
Abstract
Testicular Sertoli cells make a niche for the division and differentiation of germ cells. Sertoli cells respond to increased follicle-stimulating hormone (FSH) and testosterone (T) levels at the onset of puberty by producing paracrine factors which affect germ cells and trigger robust onset of spermatogenesis. Such paracrine support to germ cells is absent during infancy, despite Sertoli cells being exposed to high FSH and T within the infant testis. This situation is similar to certain cases of male idiopathic infertility where post-pubertal Sertoli cells fail to support germ cell division and differentiation in spite of endogenous or exogenous hormonal support. Defective Sertoli cells in such individuals may fail to express the full complement of their paracrine repertoire. Identification and supplementation with such factors may overcome Sertoli cells deficiencies and help trigger quantitatively and qualitatively normal differentiation of germ cells. To this end, we compared the transcriptome of FSH- and T-treated infant and pubertal monkey Sertoli cells by DNA microarray. Expression of Wnt3, a morphogen of the Wnt/β-catenin pathway, was higher in pubertal Sertoli cells relative to infant Sertoli cells. Transgenic mice were generated by us in which Wnt3 expression was curtailed specifically in post-pubertal Sertoli cells by shRNA. Subfertility and oligozoospermia were noticed in such animals with low Wnt3 expression in post-pubertal Sertoli cells along with diminished expression of Connexin43, a gap-junctional molecule essential for germ cell development. We report that the FSH- and T-targetedf Wnt3 governs Sertoli cell-mediated regulation of spermatogenesis and hence is crucial for fertility.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alarming declines in sperm counts and increasing incidences of male infertility reported from several countries have become a major concern across the globe (Irvine 1998; Krausz 2011; Sharpe 2012). This remains an unresolved crisis because a large proportion of male infertility and the vast majority of subfertility, with or without diminished sperm counts, are not treatable by hormonal supplementation, and these individuals remain beyond the scope of therapeutic intervention (Irvine 1998; Hamada et al. 2012; Anawalt 2013; Lee and Foo 2014).
Spermatogenesis is a complex developmental process wherein male spermatogonial cells undergo precise division and differentiation within the niche provided by the somatic component of the testicular seminiferous epithelia, mainly the Sertoli cells, to generate functional haploid sperm (Griswold 1998). Sertoli cells are the major source of such somatic support to germ cells for their division and differentiation (Griswold 1998; Sharpe et al. 2003; Rossi and Dolci 2013). Upon attainment of puberty, Sertoli cells provide developmental cues necessary for the onset of germ cell differentiation. Crosstalk between germ cells and Sertoli cells involves multitudes of cellular and molecular factors, their precise and subtle regulation being an essential prerequisite for functional spermatogenesis (Griswold 1998; Matzuk and Lamb 2008). Primordial germ cells in the embryonic epiblast acquire functional competence to differentiate into sperm (Chuma et al. 2005). However, the process of germ cells differentiation into sperm is only witnessed at and after puberty. Germ cells in postnatal infant testis are restricted to self-renewal due to a lack of morphogenic induction by the somatic niche, predominantly generated by Sertoli cells. Sertoli cell maturation is a key stage in testicular development which coincides with the pubertal triggering of hypothalamic GnRH secretion which occurs approximately at 10–12 years of age in humans and 3–4 years after birth in monkeys. We and others have shown that. upon attainment of maturity, Sertoli cells undergo a developmental switch in FSH and T response and express factors which are essential for germ cell division and differentiation (Griswold 1998; Sharpe et al. 2003; Majumdar et al. 2012; Bhattacharya et al. 2012; Rossi and Dolci 2013; Bhattacharya et al. 2015). Augmented hormonal levels and fully functional Sertoli cells that respond to the surge of reproductive hormones trigger the division and differentiation of germ cells, leading to the initiation of spermatogenesis at puberty. Sertoli cells from young boys (up to 6 months of age) and infant rhesus monkeys (up to 3 months of age) fail to induce robust spermatogonial differentiation despite adequate levels of circulating FSH and T as well as their respective receptors on Sertoli cells (Rappaport and Amselem 2001; Majumdar et al. 2012). Defects of Sertoli cell differentiation and lack of Sertoli cell mediated paracrine support to germ cells has previously been shown to limit spermatogenesis (Matzuk and Lamb 2008; Juul et al. 2014). While several Sertoli cell factors necessary for the pubertal stimulation of germ cell division and differentiation have been identified using rodents, such information for human and primate Sertoli cells are limited (Holmes et al. 1984; Devi et al. 2006; Majumdar et al. 2012). Unlike rodents, testicular development in monkeys is similar to that in humans where a long hypogonadotrophic juvenile phase separates the phases of infancy and puberty (Plant et al. 2005). In both infant and pubertal phases, circulating levels of hormones are higher and similar, in contrast to those observed during the juvenile phase.
The inability of defective Sertoli cells to produce paracrine factors important for germ cell differentiation has been suggested to be responsible for compromised male fertility (Schaison et al. 1993; Steger et al. 1996; De Kretser and Baker 1999; Bar-Shira Maymon et al. 2000; Maymon et al. 2002; Plotton et al. 2006). Incompetence of infant Sertoli cells to support robust germ cell differentiation in the face of adequate hormonal exposure resembles this situation. We have used a DNA microarray-based approach to compare the gene expression profiles of immature (infant) and mature (pubertal) primate Sertoli cells, which were exposed to similar hormonal milieu. Our objective was to identify important paracrine factors produced by Sertoli cells which may potentially promote germ cell differentiation during puberty. Since it is difficult to precisely ascertain onset of puberty in monkeys, we experimentally induced puberty in juvenile monkeys by intermittent intravenous infusion of GnRH for about 4 weeks, which is known to induce robust germ cell differentiation in pubertal testis (Marshall and Plant 1996; Devi et al. 2006). We found that, along with several other paracrine morphogens, expression of Wnt3 was upregulated many fold in Sertoli cells of pubertal monkey (showing active spermatogenesis) as compared to Sertoli cells of infant monkeys (showing restricted spermatogenesis).
Wnt3 is known to activate Wnt/β-catenin signaling and affect crucial stages of development and differentiation (Logan and Nusse 2004a). The importance of optimal Wnt/β-catenin signaling in regulation of spermatogenesis is increasingly becoming apparent. However, little information is available on Wnt ligands that activate the Wnt/β-catenin pathway and the age-specific regulation of Wnt signaling in testis (Lombardi et al. 2013; Kerr et al. 2014). We knocked-down the expression of Wnt3 in Sertoli cells at puberty using transgenic mice to determine its role in the regulation of spermatogonial differentiation, and found that compromised Wnt3 expression in Sertoli cells during or after puberty may lead to subfertility and oligozoospermia via diminished expression of Connexin43, a gap-junctional molecule essential for germ cell development.
Materials and methods
Animals
All experimental animals, monkeys as well as mice, were reared in the animal facility of our institute following the National guidelines of the Committee for the Purpose of Control and Supervision of the Experiments on Animals (CPCSEA) in India. All animal experiments in this study were performed following protocols approved by CPCSEA and the Institutional Animal Ethics Committee (IAEC Number 49/99, 187/08, 249/10) of the National Institute of Immunology, New Delhi, India. FVB/J mice were procured from the Small Animal Facility of the National Institute of Immunology. The mice were sacrificed, when required, by cervical dislocation, as approved by the Institutional Animal Ethics Committee of National Institute of Immunology. Rhesus male monkeys (Macaca mulatta) born and raised at the Primate Research Center of National Institute of Immunology were used for this study. The selected monkeys were members of a captive breeding group that lived within open enclosures. Monkeys were anesthetized with pentobarbital sodium (25 mg/kg body weight, i.v., plus 5-mg supplements as required) before catheterization or castration. Postoperatively, each monkey received cefotaxime antibiotic (75 mg/kg body weight) and an analgesic, diclofenac sodium (1 mg/kg body weight), i.m., twice daily for 5 days. No monkey was sacrificed for this study.
Preparation of pubertal monkeys
Precocious puberty was induced in 5 juvenile monkeys by stimulating the dormant pituitary–testicular axis by pulsatile GnRH treatment as shown by us previously (Devi et al. 2006; Majumdar et al. 2012). To achieve this, chronic indwelling catheters were surgically implanted through the femoral or internal jugular vein under sterile conditions. The catheter was externalized in the midscapular region, protected by a nylon jacket and stainless steel tubing (36 in. long, 0.5 in. diameter; c. 91.4 mm, 12.7 mm) attached to a freely movable swivel joint at the top of the cage. Such an arrangement allowed for free movement of the monkey without disturbing access to circulation via catheter. Administration of pulsatile GnRH (0.3 μg GnRH in 2 ml saline over 2 min every 3 h) through catheter was continued for 4–5 weeks, until serum T levels reached the adult range and their testis size increased (Devi et al. 2006). Note that six different sets of infant and pubertal Sertoli cell cultures were used for transcriptomic studies in the monkeys, three independent sets for microarray and three independent sets for qRT PCR analyses.
Isolation and culture of Sertoli cells
Testes from infant (3 months old) and pubertal monkeys were surgically removed under general anesthesia and Sertoli cells were isolated and cultured following the protocol described by us previously (Devi et al. 2006; Majumdar et al. 2012). Testes were dissected from the FVB/J mice of two separate postnatal ages, 5 days (neonatal) and 20 days (pubertal) from which Sertoli cells were isolated following the previously described procedure (Welsh and Wiebe 1975). Uniform amounts of Sertoli cell clusters (0.5 × 104) were plated in each well on day 1 of culture, in DMEM/HAM F12 media supplemented with 1% fetal calf serum and kept in a humidified incubator at 34 °C and 5% CO2.
On day 4 of culture, hypotonic shock was given for 3 min to remove germ cells, if any (Galdieri et al. 1983). On day 5 of culture, the monkey Sertoli cells were treated with recombinant monkey follicle-stimulating hormone (FSH) (5 ng/ml) and testosterone (T) (10−7 M) since Sertoli cells are exposed to both hormones in vivo. Sertoli cells were isolated from three separate infant and three separate pubertal animals. Cultured Sertoli cells derived from two individual infants and two pubertal monkeys were treated with FSH and T for 24 h. One additional set of infant and pubertal Sertoli cell culture was treated with FSH and T for 12 h. In the case of mice Sertoli cells, ovine FSH was used for treatment instead of recombinant monkey FSH. An outline for the in vitro culture protocol is shown in Fig. 1a.

Outline of Sertoli cells culture and microarray experiment. a Outline of monkey Sertoli cells culture, individual monkey numbers given as #. Up to day 4 all cultures were treated equally. One set of infant Sertoli cells culture (#306) and pubertal Sertoli cells culture (#479) were given FSH and T for 12 h before proceeding for micro-array. Two setd of infant (#380 and #403) and two setd of pubertal (#453 and #454) Sertoli cells culture were treated with FSH and T for 24 h before sample preparation for micro-array. Rest cultures were treated for 24 h with FSH and T and RNA used for qRT PCR. b Outline for micro-array. For 24 h (#380 vs. #454 and #403 vs. #453)-treated samples, two color hybridisation protocols were used. Dye swapping was carried out to account for bias in the fluorolabeling. For 12-h FSH- and T-treated samples, a single color experiment was used (#306 vs. #479). Differential gene expression readouts from both 24-h and 12-h treatment groups were compared
RNA extraction and cDNA microarray analysis
Three sets (each set consisting of one infant monkey- and one pubertal monkey-derived Sertoli cell cultures) of differential cDNA micro-array was performed using RNA from cultured hormone-treated Sertoli cells. For this, RNA was extracted from cultured infant and pubertal monkey Sertoli cells using Trizol (Chomczynski and Sacchi 1987). The purity of the RNA was assessed on a Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA, USA) and found suitable for fluorolabeling. A two-color hybridization method was used for the 24-h FSH and T treatment groups. Briefly, Cy3/Cy5-labeled cRNA probes were prepared and hybridized to an Agilent Whole Human Genome Microarray Kit, 4 × 44K (G4112F) using Agilent’s In situ Hybridization Kit (Cat #5184-3568) following the manufacturer’s instruction. Images were quantified using Agilent Feature Extraction Software (v.A.9.5.1.1). Extracted features were normalized per spot per chip by the intensity-dependent Lowess method. A one-color hybridisation protocol was used for differential micro-array of one set of infant and pubertal monkey-derived Sertoli cell culture which was treated with FSH and T for 12 h. Briefly, isolated RNA were flurolabeled by Agilent’s Quick-Amp Labeling Kit (p/n 5190-0442). Labeled samples were hybridized to the Agilent Whole Human Genome Microarray Kit 8 × 60 K (AMADID:27,144). Images were quantified using Agilent Feature Extraction Software (v.A.10.5.1.1). Extracted features were normalized per spot per chip by the percentile shift method.
Log2-transformed normalized intensity ratios of pubertal Sertoli cells versus infant Sertoli cells were plotted against log-processed signals per spot per chip to determine the genes which had significantly high or low signals in all three sets of the micro-arrays. Fold change in the expression levels of each gene was calculated using expression levels in infant Sertoli cells as reference. Genes with greater than 1.4-fold expression in Log2 scale were taken to be upregulated. The normalized micro-array data has been submitted to the NCBI-GEO database. Accession numbers of submitted data are GSE57937 and GSE66129 for the two-color and one-color experiments, respectively (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=axqpaiyshronhmn&acc=GSE57937 and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ytoxcyqgvzqflcz&acc=GSE66129).
An outline of the microarray experiment is shown in Fig. 1b.
Quantitative RT PCR
For qRT-PCR, total RNA was extracted from testicular tissues and cultured cells using Trizol reagent (Chomczynski and Sacchi 1987). Isolated RNA was treated with DNaseI (Life Technologies, USA) as per the manufacturer’s protocol, and cDNA was prepared with the MMLV reverse transcriptase kit (Eurogentec, Belgium) with 1 μg RNA using oligo dT primer (Promega, USA). qRT PCR was set up with mesa green (Eurogentec), 1 μl cDNA and 0.5 μl of forward and reverse primers each. PCR conditions used were melting of cDNA at 95 °C for 10 min, followed by 40 amplification cycles (20 s at 95 °C, 30 s at Tm and 30 s at 72 °C). Melt curve analysis was performed after each run to ensure that a specific amplicon was generated.
Endogenous 60S ribosomal protein L32 was used for normalizing the expression of RNA derived from monkey Sertoli cells as per a previous report (Ahn et al. 2008). In the case of RNA derived from mice, peptidyl prolyl isomerase A) was used as an internal control. The 2-∆∆Ct method was used to calculate relative gene expression (Schmittgen and Livak 2008). Mean (±SEM) data representing at least three separate experiments were used to generate histograms for graphical presentation. Primer sequences and details are given in Table S1.
Generation of transgenic mice
A transgenic cassette was designed to abrogate Wnt3 expression in pubertal Sertoli cells by RNA interference. shRNA against Wnt3 and bacterial LacZ (as a control) were designed and cloned under the Pem promoter which is a Pol II promoter and reported to drive shRNA expression specifically in pubertal Sertoli cells (Lindsey and Wilkinson 1996; Sutton et al. 1998; Rao and Wilkinson 2006). Details are given in Fig. S4a. We designed shRNA targeting Wnt3 mRNA to knockdown Wnt3 expression in pubertal Sertoli cells. We determined the specificity of shRNA against Wnt3 by the BLASTn tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch). Sequences of shRNA which were designed are given in Table S2. We generated transgenic mice for this study using testicular transgenesis (Usmani et al. 2013). Briefly, transgenic cassettes were linearized by digestion with appropriate restriction enzymes. Linear DNA was electroporated into testis of male fore founders. Fore founder males for each specific cassette were cohabited with wild-type females to generate progeny termed G1. Several individuals from the G1 generation were transgenic. Transgenic males and females from the G1 generation were interbred to generate a G2 generation. All subsequent studies were carried out on transgenic males from the G2 generation.
Histology
Testis of mice and monkeys were surgically removed and a fraction was immersion-fixed in Bouin’s fluid for 10–12 h at room temperature. Fixed tissues were embedded in paraffin before sectioning them at 4 μm thickness. Sections were stained with Hematoxylin and Eosin for assessing the status of spermatogenesis.
Immunohistochemistry, immunocytochemistry and immunoblot analysis
Tissue sections were blocked with antisera or 5% BSA for 20 min followed by incubation with primary antibody. Fluorophore-conjugated secondary antibody was used to detect the bound primary antibody. Anti-goat (Molecular Probes, Life technologies, USA) IgG was used as secondary antibody for Wnt3. Epifluorescence microscopy was used to detect Wnt3 staining. For immunohistochemical detection of Connexin43, Horse Radish Peroxidase-Labeled anti-rabbit (Epitomics 3053-1; Thermo Scientific, USA) IgG were used as secondary antibodies. A DAB-based immunoperoxidase method was used to visualize Connexin43 immunostaining under light microscope (Mason and Taylor 1978).
Immunocytochemistry was performed on cultured Sertoli cells. Cells were blocked with 3% BSA for 30 min at 37 °C followed by incubation with primary antibodies. Fluorophore-conjugated secondary antibodies were used to detect bound primary antibodies. Anti-goat (Molecular Probes) and anti-mouse (Molecular Probes) IgG were used as secondary antibodies for Wnt3 and Vimentin, respectively. Epifluorescence microscopy was used to detect staining.
Immunoblot analysis was performed from tissue lysates of testis from normal and transgenic mice for estimating expression of Wnt3, phospho β-catenin, β-catenin and Connexin43. Testicular tissue were lysed separately with ice-cold RIPA buffer containing phosphatase and protease inhibitors (1 mM PMSF, 1 mg/ml aprotinin and 1 mg/ml leupeptin). Protein concentration was determined by Bradford’s method (Bradford 1976). Then, 20 μg protein was resolved by one-dimensional SDS PAGE (10-12% acrylamide) under reducing conditions and electrophoretically transferred to a nitrocellulose membrane. Blots were blocked with 3% skimmed milk in PBST for Wnt3 and Connexin43 and with 5% BSA in PBST for phospho β catenin for 4 h at room temperature. For β-catenin, the blots were blocked with 5% BSA in PBST overnight at 4 °C. Subsequently, blots were incubated overnight at 4 °C with primary antibody. β actin blotting was used to check equal loading in each lane. Horse Radish Peroxidase-conjugated secondary antibodies were used to detect bound primary antibodies. Anti–rabbit (Epitomics 3053-1; Thermo Scientific) IgG was used as secondary antibody for phospho β-catenin and Connexin43. Anti-mouse (Pierce 31430; Thermo Scientific) and anti-goat (Pierce 31402; Thermo Scientific) IgG were used as secondary antibodies for β-catenin and Wnt3, respectively. Bands were detected by chemiluminiscence using the SuperSignal West Pico Chemiluminiscent substrate (Thermo Scientific) and chemiluminescence film (GE Amersham, Piscataway, NJ, USA). ImageJ software was used to assess level of β-catenin phospho Ser45 relative to β actin for all LacZ KD and Wnt3 KD samples. A list of all antibodies that were used in the study is given in Table S3.
Sperm count and fertility assessment
Testes of Wnt3 KD and LacZ KD mice were taken from euthanized transgenic male mice of 10 weeks age. The 10-week-old male mice were cohabited with a pair of female mice for 3 weeks. In the case of a lack of conception, we changed the females and cohabitated them for another 3 weeks. Males were checked for breeding at 12–15 weeks. Testis weight was recorded. The epididymis of each mouse was punctured to release the sperm in 1 ml of PBS kept at 37 °C. Total sperm count was determined by using a hemocytometer. Approximately 10-week-old Wnt3 KD or LacZ KD male were cohabited with wild-type females for 3 weeks. Litter size was determined after the delivery of the progeny to assess the fertility of the sire.
Statistical analysis
All statistical analyses for gene expression studies were performed using Graphpad Prism 5 (Graphpad Software, Lajolla, CA, USA). Data are represented as means ± SEM of at least three monkeys and at least six mice belonging to a particular group. Significance was determined by the Mann–Whitney test and a P value <0.05 was taken as significant.
Results and discussion
Differential Wnt3 expression by pubertal and infant Sertoli cells
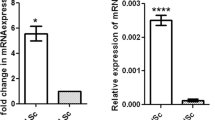
In agreement with previous reports (Devi et al. 2006; Majumdar et al. 2012), the puberty-like situation was generated upon 4–5 weeks of pulsatile GnRH administration to juvenile monkeys. These monkeys showed morphological changes in seminiferous epithelia, suggesting the onset of germ cell differentiation similar to that found in pubertal monkeys (Fig. S2a and b). Based on cDNA micro-array analysis, several genes were found to be differentially expressed by infant and pubertal monkey Sertoli cells (Fig. 2a). A gene was considered to be upregulated during puberty if normalized signals of its expression were at least 1.4-fold higher in pubertal Sertoli cells than that in infant Sertoli cells. WNT3, one of several morphogens with puberty-specific expression, was significantly (P < 0.05) upregulated in pubertal Sertoli cells relative to infant Sertoli cells in all three micro-array datasets. WNT3 was upregulated 2.03-fold and 1.61-fold in the two sets treated with FSH and T for 24 h. Wnt3 was upregulated 1.49-fold in the set treated with FSH and T for 12 h (Fig. 2a, b). An age-specific difference in the expression of several other Wnt ligands was also apparent. However, the expression of only WNT3 was found to be clearly augmented in pubertal Sertoli cells in each of the three micro-array data- sets (Fig. 2a; Fig. S3a). The expression levels of other Wnt signaling components, such as Wnt receptors, β-catenin destabilization complex proteins and β-catenin-associated responsive transcription factors were not different in the two age groups (Fig. S3b, c). A 24-h treatment with FSH and T, significantly augmented (P < 0.05) WNT3 mRNA expression by pubertal monkey Sertoli cells but not by infant Sertoli cells by qRTPCR (Fig. 2b). The micro-array revealed several other genes apart from Wnt3 that showed altered expression levels between infant and pubertal Sertoli cells when exposed to FSH and T for 24 h. The differential expression of these genes was validated by qRTPCR (Fig. S1).

Differential expression of Wnt3 in infant and pubertal Sertoli cells. a Heat map of genes found differentially expressed in pubertal Sertoli cells as compared to infant Sertoli cells: green upregulated in puberty and red downregulated in puberty. b Quantitative real-time PCR analysis of the Wnt3 expression in infant and pubertal Sertoli cells cultured from monkey and mice. Wnt3 expression was found to be upregulated in pubertal Sertoli cells in both species. Sertoli cells was cultured from 5-day-old mice (for infant) and 20-day-old mice (for pubertal) (n = 6). Five independent infant and pubertal monkey samples were combined for analyses. Histograms depict mean ± SEM. *P < 0.05. WNT3 was upregulated 2.03-fold and 1.61 in the 24-h sets (A) and (B). Wnt3 was upregulated 1.49-fold in the 12-h set. c Immunocytochemical analysis of cultured Sertoli cells, isolated from infant and pubertal mice, stained with anti-Vimentin (Sertoli cell marker) and Wnt3 antibody for detection of Wnt3 expression. Wnt3 expression was found to be present in pubertal Sertoli cells and absent in infant Sertoli cells. For detection of Wnt3 and Vimentin expression,the secondary antibody was tagged to Alexa flour 488 and Alexa flour 546, respectively. The nucleus was stained with DAPI. c, dImage under UV with FITC filter. c’, d’ Image under blue/cyan filter. c”, d” Image under UV with TRITC filter. c”’, d”’ Merged images for Vimentin and Wnt3. Scale bar 50 μm
Since in vivo studies of functional genomics were to be undertaken using transgenic mouse model, we checked the status of Wnt3 expression in Sertoli cells from 5-day-old (infant) and 20-day-old (pubertal) wild-type FVB/J mice. A 24-h exposure to FSH and T significantly (P < 0.05) augmented Wnt3 mRNA expression by Sertoli cells of pubertal mice as compared to that of infant mice (Fig. 2b). Immunofluorescence microscopy of infant and pubertal mice Sertoli cells also revealed higher expression of Wnt3 by pubertal Sertoli cells (Fig. 2c).
Wnt signaling is known to be essential for female gonadal development and oogenesis (Tevosian and Manuylov 2008). Its role in spermatogenesis has emerged only recently (Lombardi et al. 2013). Wnt signaling is necessary at embryonic age, since its disruption leads to malformation of the male reproductive tract (Jeays-Ward et al. 2004; Warr et al. 2009; Chawengsaksophak et al. 2012). β-catenin is a secondary messenger of Wnt signaling (Clevers and Nusse 2012). It has been reported recently that loss or gain of β-catenin activity within the seminiferous epithelia during adulthood leads to disruption of germ cell differentiation suggesting that spermatogenesis is affected due to disturbed β-catenin activity (Kerr et al. 2014). Dickkopf 3, a Wnt signaling pathway inhibitor mediating suppression of Wnt signaling has also been shown by us to interfere with spermatogenesis (Das et al. 2013). Recently, Wnt5a has been reported as a cell-extrinsic factor involved in the maintenance of stemness of germinal stem cells (Yeh et al. 2011). However, specific Wnt ligand(s) which activate the Wnt signaling pathway in testis, leading to maintenance and differentiation of germ cells, remain largely unknown. Recent studies in mice suggest increased expression of several Wnt ligands during puberty (Kerr et al. 2014). A recent report (Takase and Nusse 2016) has identified Wnt6 as a Sertoli cell-expressed paracrine factor necessary for the regulation of Wnt signaling within different lineages of spermatogonia including SSCs. However, in monkeys (which are closer to humans in ancestry), we found WNT3 to be the sole Wnt ligand consistently upregulated in pubertal Sertoli cells. FSH- and T-mediated augmentation of Wnt3 expression by pubertal monkey as well as mouse Sertoli cells raises the possibility of Wnt3-mediated regulation of Wnt/ β-catenin signaling which plays a major role in spermatogenesis. Testosterone has been previously shown to interact with β-catenin and TCF, as a component of the AR nuclear receptor complex, in prostate cancer and in bone metabolism (Callewaert et al. 2010; Antony et al. 2014). Similarly, FSH and classical Wnt signaling has been shown to work synergistically in rat granulosa cells to establish Gap junctions (Wang et al. 2013). Wnt signaling also regulates the selection of dominant follicles during oogenesis which is governed by FSH (Gupta et al. 2014). We found that FSH and T stimuli to Sertoli cells lead to augmentation of Wnt3 expression, selectively during puberty in non-human primates as well as rodents.
Generation and analysis of Wnt3 knockdown (Wnt3 KD) transgenic mice
In vivo abrogation of a gene in a cell and developmental stage-specific manner often reveals important aspects of a gene’s function (Capecchi 2005). Transgenic animal models with RNA interference-based targeted cell-specific gene knockdown with varied degrees of suppression are useful for studies of functional genomics (Coumoul and Deng 2006; Kleinhammer et al. 2011; Livshits and Lowe 2013). We expressed shRNA directed against Wnt3 transcript under the control of the Pem promoter, which is a PolII promoter known to drive shRNA expression in mice Sertoli cells, specifically after 9 days of age and onwards (Lindsey and Wilkinson 1996; Sutton et al. 1998; Rao and Wilkinson 2006). We used Pem-driven LacZ shRNA-expressing mice as a control for comparing results from in vivo shRNA-mediated knockdown (Yi et al. 2005). Transgenic mice expressing Wnt3 shRNA and LacZ shRNA were identified by PCR and slot blot analysis (Fig. 3a–d; Fig. S4b, c). Mice expressing shRNA against Wnt3 are henceforth referred to as Wnt3 KD. Similarly, mice expressing shRNA against LacZ are referred to as LacZ KD.

Generation of transgenic mice, abrogating Wnt3 expression specifically in Sertoli cells at puberty. a Genotyping of progeny (G1) generated by crossing electroporated male and wild-type female mice by PCR analysis of gDNA obtained from tail biopsy. Lanes 1–8 indicates gDNA of pups from G1 progeny; Wt wild-type gDNA; +ve plasmid DNA bearing shRNA cassette. b Slot blot analysis of gDNA isolated from pups generated in G1 progeny. Spots 1–8 indicate gDNA of G1 progeny pups. Wt iwild-type gDNA as negative control; +ve template used to design probe. Lane with Wt gDNA indicated by the box inset. c Genotyping of progeny (G2) generated by crossing transgene-positive male and transgene-positive female mice by PCR analysis of gDNA obtained from tail biopsy. Lanes 1–11 indicate gDNA of pups from G2 progeny; Wt wild-type gDNA; +ve plasmid DNA bearing shRNA cassette. d Slot blot analysis of gDNA isolated from pups generated in G2 progeny. Spots 1–11 indicate gDNA of G2 progeny pups; Wt wild-type gDNA as negative control; +ve template used to design probe. Lane with Wt gDNA indicated by the box inset. e Relative expression of Wnt3 was highly reduced in testis of Wnt3 KD mice as opposed to age-matched LacZ KD mice as detected by quantitative real-time PCR. Histogram depicts mean ± SEM, n ≥ 9. *P < 0.05. f Immunoblot analysis of Wnt3 expression in total protein isolated from testis. Expression of Wnt3 was found to be reduced drastically in testis of Wnt3 KD animals as opposed to age-matched control LacZ KD mice. β actin level was measured as loading control. Wnt3 KD and LacZ KD mice were 10–12 weeks old when protein was extracted from testis
Level of Wnt3 mRNA was significantly (P < 0.05) low in transgenic 10-week-old Wnt3KD mice with ~85% reduction in testicular Wnt3 mRNA compared to age-matched LacZ KD controls (Fig. 3e). Western blot analysis also showed a remarkable decrease in testicular Wnt3 levels in Wnt3 KD mice (Fig. 3f). Poor expression of Wnt3 in Sertoli cells of Wnt3 KD mice as compared to Sertoli cells of LacZ KD mice was also apparent in the immunostaining of testicular sections (Fig. S4d–g).
Regulation of Wnt/β-catenin signaling by Wnt3
Wnt3 elicits classical Wnt/β-catenin signaling upon binding to its cognate receptors (Clevers and Nusse 2012). Wnt3 binds at the cell surface leading to inactivation and sequestration of inhibitory kinases, GSK3β and CKIα, preventing phosphorylation at Ser 45 of β-catenin, stopping ubiquitination and leading to nuclear localization of β-catenin and expression of β-catenin responsive genes (Logan and Nusse 2004b). Western blot of testicular proteins showed that Wnt3 KD mice had higher phosphorylation at Ser 45 of β-catenin as compared to LacZ KD control mice (Fig. 4a). Total β-catenin levels also declined in Wnt3KD mice (Fig. 4a). The ratio of phospho β-catenin to β-catenin increased significantly (P < 0.05) in Wnt3 KD mice, correspondingly causing increased β-catenin degradation in these mice (Fig. 4b). Expression levels of β-catenin mRNA were also found to be significantly (P < 0.05) low in Wnt3KD mice as compared to LacZ KD mice (Fig. 4c). β-catenin is known to regulate expression levels of several target genes, such as Axin2 and Pitx2 (Clevers and Nusse 2012). Significant (P < 0.05) decrease in levels of Axin2 and Pitx2 in Wnt3KD mice relative to age-matched LacZ KD controls (Fig. 4d, e) provided substantial evidence that loss of Wnt3 affects the status of canonical Wnt/β catenin signaling in testis. Axin2 has been recently shown to be expressed in spermatogonial cells with Wnt6, provided by Sertoli cells, as a regulator of its expression (Kerr et al. 2014; Takase and Nusse 2016). Given the decrease in axin2 expression and the deleterious effects on spermatogenesis, which we shall address below, it is reasonable to believe that abrogation of Sertoli cell-derived Wnt3 expression affects Wnt signaling within the spermatogonial niche which is vital for normal spermatogenesis.

Wnt3 knockdown mediated effects on β-catenin. a Top phosphorylated β- catenin at Ser 45 in LacZ KD and Wnt3 KD mice; middle β-catenin levels in LacZKD and Wnt3KD mice; bottom β actin was used to check equal loading. A single representative blot is shown. b β-catenin phophorylation in Wnt3 KD mice as opposed to age-matched LacZ KD mice. c–e Relative mRNA expression of β-catenin, and its transcriptional targets Axin2 and Pitx2 in Wnt3 KD mice as opposed to LacZ KD mice. f Wnt3 knockdown affecting Connexin43 expression as compared to LacZ KD mice. g Immunoblot analysis of Connexin43 protein in testis of Wnt3 KD mice as compared to age-matched LacZ KD mice. β actin was used to check equal loading. h Localization of Connexin43 at the basal compartment of seminiferous tubule in LacZ KD mice and i Wnt3 KD mice. Magnification ×60. Scale bar 20 μm. Histogram depicts mean ± SEM, n ≥ 9. *P < 0.05
Regulation of Connexin 43 expression by Wnt3
Connexin43 is a member of the gap junctional complex that is crucial for formation and maintenance of the blood–testis barrier as well as normal spermatogenesis at puberty (Pointis and Segretain 2005; Weider et al. 2011). Seminiferous tubules of men showing spermatogenic arrest at the level of spermatogonia as well as those afflicted with Sertoli cell-only syndrome, exhibit a reduction and sometimes loss of Connexin43 in germ cells and Sertoli cells (Steger et al. 1999; Brehm et al. 2002; Defamie et al. 2003; Chevallier et al. 2013). Wnt signaling has been shown to regulate Connexin43 expression in vitro (van der Heyden et al. 1998). β-catenin has been shown to activate Connexin43 in bone metabolism and cardiac pathophysiology as well as in several ovarian cancers which have constitutive β-catenin activity (Zhai et al. 2002; Dawson et al. 2013; Lloyd et al. 2014). Since classical Wnt signaling has been reported to regulate Connexin43 expression in other systems, we evaluated the status of Connexin43 in testis of Wnt3KD mice. Mice in which Wnt3 was substantially knocked-down displayed significantly (P < 0.05) diminished levels of Connexin 3 mRNA compared to age-matched LacZ KD mice (Fig. 4f). Western blot analysis of testicular proteins from Wnt3KD mice and age-matched LacZ KD controls revealed that the expression of Connexin43 was severely reduced in Wnt3KD mice (Fig. 4g). Connexin43 was distributed evenly across the entire basal circumference of seminiferous tubules in sections from LacZ KD control mice (Fig. 4h), whereas the distribution of Connexin43 in the seminiferous tubules of age-matched Wnt3 KD mice was discontinuous and lacked uniformity (Fig. 4i). Observations from Sertoli cell-specific Wnt3 knockdown suggested that elevation of Wnt3 expression during puberty is necessary for augmenting the levels of Connexin43 and the establishment of functional gap junctions between Sertoli cells and germ cells at the onset of puberty. Previous reports of infertility caused due to Sertoli cell-specific knockout of Connexin43 strengthened our observations (Brehm et al. 2007; Weider et al. 2011; Giese et al. 2012).
Fertility status in Wnt3KD mice
Wnt3 KD mice were grouped into two categories based on the extent of the reduction in Wnt3 expression as detected by qRT PCR. In group Wnt3 KD(I), Wnt3 expression was reduced by ~80% and in group Wnt3 KD(II) by ~90% as compared to LacZ KD control. In both groups of Wnt3KD, litter size was significantly (P < 0.05) reduced as compared to that of age-matched (10–12 weeks old) control males expressing LacZ shRNA; litter size was smaller in group II which had greater loss of Wnt3 expression (Fig. 5a). Sperm counts of Wnt3 KD mice were significantly (p < 0.05) reduced as compared to age matched controls. Decline in sperm count was more pronounced in Wnt3KD mice which had higher ablation of Wnt3 mRNA (Fig. 5b). A slight but statistically significant (P < 0.05) reduction of testicular weight was also observed in adult Wnt3 KD mice as compared to age-matched controls (Fig. 5c). It is possible that reduced release of germ cells from the seminiferous epithelium may explain the significant loss of epididymal sperm count in Wnt3KD mice but not a commensurate decrease in testis weight. Interestingly, sperm motility was unaffected in Wnt3 KD mice. Comparison of testicular sections from 10-week-old Wnt3 KD (Fig. 5d) and control mice (Fig. 5e) revealed abnormal tubular structure, diminished presence of sperm and sloughed-off germ cells in tubular lumen of Wnt3KD mice. β-catenin and connexin43 have previously been shown to be necessary for normal spermatogenesis (Brehm et al. 2007; Weider et al. 2011; Giese et al. 2012; Kerr et al. 2014). Since Wnt3 is needed for regulation of β-catenin activity which is known to control Connexin43 expression, it is reasonable to believe that compromised Wnt3 expression led to reduced Connexin43 expression which compromised male fertility. Step-by-step identification of such differentially expressed factors necessary for the regulation of spermatogenesis may help in revealing unknown causes of male infertility which may act together to determine the status of spermatogenesis and fertility in males.

Compromised fertility of Wnt3 knockdown mice. a Greater reduction of Wnt3 expression: Wnt3 KD(II) showed relatively greater effect on fertility as compared to that seen in Wnt3 KD(I). (b) Reduction in epididymal sperm count. (c) Reduction in testis weight. d H/E staining of testicular sections from Wnt3 KD mice show sloughed-off germ cells (red arrowhead) in tubular lumen (d, d’), and decreased sperm levels( blue arrowhead) within lumen (d’). e In LacZ KD mice, functional spermatogenesis is indicated by yellow arrow-head. Note: Wnt3 expression was ~80% reduced in Wnt3 KD(I), and ≥90% reduced in the group Wnt3 KD(II) as compared to LacZ KD control. Magnification ×20. Scale bar 50 μm. Histograms depict mean ± SEM. *P < 0.05
References
Ahn K, Huh J-W, Park S-J et al (2008) Selection of internal reference genes for SYBR green qRT-PCR studies of rhesus monkey (Macaca mulatta) tissues. BMC Mol Biol 9:78. https://doi.org/10.1186/1471-2199-9-78
Anawalt BD (2013) Approach to male infertility and induction of spermatogenesis. J Clin Endocrinol Metab 98:3532–3542. https://doi.org/10.1210/jc.2012-2400
Antony L, Van Der Schoor F, Dalrymple SL, Isaacs JT (2014) Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/??-catenin/TCF-4 complex inhibition of c-MYC transcription. Prostate 74:1118–1131. https://doi.org/10.1002/pros.22828
Bar-Shira Maymon B, Paz G, Elliott DJ et al (2000) Maturation phenotype of Sertoli cells in testicular biopsies of azoospermic men. Hum Reprod 15:1537–1542
Bhattacharya I, Pradhan BS, Sarda K et al (2012) A switch in Sertoli cell responsiveness to FSH may be responsible for robust onset of germ cell differentiation during prepubartal testicular maturation in rats. Am J Physiol Endocrinol Metab 303:E886–E898. https://doi.org/10.1152/ajpendo.00293.2012
Bhattacharya I, Basu S, Sarda K et al (2015) Low levels of Gαs and Ric8b in testicular sertoli cells may underlie restricted FSH action during infancy in primates. Endocrinology 156:1143–1155. https://doi.org/10.1210/en.2014-1746
Bradford M (1976) A rapid and sensitive method for the quantiWcation of microgram quantities of protein utilizing the principle of protein– dye binding. Anal Biochem 72:248–254
Brehm R, Marks A, Rey R et al (2002) Altered expression of connexins 26 and 43 in Sertoli cells in seminiferous tubules infiltered with carcinoma-in-situ or seminoma. J Pathol 197:647–653. https://doi.org/10.1002/path.1140
Brehm R, Zeiler M, Rüttinger C et al (2007) A sertoli cell-specific knockout of connexin43 prevents initiation of spermatogenesis. Am J Pathol 171:19–31. https://doi.org/10.2353/ajpath.2007.061171
Callewaert F, Bakker A, Schrooten J et al (2010) Androgen receptor disruption increases the osteogenic response to mechanical loading in male mice. J Bone Miner Res 25:124–131. https://doi.org/10.1359/jbmr.091001
Capecchi MR (2005) Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet 6:507–512. https://doi.org/10.1038/nrg1619
Chawengsaksophak K, Svingen T, Ng ET et al (2012) Loss of Wnt5a disrupts primordial germ cell migration and male sexual development in mice. Biol Reprod 86:1–12. https://doi.org/10.1095/biolreprod.111.095232
Chevallier D, Carette D, Gilleron J et al (2013) The emerging role of Connexin 43 in testis pathogenesis. Curr Mol Med 13:1331–1344
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159. https://doi.org/10.1016/0003-2697(87)90021-2
Chuma S, Kanatsu-Shinohara M, Inoue K et al (2005) Spermatogenesis from epiblast and primordial germ cells following transplantation into postnatal mouse testis. Development 132:117–122. https://doi.org/10.1242/dev.01555
Clevers H, Nusse R (2012) Wnt/β-catenin signaling and disease. Cell 149:1192–1205. https://doi.org/10.1016/j.cell.2012.05.012
Coumoul X, Deng C-X (2006) RNAi in mice: a promising approach to decipher gene functions in vivo. Biochimie 88:637–643. https://doi.org/10.1016/j.biochi.2005.11.010
Das DS, Wadhwa N, Kunj N et al (2013) Dickkopf homolog 3 (DKK3) plays a crucial role upstream of WNT/β-CATENIN signaling for Sertoli cell mediated regulation of spermatogenesis. PLoS ONE 8:e63603. https://doi.org/10.1371/journal.pone.0063603
Dawson K, Aflaki M, Nattel S (2013) Role of the Wnt-frizzled system in cardiac pathophysiology: a rapidly developing, poorly understood area with enormous potential. J Physiol 591:1409–1432. https://doi.org/10.1113/jphysiol.2012.235382
De Kretser DM, Baker HW (1999) Infertility in men: recent advances and continuing controversies. J Clin Endocrinol Metab 84:3443–3450. https://doi.org/10.1210/jcem.84.10.6101
Defamie N, Berthaut I, Mograbi B et al (2003) Impaired gap junction connexin43 in Sertoli cells of patients with secretory azoospermia: a marker of undifferentiated Sertoli cells. Lab Investig 83:449–456. https://doi.org/10.1097/01.LAB.0000059928.82702.6D
Devi YS, Sarda K, Stephen B et al (2006) Follicle-stimulating hormone-independent functions of primate Sertoli cells: potential implications in the diagnosis and management of male infertility. J Clin Endocrinol Metab 91:1062–1068. https://doi.org/10.1210/jc.2005-2072
Galdieri M, Zani BM, Monaco L et al (1983) Changes of Sertoli cell glycoproteins induced by removal of the associated germ cells. Exp Cell Res 145:191–198
Giese S, Hossain H, Markmann M et al (2012) Sertoli-cell-specific knockout of connexin 43 leads to multiple alterations in testicular gene expression in prepubertal mice. Dis Model Mech 5:895–913. https://doi.org/10.1242/dmm.008649
Griswold MD (1998) The central role of Sertoli cells in spermatogenesis. Semin Cell Dev Biol 9:411–416. https://doi.org/10.1006/scdb.1998.0203
Gupta PSP, Folger JK, Rajput SK et al (2014) Regulation and regulatory role of WNT signaling in potentiating FSH action during bovine dominant follicle selection. PLoS ONE. https://doi.org/10.1371/journal.pone.0100201
Hamada AJ, Montgomery B, Agarwal A (2012) Male infertility: a critical review of pharmacologic management. Expert Opin Pharmacother 13:2511–2531. https://doi.org/10.1517/14656566.2012.740011
Holmes SD, Lipshultz LI, Smith RG (1984) Regulation of transferrin secretion by human Sertoli cells cultured in the presence or absence of human peritubular cells. J Clin Endocrinol Metab 59:1058–1062. https://doi.org/10.1210/jcem-59-6-1058
Irvine DS (1998) Epidemiology and aetiology of male infertility. Hum Reprod 13:33–44. https://doi.org/10.1093/humrep/13.1.33
Jeays-Ward K, Dandonneau M, Swain A (2004) Wnt4 Is required for proper male as well as female sexual development. Dev Biol 276:431–440. https://doi.org/10.1016/j.ydbio.2004.08.049
Juul A, Almstrup K, Andersson A-M et al (2014) Possible fetal determinants of male infertility. Nat Rev Endocrinol 10:553–562. https://doi.org/10.1038/nrendo.2014.97
Kerr GE, Young JC, Horvay K et al (2014) Regulated Wnt/beta-catenin signaling sustains adult spermatogenesis in mice. Biol Reprod 90:3,1–312. https://doi.org/10.1095/biolreprod.112.105809
Kleinhammer A, Deussing J, Wurst W, Kühn R (2011) Conditional RNAi in mice. Methods 53:142–150
Krausz C (2011) Male infertility: pathogenesis and clinical diagnosis. Best Pract Res Clin Endocrinol Metab 25:271–285. https://doi.org/10.1016/j.beem.2010.08.006
Lee LK, Foo KY (2014) Recent insights on the significance of transcriptomic and metabolomic analysis of male factor infertility. Clin Biochem 47:973–982. https://doi.org/10.1016/j.clinbiochem.2014.05.053
Lindsey JS, Wilkinson MF (1996) Pem: a testosterone- and LH-regulated homeobox gene expressed in mouse Sertoli cells and epididymis. Dev Biol 179:471–484. https://doi.org/10.1006/dbio.1996.0276
Livshits G, Lowe SW (2013) Accelerating cancer modeling with RNAi and nongermline genetically engineered mouse models. Cold Spring Harb Protoc 2013:991–1005. https://doi.org/10.1101/pdb.top069856
Lloyd SA, Loiselle AE, Zhang Y, Donahue HJ (2014) Shifting paradigms on the role of connexin43 in the skeletal response to mechanical load. J Bone Miner Res 29:275–286
Logan CY, Nusse R (2004a) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810. https://doi.org/10.1146/annurev.cellbio.20.010403.113126
Logan CY, Nusse R (2004b) The wnt signaling pathway in development and disease
Lombardi APG, Royer C, Pisolato R et al (2013) Physiopathological aspects of the Wnt/β-catenin signaling pathway in the male reproductive system. Spermatogenesis 3:e23181. https://doi.org/10.4161/spmg.23181
Majumdar SS, Sarda K, Bhattacharya I, Plant TM (2012) Insufficient androgen and FSH signaling may be responsible for the azoospermia of the infantile primate testes despite exposure to an adult-like hormonal milieu. Hum Reprod 27:2515–2525. https://doi.org/10.1093/humrep/des184
Marshall GR, Plant TM (1996) Puberty occurring either spontaneously or induced precociously in rhesus monkey (Macaca mulatta) is associated with a marked proliferation of Sertoli cells. Biol Reprod 54:1192–1199
Mason DY, Taylor CR (1978) Distribution of transferrin, ferritin, and lactoferrin in human tissues. J Clin Pathol 31:316–327
Matzuk MM, Lamb DJ (2008) The biology of infertility: research advances and clinical challenges. Nat Med 14:1197–1213. https://doi.org/10.1038/nm.f.1895
Maymon BB-S, Yogev L, Paz G et al (2002) Sertoli cell maturation in men with azoospermia of different etiologies. Fertil Steril 77:904–909
Plant TM, Ramaswamy S, Simorangkir D, Marshall GR (2005) Postnatal and pubertal development of the rhesus monkey (Macaca mulatta) testis. Ann N Y Acad Sci 1061:149–162. https://doi.org/10.1196/annals.1336.016
Plotton I, Sanchez P, Durand P, Lejeune H (2006) Decrease of both stem cell factor and clusterin mRNA levels in testicular biopsies of azoospermic patients with constitutive or idiopathic but not acquired spermatogenic failure. Hum Reprod 21:2340–2345. https://doi.org/10.1093/humrep/del158
Pointis G, Segretain D (2005) Role of connexin-based gap junction channels in testis. Trends Endocrinol Metab 16:300–306
Rao MK, Wilkinson MF (2006) Tissue-specific and cell type-specific RNA interference in vivo. Nat Protoc 1:1494–1501. https://doi.org/10.1038/nprot.2006.260
Rappaport R, Amselem S (eds) (2001) Hypothalamic–pituitary development. Karger, Basel
Rossi P, Dolci S (2013) Paracrine mechanisms involved in the control of early stages of mammalian spermatogenesis. Front Endocrinol (Lausanne) 4:1–8. https://doi.org/10.3389/fendo.2013.00181
Schaison G, Young J, Pholsena M et al (1993) Failure of combined follicle-stimulating hormone-testosterone administration to initiate and/or maintain spermatogenesis in men with hypogonadotropic hypogonadism. J Clin Endocrinol Metab 77:1545–1549. https://doi.org/10.1210/jcem.77.6.8263139
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108
Sharpe RM (2012) Sperm counts and fertility in men: a rocky road ahead. Science & Society Series on sex and science. EMBO Rep 13:398–403. https://doi.org/10.1038/embor.2012.50
Sharpe RM, McKinnell C, Kivlin C, Fisher JS (2003) Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood. Reproduction 125:769–784
Steger K, Rey R, Kliesch S et al (1996) Immunohistochemical detection of immature Sertoli cell markers in testicular tissue of infertile adult men: a preliminary study. Int J Androl 19:122–128. https://doi.org/10.1111/j.1365-2605.1996.tb00448.x
Steger K, Tetens F, Bergmann M (1999) Expression of connexin 43 in human testis. Histochem Cell Biol 112:215–220. https://doi.org/10.1007/s004180050409
Sutton KA, Maiti S, Tribley WA et al (1998) Androgen regulation of the Pem homeodomain gene in mice and rat Sertoli and epididymal cells. J Androl 19:21–30
Takase HM, Nusse R (2016) Paracrine Wnt/β-catenin signaling mediates proliferation of undifferentiated spermatogonia in the adult mouse testis. Proc Natl Acad Sci U S A 113(11):E1489–E1497
Tevosian SG, Manuylov NL (2008) To beta or not to beta: canonical beta-catenin signaling pathway and ovarian development. Dev Dyn 237:3672–3680. https://doi.org/10.1002/dvdy.21784
Usmani A, Ganguli N, Sarkar H et al (2013) A non-surgical approach for male germ cell mediated gene transmission through transgenesis. Sci Rep 3:3430. https://doi.org/10.1038/srep03430
van der Heyden MA, Rook MB, Hermans MM et al (1998) Identification of connexin43 as a functional target for Wnt signalling. J Cell Sci 111(Pt 1):1741–1749
Wang HX, Gillio-Meina C, Chen S et al (2013) The canonical WNT2 pathway and FSH interact to regulate gap junction assembly in mouse granulosa cells. Biol Reprod 89:39. https://doi.org/10.1095/biolreprod.113.109801
Warr N, Siggers P, Bogani D et al (2009) Sfrp1 And Sfrp2 are required for normal male sexual development in mice. Dev Biol 326:273–284. https://doi.org/10.1016/j.ydbio.2008.11.023
Weider K, Bergmann M, Brehm R (2011) Connexin 43: its regulatory role in testicular junction dynamics and spermatogenesis. Histol Histopathol 26:1343–1352
Welsh MJ, Wiebe JP (1975) Rat sertoli cells: a rapid method for obtaining viable cells. Endocrinology 96:618–624. https://doi.org/10.1210/endo-96-3-618
Yeh JR, Zhang X, Nagano MC (2011) Wnt5a Is a cell-extrinsic factor that supports self-renewal of mouse spermatogonial stem cells. J Cell Sci 124:2357–2366. https://doi.org/10.1242/jcs.080903
Yi R, Doehle BP, Qin Y et al (2005) Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA 11:220–226. https://doi.org/10.1261/rna.7233305
Zhai Y, Wu R, Schwartz DR et al (2002) Role of beta-catenin/T-cell factor-regulated genes in ovarian endometrioid adenocarcinomas. Am J Pathol 160:1229–1238
Acknowledgements
This work was supported by Grant BT/PR11313/AAQ/01/376/2008 from the Department of Biotechnology, Govt. of India, India, Grant BT/HRD/35/01/01/2010 from TATA Innovation Award and Grant 5/10/FR/8/2010-RHN from Indian Council of Medical research, Govt of India, India. We are grateful to the Director of National Institute of Immunology for her support. We acknowledge the technical support from Birendra N. Roy, Dharamveer and Ram Singh. We acknowledge the help from staff of Primate Research Centre and Small Animal Facility of National Institute of Immunology.
Author information
Authors and Affiliations
Contributions
SSM and SB planned the study and designed experiments. SB, SPA, AU, BSP, RKS, MS and KS performed experiments. SSM, KS and SS worked on monkeys. SSM, SB and KM analyzed the results. SSM, SB, MS and KM wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts interests.
Rights and permissions
About this article

Cite this article
Basu, S., Arya, S.P., Usmani, A. et al. Defective Wnt3 expression by testicular Sertoli cells compromise male fertility. Cell Tissue Res 371, 351–363 (2018). https://doi.org/10.1007/s00441-017-2698-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-017-2698-5