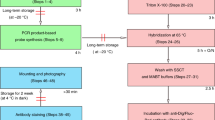
Workflow
Preparation Day 1 1× PBT, nuclease free: 1× PBS + 0.1% Tween 20 4% PFA in PBS, pH 7 Sterile nuclease free 1× PBS Perfusion materials MeOH solutions in PBT (50%, 75%, 100%) Heat water bath or incubator to 40 °C with gentle agitator/rocker if available Sample Collection/Fixation Transcardially perfuse mice with 4% PFA in PBS Remove and microdissect tissue of interest in 0.4% PFA being sure to remove all extraneous tissue Immersion-fix tissue in 4% PFA/PBS for 1 h (ear) or 2 h (thick brain sections) at room temperature (RT) on rocker Transition Wash tissue 3 × 10 min in PBT Pretreatment/Dehydration Dehydrate tissue through a graded MeOH/PBT series: 50%, 75% for at least 5 min each at RT on rocker Transfer tissue to 100% MeOH for at least 5 min. Note: Can store tissue in 100% MeOH at -20 °C if needed Rehydrate Reverse MeOH/PBT series as above Transition Wash tissue 3 × 5 min in PBT at RT on rocker Protease Digestion Protease III 1× solution (see Table 1 for incubation times) at RT on rocker Stop Digestion Wash tissue 3 × 5 min in PBT at RT on rocker. Start to warm probes (see below) Prepare Probe(s) Pre-warm probes to 40 °C (10 min) and then cool down to RT (~10 min). This ensures there is no precipitation Invert probe solutions to mix. Briefly spin the C2 and/or C3 probes to collect liquid at the bottom of tubes Mix Probes: Channel-1 probe (C1; 1×), Channel-2 (C2; 50×), Channel-3 (C3; 50×). To make a multiplex mixture at 1× concentration, mix C2 and/or C3 probes 1:50 with C1 probe Hybridization Incubate tissue in probe mix at 40 °C with light rocking. See suggested hybridization times in Table 1 Preparation Day 2 0.2× SSC: filute 20× SSC with sterile nuclease free dH2O Heat water bath or incubator to 40 °C Equilibrate FL-AMP1–4 reagents (1×) to RT and invert to mix before use If using, remove Prolong Diamond Mountant from −20 °C and place at 4 °C for 24h or RT for ~6h Probe Removal Recover the probe solution in a new tube; these can be reused. Label and store at 4 °C for up to 6 months Wash Wash the tissue 3 × 15 min with 0.2× SSC at RT on rocker Postfixation Fix tissue again with 4% PFA in PBS at RT for 10 min Wash Wash the tissue 3 × 5 min with 0.2× SSC at RT on rocker Preamplifier Hybridization Remove 0.2× SSC and replace it with Amp1 solution (1×) Incubate at 40 °C for 35 min with gentle rocking (best practice) or invert tube regularly Wash Wash the tissue 3 × 15 min with 0.2× SSC at RT on rocker. Signal Enhancement Remove 0.2× SSC and replace it with Amp2 solution (1×) Incubate at 40 °C for 20 min with gentle rocking (best practice) or invert tube regularly Wash Wash the tissue 3 × 15 min with 0.2× SSC at RT on rocker Amplifier Hybridization Remove 0.2× SSC and replace it with Amp3 solution (1×) Incubate at 40 °C for 35 min with gentle rocking (best practice) or invert tube regularly Wash Wash the tissue 3 × 15 minutes with 0.2× SSC at RT on rocker Label Probe Hybridization Remove 0.2× SSC and replace it with Amp4 solution (1×). [Use desired reagent, Alt A, B, or C]. LIGHT SENSITIVE! Protect from light! Remember the combos: ALTA=C1(488), C2(550), C3(647); ALTB=C1(550), C2(488), C3(647); ALTC=C1(550), C2(647), C3(488) Incubate at 40 °C for 20 min with gentle rocking (or invert tube occassionally) Wash Wash the tissue 3 × 5 min with 0.2× SSC at RT on rocker. Note: Temporary storage (1 day) in 0.2× SSC is possible before mounting IF/Counterstain Remove 0.2× SSC and add fluorescent nuclear staining solution if desired and/or proceed with immunofluorescence (IF) according to protocol Wash Wash the tissue 3 × 5 min with 1× PBS at RT on rocker Mount/View Warm the bottle of ProLong® Diamond Antifade Mountant to room temperature before use Remove excess moisture from the slide before ProLong® Diamond Antifade Mountant is added. Flat mount samples Cure the sample: Place the mounted sample/slide on a flat, dry surface for 24h at room temperature protected from light before viewing |
Trouble shooting
The most critical issue is the length of fixation and proteinase digestion of tissue. The latter is crucial for complete penetration of WH-FISH and also defines how much residual fluorescence remains of the fluorescent proteins we tested here (tdTomato). We found that digestion exceeding 7 min resulted in loss of endogenous fluorescence of dtTomato. We recommend comparing density of puncta at various depths within the tissue to confirm uniform expression of a control gene that is known to be expressed at a similar level throughout the tissue (we used Cdc42 as a uniform marker) to verify complete penetration of the tissue. Irrespective to this possible penetration problem, we noted that puncta comparison of overlapping mRNA expression is always possible as penetration issues appear to be similar for all probes that we tested. Sections revealed that penetration was variable in sections over 200 μm thickness. Our protocol should not be used for thicker sections without modifying digestion and hybridization times.
Reagent order information
Company | REF/product no. | Description |
|---|---|---|
ACD | 462771-C2 | Mm-Myo7a-C2 RNAscope probe |
ACD | 312251 | Mm-Ntf3 C1 RNAscope probe |
ACD | 473041-C3 | Mm-Cdc42-C3 RNAscope probe |
ACD | 423611-C3 | Mm-Ntrk2-C3 RNAscope probe (TrkB) |
ACD | 423621 | Mm-Ntrk3 RNAscope probe (TrkC) |
ACD | 322337 | Protease III solution (1×) |
ACD | 320851 | RNAscope fluorescent multiplex detection reagents (1×); FL-AMP1 (320852), FL-AMP2 (320853), FL-AMP3 (320854), FL-AMP4-Alt A (320855), FL-AMP4-Alt B (320856), FL-AMP4-Alt C (320857), DAPI |
Polysciences Inc. | 09460 | Hoechst nuclear stain |
Invitrogen | P36961 | Prolong Diamond Anti-fade Mountant |
Proteus Biosciences Inc. | 25-6790 | Myosin-VIIa rabbit polyclonal antibody |
Millipore-Sigma | T7451 | Anti-acetylated mouse monoclonal tubulin antibody |
Eppendorf | 022600044 | 2.0 ml Eppendorf safe-lock tubes, Sterile, Rnase-free |
MBP | 7003 | Rnase Away |
IDT | 11-05-02-02 | Nuclease-free water |
Thermo Fisher Scientific | BP3994 | 10× PBS |
RPI | P20370 | Tween 20 |
Millipore-Sigma | T8787 | Triton X-100 |
MP Biomedicals | 191,356 | Normal Goat Serum (NGS) |
Corning Inc. | 351,147 | Falcon polystyrene microplates, non-treated, 24 wells |
Affymetrix Inc. | 19,943 | Paraformaldehyde solution, 4% in PBS |
Roche | 11666681001 | 20× SSC buffer |
Thermo Fisher Scientific | A32731 A32727 A32728 | Alexa Fluor conjugated secondary antibodies 488, 555, and 647 nm |
Results
RNAscope WH-FISH allows multicolor imaging of overlapping gene expression in the whole mounted cochlea
Multiplex RNAscope for Myo7a, Ntf3 and Cdc42 not only shows expression in the organ of Corti as expected (Dabdoub et al. 2016; Jahan et al. 2010; Ueyama et al. 2014) but also reveals the distinct distribution in different cell types (Figs. 1 and 2). At the stage analyzed (P2), the neurotrophin Ntf3 was most profoundly expressed in supporting cells adjacent to inner hair cells (Figs. 1 and 2) with somewhat lesser expression surrounding the outer hair cells. RNAscope signal was not related to cell packing density as the greater epithelial ridge has the highest nuclear density revealed with Hoechst staining (Figs. 1 and 2). Both overviews (Fig. 1) and cellular details (Fig. 2) can be imaged.

Overview images of z-axis collapsed stacks of RNAscope WH-FISH of the organ of Corti (OC). Note that all three probes (Myo7a in a; Ntf3 in b; Cdc42 in c) show specific targeted expression in the organ of Corti (OC). However, Cdc42 as an actin polymerization regulating protein is present in all cells at variable levels. Combining all three labelings shows overlap with discrete cellular distribution that is not resolvable at this level of magnification (d). WH-FISH for Ntf3 (green) can be combined with anti-Myo7a immunofluorescence (lilac in e) and Hoechst nuclear staining (blue in e) but we noticed a slight reduction in overall WH-FISH signal (compare Ntf3 signal in b and e) leaving more prominent labeling around inner hair cells in the GER. Abbreviations: Cdc42 cell division cycle 42, GER greater epithelial ridge, IHC inner hair cell row, Myo7a myosin VIIa, Ntf3 Neurotrophin 3, OC organ of Corti, OHC outer hair cell row

RNAscope WH-FISH can be combined with Hoechst nuclear staining (a). The in situ signal for Myo7a (b), Ntf3 (c) and Cdc42 (d) is not associated with cellular density that is highest in the greater epithelial ridge (GER, shown with nuclear staining in a and e). Myo7a (b, e) labels individual hair cells of the organ of Corti (OC) shown as three rows of outer hair cells and one row of inner hair cells. At P2, the neurotrophin Ntf3 is predominantly distributed in the supporting cells around the inner hair cells, forming a red line separating the single row of inner hair cells from the three rows of outer hair cells (c). There is a strong Ntf3 expression in a row of supporting cells flanking the outermost row of outer hair cells (c, e). Note that Hoechst stain is false colored as cyan in the single image (a) but is blue in the combined image (e) for better visibility of the combined colors. See Fig. 1 for abbreviations
RNAscope can be combined with certain antibodies. Here we show that anti-tubulin and anti-myosin-VIIa (Myo7a) staining can be combined with RNAscope in situ hybridization allowing visualization for both the RNAscope signal in neurons and immunostaining of processes to reveal their projection to distinct targets such as the hair cells of the organ of Corti (Fig. 3).

Whole mount RNAscope in situ hybridization (WH-FISH) can be combined with immunofluorescence and endogenous fluorescent protein such as tdTomato. The overview (a) and details (c, d) show that TrkC-tdTomato fluorescent protein can be combined with immunofluorescence using antibodies against tubulin. Note that the spiral ganglion neuron (SGN) cell bodies are nearly exclusively labeled with RNAscope (b) and that the RNAscope granules overlap with the tdTomato labeled spiral ganglion neurons cell bodies (SGN in a, d). Rendering RNAscope signal in such overlapping labeling into white dots (d) allows easier quantification. Note that anti-tubulin staining shows the radial fibers (RF) extending to the organ of Corti (OC) and also shows the intraganglionic spiral bundle (IGSB) at the borders of the spiral ganglion neurons. Abbreviations: IGSB intraganglionic spiral bundle, OC organ of Corti, RF radial fibers, SGN spiral ganglion neurons. Squares in a indicate approximate location of images c and d
The fluorescent protein tdTomato is affected by the needed digestion with a loss of much of the signal except for labeled cell bodies. We obtained this residual labeling only after we slightly reduced the digestion time to 7 min while maintaining a similar level of penetration of the RNAscope reagents.
The importance of whole mount imaging of two or more RNAscope signals in whole mounts can be revealed in cases with apparent longitudinal expression level changes. For example, neurotrophins are known to go through rapid embryonic and neonatal expression level changes (Fariñas et al. 2001; Pirvola et al. 1992), whereas no such changes have been reported previously for neurotrophin receptors (Fritzsch et al. 2016; Ylikoski et al. 1993). We show here that TrkB and TrkC receptors seemingly display both a longitudinal (Fig. 4 a, b) and a local radial (Fig. 4 c, c’, c”) expression variation. Our preliminary quantification using ImageJ supports this impression through puncta counts (Figs. 4 and 5). Moreover, using similar criteria, the number of puncta varies with the magnification of the lens used (Figs. 4 and 5). These preliminary data need further exploration at different stages to see how it compares to the well-known ligand expression dynamics of neurotrophin ligands in the ear as revealed by qPCR (Bailey and Green 2014).

Comparison of co-localized neurotrophin receptors using different magnifications. Note that the WH-FISH double labeling shows an apparent gradient within the spiral ganglion neuron populations (SGN). Specifically, at E18.5, there appears to be a stronger expression of neurotrophic tyrosine kinase receptor 2 (Ntrk2, here labeled as TrkB) in the apex (a, more green dots), whereas neurotrophic tyrosine kinase receptor 3 (Ntrk3 here labeled as TrkC) is more prominent in the base (b, more lilac dots). Note that many green (TrkB) dots are outside the area of the SGN and thus should not be quantified. In the middle turn (c), there appears to be also a minor advantage for TrkC dots (c’, c”). Importantly, the total number of dots recognized will co-vary with the magnification and resolution of the lens (20 × 0.75 NA for a, b; 40 × 1.35 NA for c, c’, c”). Square in “b” shows one of three areas used for detailed quantification of dots in Fig. 5

Quantification of TrkB and TrkC mRNA (dots) detected by the RNAscope WH-FISH method in E18.5 mouse cochlea. Note that the apical (apex) and basal (base) spiral ganglion cochlear regions seemingly have different overall expression levels at this stage (left, 20 × 0.75 NA, “a” in Fig. 4). An increase in microscope lens magnification and resolution (NA) dramatically increases the number of detectable dots for the same area by an order of magnitude (right, 40 × 1.35 NA, “b” in Fig. 4). Therefore, use of the highest possible resolution lenses is recommended for all quantification
We also show that the RNAscope WH-FISH method is applicable to other tissues. Specifically, we show that the distribution of the probes shown above for the whole mount organ of Corti works for thick sections of the cerebellum (Fig. 6). In addition, combining RNAscope with Hoechst nuclear staining allows the identification of RNA signal distribution in the highly ordered cerebellar cortex (Fig. 6).

Ntf3 expression in a thick sagittal section of the cerebellum. The RNAscope technique developed here works equally well on thick sections to show the expression of the neurotrophin Ntf3 in the molecular layer (ML) of the cerebellum. The proliferating granule cell precursors in the external granule layer (EGL) show very limited expression compared to the cells in the molecular layer (ML) that at this stage includes Purkinje cells
Discussion
The increased insights into expression differences in adjacent cells and their complex regulation at the RNA and protein level (Booth et al. 2018) require novel techniques that go beyond expression profiles of isolated cells (Burns and Stone 2017) to establish variation of nearby cells. We use the distinct topological distribution of the nearly two-dimensional mammalian organ of Corti (Jahan et al. 2015a) to demonstrate that expression of different genes can be visualized (Figs. 1, 2, 3 and 4) and quantified (Figs. 4 and 5). In addition, we show that the basic principle developed in the whole mounted ear can be expanded to thick brain sections (Fig. 6). Data generated with WH-FISH approaches can establish locally distinct levels of expression of one or more genes needed to understand how local cell-specific expression levels contribute to global cellular variability and its orchestrated development. Ideally, one not only likes to see the expression of a given gene in a given cell, such as a spiral ganglion neuron but also analyze at the same time its peripheral innervation to verify how a given profile relates to distinct projection development such as nerve fibers growing to outer hair cells (Fig. 3).
Our data show that as with traditional in situ hybridization (Duncan et al. 2015), RNAscope WH-FISH can be used for multiplexed fluorescence and can be combined with immunofluorescence and endogenous fluorescence from reporter proteins labeling. Whole mounts of the ear allow both an overview of expression profiles (Figs. 1 and 3) and detailed expression profiles (Figs. 2, 3 and 4) that allow quantitative analysis of RNAscope puncta (dots) using various techniques for spot counting previously described (Morrison et al. 2017; Yin 2018). Antigens previously identified to be retained after proteinase K digestion (Duncan et al. 2015) were also found to be retained after RNAscope (Protease III) digestion as shown by myo7a and tubulin immunofluorescent labeling (Figs. 1 and 3).
RNAscope can be combined with endogenous fluorescence from reporter proteins like tdTomato (Fig. 3). However, we noted a signal reduction allowing only to image the cell bodies while fibers that were clearly visible in control animals did not retain enough fluorescent protein to be imaged. Modifications of the digestion beyond the range we tested (7–17 min) could allow finding a compromise that allows retaining such proteins without compromising penetration of the in situ probes.
The importance of whole mount RNAscope (WH-FISH) labeling becomes apparent when comparing the gradients of expression level differences of two neurotrophin receptors (TrkB and TrkC) in all spiral ganglion neurons but also local variation in intensity (Figs. 4 and 5). Quantification of puncta (dots) using readily available ImageJ software supports this notion by showing slightly more TrkC and TrkB dots in the middle and the base (Fig. 5) but obviously more TrkB compared to TrkC dots in the apex (Fig. 5). We also noted that the same settings for quantification could generate large differences in total dots correlated if resolution was not considered. Higher resolution power of the microscope lens used is the reason for such differences (20 × 0.75 NA versus 40 × 1.4 NA).
For unbiased quantification, we suggest using at least three (preferentially six) equally sized squares of a given region imaged per each of at least three biological samples to mimic the suggested quantification of the MIQE protocol for qPCR (Bustin et al. 2010). Ideally, one should be able to compare the mean expression levels obtained with a normalized approach for RNAscope with those obtained from qPCR assays of the same tissue type. More quantification is needed to consolidate this initial finding and verify if Trks show rapid changes in expression level comparable to those of neurotrophins in the ear (Bailey and Green 2014; Fritzsch et al. 2016) that were previously unrecognized due to technique restraints (Ylikoski et al. 1993).
Conclusion
We modified previous RNAscope whole mount (WH-FISH) approaches to allow multiplex staining of various gene expressions in the neonatal mouse ear and thick brain sections. We show that the RNAscope assay can be combined with certain antibodies to label nerve fibers and cochlear hair cells but may reduce endogenous fluorescent proteins due to proteinase digestion. It is possible that other epitopes will remain immunopositive after digestion and can be used for immunofluorescent labeling in combination with RNAscope WH-FISH and nuclear staining. We demonstrate the importance of whole mount approaches to mRNA expression profiling that should be adaptable to various tissue types and fields of research.
References
Alvarez-Dominguez JR, Lodish HF (2017) Emerging mechanisms of long noncoding RNA function during normal and malignant hematopoiesis. Blood 130:1965–1975
Bai L, Lehnert BP, Liu J, Neubarth NL, Dickendesher TL, Nwe PH, Cassidy C, Woodbury CJ, Ginty DD (2015) Genetic identification of an expansive mechanoreceptor sensitive to skin stroking. Cell 163:1783–1795
Bailey EM, Green SH (2014) Postnatal expression of neurotrophic factors accessible to spiral ganglion neurons in the auditory system of adult hearing and deafened rats. J Neurosci 34:13110–13126
Bingham V, McIlreavey L, Greene C, O’Doherty E, Clarke R, Craig S, Salto-Tellez M, McQuaid S, Lewis C, James J (2017) RNAscope in situ hybridization confirms mRNA integrity in formalin-fixed, paraffin-embedded cancer tissue samples. Oncotarget 8:93392–93403
Booth KT, Azaiez H, Smith RJH, Jahan I, Fritzsch B (2018) Intracellular regulome variability along the organ of Corti: evidence, approaches, challenges and perspective. Front Genet 9:156
Burns JC, Stone JS (2017) Development and regeneration of vestibular hair cells in mammals. Semin Cell Dev Biol 65. Elsevier:96–105
Bustin SA, Beaulieu J-F, Huggett J, Jaggi R, Kibenge FS, Olsvik PA, Penning LC, Toegel S (2010) MIQE precis: practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. Springer
Dabdoub A, Fritzsch B, Popper AN, Fay RR (2016) The primary auditory neurons of the mammalian cochlea. Springer, New York
Duncan JS, Elliott KL, Kersigo J, Gray B, Fritzsch B (2015) Combining whole-mount in situ hybridization with neuronal tracing and immunohistochemistry. In Situ Hybridization Methods 339–352
Fariñas I, Jones KR, Tessarollo L, Vigers AJ, Huang E, Kirstein M, De Caprona DC, Coppola V, Backus C, Reichardt LF (2001) Spatial shaping of cochlear innervation by temporally regulated neurotrophin expression. J Neurosci 21:6170–6180
Fritzsch B, Duncan JS, Kersigo J, Gray B, Elliott KL (2016) Neuroanatomical tracing techniques in the ear: history, state of the art, and future developments. Auditory and Vestibular Research: Methods and Protocols 243–262
Gross-Thebing T, Paksa A, Raz E (2014) Simultaneous high-resolution detection of multiple transcripts combined with localization of proteins in whole-mount embryos. BMC Biol 12:55
Hauptmann G (2015) In situ hybridization methods. Springer
Hauptmann G, Lauter G, Söll I (2016) Detection and signal amplification in zebrafish RNA FISH. Methods 98:50–59
Imayoshi I, Kageyama R (2014) bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron 82:9–23
Isomura A, Kori H, Kageyama R (2017) Segmentation genes enter an excited state. Dev Cell 43:121–123
Jahan I, Kersigo J, Pan N, Fritzsch B (2010) Neurod1 regulates survival and formation of connections in mouse ear and brain. Cell Tissue Res 341:95–110
Jahan I, Pan N, Elliott KL, Fritzsch B (2015a) The quest for restoring hearing: understanding ear development more completely. Bioessays 37:1016–1027
Jahan I, Pan N, Kersigo J, Fritzsch B (2015b) Neurog1 can partially substitute for Atoh1 function in hair cell differentiation and maintenance during organ of Corti development. Development 142:2810–2821
Kersigo J, D'Angelo A, Gray BD, Soukup GA, Fritzsch B (2011) The role of sensory organs and the forebrain for the development of the craniofacial shape as revealed by Foxg1-cre-mediated microRNA loss. Genesis 49:326–341
Morrison JA, McKinney MC, Kulesa PM (2017) Resolving in vivo gene expression during collective cell migration using an integrated RNAscope, immunohistochemistry and tissue clearing method. Mech Dev 148:100–106
Nakano Y, Jahan I, Bonde G, Sun X, Hildebrand MS, Engelhardt JF, Smith RJ, Cornell RA, Fritzsch B, Bánfi B (2012) A mutation in the Srrm4 gene causes alternative splicing defects and deafness in the Bronx waltzer mouse. PLoS Genet 8:e1002966
Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, Ginty DD (2016) Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell 166:299–313
Pirvola U, Ylikoski J, Palgi J, Lehtonen E, Arumäe U, Saarma M (1992) Brain-derived neurotrophic factor and neurotrophin 3 mRNAs in the peripheral target fields of developing inner ear ganglia. Proc Natl Acad Sci 89:9915–9919
Soares RJ, Maglieri G, Gutschner T, Diederichs S, Lund AH, Nielsen BS Holmstrøm K (2017) Evaluation of fluorescence in situ hybridization techniques to study long non-coding RNA expression in cultured cells. Nucleic Acids Res 46:e4
Ueyama T, Sakaguchi H, Nakamura T, Goto A, Morioka S, Shimizu A, Nakao K, Hishikawa Y, Ninoyu Y, Kassai H Suetsugu S, Koji T, Fritzsch B, Yonemura S, Hisa Y, Matsuda M, Aiba A, Saito N (2014) Maintenance of stereocilia and apical junctional complexes by Cdc42 in cochlear hair cells. J Cell Sci 127:2040–2052
Wang F, Flanagan J, Su N, Wang L-C, Bui S, Nielson A, Wu X, Vo H-T, Ma X-J, Luo Y (2012) RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 14:22–29
Wang H, Su N, Wang L-C, Wu X, Bui S, Chang K-J, Nielsen A, Vo H-T, Luo Y, Ma X-J (2015) Multiplex fluorescent RNA in situ hybridization via RNAscope. In Situ Hybridization Methods 405–414
Weston MD, Pierce ML, Jensen-Smith HC, Fritzsch B, Rocha-Sanchez S, Beisel KW, Soukup GA (2011) MicroRNA-183 family expression in hair cell development and requirement of microRNAs for hair cell maintenance and survival. Dev Dyn 240:808–819
Yin VP (2018) In situ detection of microRNA expression with RNAscope probes. RNA Detection Springer, pp 197–208
Ylikoski J, Pirvola U, Moshnyakov M, Palgi J, Arumäe U, Saarma M (1993) Expression patterns of neurotrophin and their receptor mRNAs in the rat inner ear. Hear Res 65:69–78
Zheng Y, Bai L, Ginty DD (2014) RNA profiling of major DRG sensory neuron subtypes: a resource for identification of genes involved in mechanosensation. J Gen Physiol 144. Rockefeller Univ Press 1114 FIRST AVE, 4TH FL, New York, NY 10021 USA:17A
Acknowledgments
This work was supported by INI (Roy J. Carver Chair in Neuroscience), R01 (DC005590), the University of Iowa (sabbatical to BF) and Decibel Therapeutics. The authors express their gratitude to Dr. David Ginty (Harvard University) for providing the TrkC-tdTomato-expressing mice used for Fig. 3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The University of Iowa, Decibel Therapeutics, or Harvard University Institutional Animal Care and Use Committee reviewed and approved all procedures used here.
Rights and permissions
About this article

Cite this article
Kersigo, J., Pan, N., Lederman, J.D. et al. A RNAscope whole mount approach that can be combined with immunofluorescence to quantify differential distribution of mRNA. Cell Tissue Res 374, 251–262 (2018). https://doi.org/10.1007/s00441-018-2864-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-018-2864-4