
Abstract
Invariant natural killer T (iNKT) cells play important roles in antimicrobial defense and immune-regulation. We have previously shown that iNKT cells express certain toll-like receptors (TLR), and that TLR co-stimulation of iNKT cells in the presence of suboptimal concentrations of T cell receptor (TCR) agonists enhances cellular activation. In the present study, we investigated the regulatory effects of CpG oligonucleotides in mouse primary hepatic and splenic iNKT cells and in DN32.D3 iNKT cells. We show that CpG treatment of iNKT cells in the presence of higher concentrations of TCR agonists (α-GalCer or anti-CD3 mAb) results in the up-regulation of TLR9 in iNKT cells with a concurrent reduction in their cellular activation, as assessed by their production of IL-2, IL-4 and IFN-γ compared with controls. CpG-mediated down-regulation of iNKT cell activation has been found to depend, at least in part, on signaling by MyD88, a critical adapter moiety downstream of TLR9 signaling. Mechanistically, iNKT cells treated with CpG in the presence of TCR agonists show inhibition of MAPK signaling as determined by the levels of ERK1/2 and p38 MAPKs. Furthermore, CpG treatment leads to an increased induction of phosphatases, DUSP1 and SHP-1, that seem to impede MAPK and TCR signaling, resulting in the negative regulation of iNKT cell activation. Our findings therefore suggest a novel regulatory role for CpG in iNKT cells in the mediation of a negative feedback mechanism to control overactive iNKT cell responses and hence to avoid undesirable excessive immunopathology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Invariant natural killer T (iNKT) cells are an important subset of T lymphocytes and express the invariant Vα14-Jα18 T cell receptor (TCR; Gong et al. 2008) in mice (Vα24-Jα18 in humans). They typically recognize glycolipid antigens presented in the context of the MHC class I-like CD1d molecule and alpha-galactosylceramide (α-GalCer), a glycolipid antigen, has extensively been used as a potent TCR agonist for iNKT cell activation (Wu and Van Kaer 2009). Activation of invariant NKT cells following the recognition of their cognate glycolipid antigen results in the production of various cytokines including interferon (IFN)-γ and interleukin (IL)-4, which are important in anti-microbial defense and in the maintenance of immunological homeostasis (Kulkarni et al. 2010; Sharif et al. 2002).
In the context of anti-microbial defense, iNKT cells can either be activated through direct TCR engagement or indirectly by the actions of cytokines secreted by antigen-presenting cells (APC) such as dendritic cells (DCs; Wu and Van Kaer 2009). The capability of sensing microbial agents through Toll-like receptors (TLR) has often been attributed to APC (Nagarajan and Kronenberg 2007; Paget et al. 2007; Tyznik et al. 2008). Evidence is also available that cytosolic peptidoglycan-sensing Nod1 and Nod2 receptor expression in DCs is necessary for optimal IFN-γ production by iNKT cells in response to bacterial infection (Selvanantham et al. 2013). However, as reviewed by us and others, several lines of evidence exist that demonstrate the ability of different T cell subsets, including conventional αβ T cells, regulatory T cells and γδ T cells, to express functional TLRs (Kabelitz 2007; Kulkarni et al. 2011). Importantly, some variation has been found among T cell subsets with regard to the expression of TLR and the cellular responses mediated by these receptors. For example, whereas TLR2 ligand, Pam3CSK4, has been shown to promote cytokine production in naive CD4+ T cells (Komai-Koma et al. 2004), the expression of functional TLR5 and 7 has been reported in memory T cells (Caron et al. 2005).
Similar to conventional T cell subsets, iNKT cells have been previously reported to also be able to express certain functional TLRs such as TLR3, 4, 5 and 9 (Gardner et al. 2010; Kulkarni et al. 2012; Villanueva et al. 2014). We have further demonstrated that TLR co-stimulation in iNKT cells that are either pre- or concurrently TCR-activated by using sub-optimal concentrations of TCR agonists can lead to enhanced iNKT cell activation (Kulkarni et al. 2012). These findings are in agreement with those of others who have investigated TLR expression and function in conventional T cells (Cottalorda et al. 2006; Gelman et al. 2004). In addition to activation, T cell responses can also be modulated by TLR-mediated signaling, thus highlighting the important biological roles of TLR in T cell responses to pathogen antigens. For example, TLR2 engagement on CD8 T cells can lower the TCR threshold for antigen-induced cellular activation (Cottalorda et al. 2006). Similarly, Gonzalez-Navajas et al. (2010) showed that TLR4 triggering on CD4+ T cells can affect their phenotype and regulate their ability to provoke intestinal inflammation in a mouse model of colitis (Gonzalez-Navajas et al. 2010). Furthermore, the authors found that lipopolysaccharide (LPS) stimulation of TLR4-bearing CD4+ T cells can inhibit ERK1/2 activation upon subsequent TCR stimulation via the induction of MAPK (mitogen-activated protein kinase) phosphatase 3 (MKP-3). In our previous studies, we observed that CpG-mediated co-stimulation of TCR-activated iNKT cells in the presence of higher concentrations of α-GalCer lead to a significant down-regulation of iNKT cell activation compared with those stimulated with α-GalCer without CpG. This observation suggests that certain TLR ligands can play a regulatory role in iNKT cells. However, to our knowledge, a detailed investigation of the TLR-mediated regulation of iNKT cell responses has not yet been reported.
In the present study, we investigate in detail the regulatory effects of CpG in iNKT cells by using mouse hepatic and splenic iNKT cells and an iNKT cell-line (DN32.D3). We show that the CpG treatment of iNKT cells in the presence of TCR agonists [α-GalCer or anti-CD3 monoclonal antibodies (mAb)] results in the iNKT cell up-regulation of TLR9 leading to decreased cell activation, as evidenced by their production of IL-2, IL-4 and IFN-γ. This effect has been found to depend, at least in part, on the signaling by MyD88, a critical adapter molecule involved in TLR signaling. Mechanistically, the CpG-mediated regulation of iNKT cell activation is associated with reduced MAPK signaling and increased phosphatase activity and, thus, results in a negative regulation of iNKT cell activation.
Materials and methods
Ex vivo iNKT cell stimulation
Animal experiments were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH), USA. The protocol was approved by the Institutional Animal Care and Use Committee of Emory University School of Medicine and the mice were housed at the Emory University Division of Animal Resources facility. Liver and spleen tissues were collected from C57BL/6 mice and single cell suspensions were prepared. Cell preparations were made by using complete RPMI (RPMI 1640; Invitrogen Life Technologies) supplemented with 10% fetal calf serum, penicillin-streptomycin, glutamine and 2-mercaptoethanol (2-ME). Livers were perfused with phosphate-buffered saline (PBS) before the isolation of the cells and subsequently mashed through a 70-mm cell strainer (BD Biosciences) and washed. The suspension was centrifuged over a 40–70% Percoll (Sigma Aldrich) gradient and mononuclear cells were isolated by harvesting the interphase. Liver mononuclear cells (LMNC) were resuspended in the appropriate amount of medium and further used for cell stimulation. Spleens were mashed and red blood cells were lysed by using red cell lysing buffer from Sigma-Aldrich, after which the cells were resuspended in complete RPMI 1640. The cell suspension was further used in cell culture.
Cells (1 × 106/well) were stimulated in 48-well tissue culture plates in the presence of 500 ng/ml α-GalCer (KRN7000, Cederlane Laboratories, ON, Canada) and with or without CpG (details below) at 10 μg/ml, together with appropriate controls for a period of 24 h. The cells were then collected for further staining and flow cytometry analysis.
Flow cytometry
To determine whether the CpG treatment of mouse LMNC and splenocytes in the presence of α-GalCer would induce TLR9 expression and regulate IFN-γ production in iNKT cells, cells were stained with the following antibodies followed by flow cytometry analysis. Anti-CD16/32, -CD3 (Pacific Blue), -TLR9 (fluorescein isothiocynate) and -IFN-γ (allophycocyanin) antibodies were purchased from BD Biosciences (San Jose, Calif., USA), whereas Live/Dead stain near IR (infrared) was purchased from Invitrogen; CD1d tetramers loaded with PBS-57 were procured from the NIH core tetramer facility, Emory University, Atlanta, Ga., USA. Briefly, cells were resuspended in staining buffer (PBS with 1% bovine serum albumin) and anti-CD16/32 was added to block nonspecific staining. Antibodies to surface markers were added and the cells were stained for 30–45 min on ice, fixed and then permeabilized by using CytoFix/CytoPerm buffer from e-Biosciences (San Diego, Calif., USA), according to the manufacturer’s protocol. The cells were then stained with anti-IFN-γ antibody at room temperature for 30 min, washed and fixed with 2% paraformaldehyde before data acquisition on a BD LSRII flow cytometer. The gating strategy included excluding doublets (larger and highly granular cells) by using forward and side scatter height and width followed by gating of the live cell population. Fluorescence minus one (FMO) and single-stain controls were used for flurochrome/stain compensation. Data analysis was carried out by using FlowJo software (Tree Star, Ashland, Ore., USA).
In vitro iNKT cell stimulation
The Vα14-Jα18+ mouse iNKT cell line DN32.D3 (Lantz and Bendelac 1994) was generously provided by Dr. Albert Bendelac (The University of Chicago, Chicago, Ill., USA). Cells were grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, nonessential amino acids, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μM 2-ME (hereafter, referred to as complete medium). All cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
To induce higher TCR reactivity, αGalCer at 500 ng/ml and plate-bound anti-CD3 monoclonal antibody (mAb; eBioscience) at 1000 ng/ml were used throughout the study. These concentrations were chosen after a pilot dose-range experiment and based on the results of our previous study (Kulkarni et al. 2012). ODN-2395 together with its non-CpG control ODN-2395C (Invivogen, San Diego, Calif., USA) was used at 10 μg/ml. ODN-2395 belongs to the type C CpGs; it combines features of both types A and B and has been shown to stimulate both DC and B cells (Marshall et al. 2005). PepinhMYD, a 26-residue peptide was purchased from Invivogen and was used at 25 μM to block MyD88 signaling in DN32.D3 iNKT cells. PD98059 MEK inhibitor and SB203580 p38 MAPK inhibitor were obtained from Invitrogen (Carlsbad, Calif., USA). Stock solutions were stored at −20 °C. Working solutions were diluted in RPMI 1640 medium with 0.1% dimethylsulfoxide (Sigma-Aldrich, St. Louis, Mo., USA), which was used at a concentration of 20 μM in the present study.
To determine whether CpG stimulation in the presence of α-GalCer or plate-bound anti-CD3 antibodies could regulate iNKT cell activation, cells were seeded at 1–2 × 105 cells/well of microtiter plates. In experiments addressing the possible molecular mechanisms of the CpG-mediated regulation of iNKT cell activation, the aforementioned inhibitors were added to the cell culture at the indicated concentrations. To determine the levels of expression of TLR9, DUSP1 (dual-specificity phosphatase 1), DUSP2, DUSP10, SHP-1 (Src homology region 2 domain-containing phosphatase-1), SHP-2 and TRAF6 (tumor necrosis factor receptor-associated factor 6) mRNA, cells were collected post-stimulation at the indicated time-points for RNA extraction and real-time polymerase chain reaction (PCR) was performed on these samples as described below.
RNA extraction and cDNA synthesis
Total RNA was extracted from iNKT cells by using TRIzol reagent (Invitrogen). RNA quality and quantity were estimated spectrophotometrically. RNA (500 ng to 1 μg) were reverse-transcribed into cDNA by using the SuperScript First-Strand kit (Invitrogen) with oligo-dT primers and according to the manufacturer’s protocol. Spleen tissue from a C57BL/6 mouse was collected and total RNA was extracted followed by cDNA synthesis for use as a positive control for gene expression.
Preparation of standard curves by using plasmid constructs
In order to determine relative gene expression, standard curves were generated for each of the genes, by using plasmid constructs containing cloned target gene fragments. Briefly, gene-specific primers were used to amplify a segment of the target gene with conventional PCR. The PCR products were purified and cloned into the pDrive cloning vector (Qiagen PCR Cloning Kit, Qiagen, ON, Canada). Plasmid constructs were then purified from recombinant Escherichia coli DH5α bacterial cells and 10-fold serial dilutions (10−1–10−9) were prepared. These plasmid dilutions were analyzed in triplicate by real-time PCR assay to obtain the standard curve.
Real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed by using the LightCycler 480 II (Roche Diagnostics, Mannheim, Germany) on 384-well or 96-well plates in a final 20-μl reaction volume. Primers for all target genes analyzed were synthesized by Sigma-Aldrich Canada (Oakville, ON, Canada) and their sequences are given in Table 1. Between 0.25–0.5 μM of each of the gene-specific primers and 5 μl of a 1:10 dilution of cDNA as the template and PCR grade water were used in the reaction. Conditions included an initial heat-denaturing step at 95 °C for 10 min, 45 cycles at 95 °C for 10 s, annealing as recommended for each of the primers and product elongation and signal acquisition (single mode) at 72 °C for 10 s. Following amplification, the melting curves of the PCR products were obtained at temperatures of 60 °C to 97 °C in a continuous acquisition mode and the temperature transition rates were set at 0.11 °C/s.
Enzyme-linked immunosorbant assay
IL-2 production by DN32.D3 iNKT cells was considered as the readout assay for cellular activation (Lantz and Bendelac 1994; Stuart et al. 2010); in addition, IL-4 and IFN-γ production by iNKT cells was measured wherever specified. The cytokine concentration in triplicate culture supernatants in each independent experiment was determined following iNKT cell stimulation with mouse IL-2, IL-4 and IFN-γ sandwich enzyme-linked immunosorbent assay (ELISA) kits (eBioscience) following the manufacturer’s instructions. Briefly, 100 μl anti-mouse cytokine capture antibody was coated overnight at 4 °C. The following day, after the blocking of the wells, the cytokine standard or 100 μl culture supernatant (diluted wherever required) was added as the antigen, followed by incubation for 2 h at room temperature. A detection antibody (biotinylated anti–mouse cytokine antibody, 100 μl) was added to the wells and incubated at room temperature for 1 h followed by avidin-horseradish peroxidase (HRP) enzyme for 30 min. TMB (3,3′,5,5′-tetramethylbenzidine) solution (100 μl, 1×) was added to each well, followed by incubation for 15 min for color development. The reaction was stopped by the addition of 50 μl of 2 N sulfuric acid to each well and the plates were read at an optical density of 450 nm.
To determine the levels of MAPKs in the stimulated DN32.D3 iNKT cell samples, the MAPK Family InstantOne sandwich ELISA Kit (eBiosciences) was used following the manufacturer’s instructions. Briefly, the cells collected after stimulation were lysed by using the kit-supplied buffer and were added to the plates pre-coated with the capture antibody together with the detection antibody. After the unbound antibodies had been washed off, substrate was added for color development and the plates were read at an optical density of 450 nm.
Western blot
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with protease and phosphatase inhibitors. The protein content of the lysates was estimated by using the Pierce BCA Protein Assay Kit (Thermo, Rockford, Ill., USA). Protein (30 μg) was loaded onto 12% polyacrylamide gels and then transferred to polyvinylidene fluoride (PVDF) membranes for blotting. The PVDF membranes were blocked with 5% skim milk in PBS with 0.05% TWEEN20 (PBST), stained with goat anti-mouse DUSP1 or SHP-1 antibody (Sigma-Aldrich) overnight at 4 °C, washed with PBST, stained with rabbit anti-goat IgG-HRP secondary antibody (Invitrogen, Carlsbad, Calif., USA) in 5% skim milk in PBST, washed with PBST followed by PBS and then soaked in Perkin Elmer Western Lightning Plus ECL chemiluminescent substrate (Perkin Elmer, Woodbridge, ON, Canada). Staining was detected on X-ray film and scanned into a computer. ImageJ software was used for densitometric analysis and background correction.
Data analysis
In experiments that evaluated the relative gene expression in iNKT cells, the PCR efficiency (E) of the plasmid standards was calculated by using the formula: E = 10–1/slope. The ratio of the target gene expression was based on their relative expression versus the level of β-actin or TBP (TATA-binding protein; house-keeping gene) expression, as a reference and was calculated by the formula: Ratio = (E T)ΔCp target [calibrator-sample] / (E R)ΔCp reference[sample-calibrator].
In this formula, E T, E R and ΔC p represent the efficiency of the target gene in the standard curve the efficiency of the reference in the standard curve, and the difference between crossing points, respectively. The relative expression of all genes of interest was calculated based on the expression of β-actin or TBP by using Pfaffl’s formula as we described previously (Pfaffl 2001).
Experimental cell cultures were performed in triplicates and sets of triplicate data were compared between the groups by using the two-tailed Student’s t-test. Differences in relative target gene expression between treatment and control culture groups were considered significant at P ≤ 0.05. The data were expressed as the means ± SEM.
The abundance of cytokines in the triplicate culture supernatants of iNKT cells was calculated by using standard curves generated for each cytokine. The data are expressed as the mean cytokine concentrations ± SD in triplicate cultures of each independent experiment. These experiments were repeated at least three times independently and the results were consistently reproducible. The results shown represent one independent experiment. Statistical comparisons were performed by using the t-test and differences were considered statistically significant at P-values ≤0.05 and ≤0.01.
Results
Ex vivo co-stimulation of mouse iNKT cells with α-GalCer and CpG leads to cellular up-regulation of TLR9 expression and decreased IFN-γ production
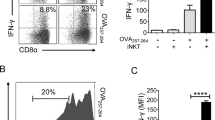
We have previously observed that certain TLR agonists such as CpG can modulate iNKT cell activation when co-stimulated in the presence of α-GalCer (Kulkarni et al. 2012). We therefore sought, by using mouse liver and splenic primary cell cultures, to investigate the co-stimulatory effects of CpG when cells were cultured for 24 h while being subject to α-GalCer-induced TCR-activation. From the results depicted in Fig. 1, three important observations were made. First, although hepatic and splenic iNKT cells were found to have a constitutive expression of TLR9, TCR signaling in these cells enhanced TLR9 expression by approximately two-fold. Furthermore, co-stimulation with CpG in the presence of α-GalCer significantly enhanced TLR9 expression in iNKT cells. This observation supports our previous finding that TLR co-stimulation of TCR-activated iNKT cells can up-regulate iNKT cell expression of TLRs (Kulkarni et al. 2012). Second, TCR activation of hepatic and splenic iNKT cells enhanced IFN-γ production by four- to six-fold compared with controls receiving no α-GalCer. However, CpG treatment significantly down-regulated the α-GalCer-induced TCR activation of iNKT cells by at least two-fold, as measured by their production of IFN-γ. This observation suggested that CpG-mediated TLR co-stimulation of iNKT cells regulated their activation status. Lastly, the down-regulation of iNKT cell activation in the CpG-treated group did not appear to occur because of cell death, since the numbers of live iNKT cells in the α-GalCer alone and α-GalCer + CpG treatment groups were similar as determined by Live/Dead staining (data not shown). Thus, we can suggest that the CpG treatment of TCR-activated iNKT cells results in the up-regulation of iNKT cell expression of TLR9 with concurrent down-modulation of their activation status.

CpG-ODN mediated invariant natural killer T (iNKT) cell expression of TLR9 and IFN-γ. Single cell suspensions were prepared by processing mouse liver and spleen tissues. Cells were stimulated with α-GalCer with or without CpG for 24 h and harvested cells post-stimulation were surface-stained with CD1d tetramers and TLR9 antibodies followed by intracellular cytokine staining by interferon-γ (IFN-γ) antibody. The control group received neither α-GalCer nor CpG. The representative flow plots show cell frequencies observed in liver (a) and spleen (b) tissue. The bar graphs adjacent to plots (a’, b’) show the frequency of positively stained cell population. Each treatment group contained three to four mice. Data from one of three independent experiments are presented. *P ≤ 0.05 (significant differences when compared with mice that received α-GalCer alone). Error bars indicate measn ± SEM (aGC α-GalCer)
In vitro co-stimulation of DN32.D3 iNKT cells in the presence of α-GalCer leads to down-regulation of iNKT activation and up-regulation of TLR9 transcription
We next sought to investigate the CpG-mediated effects in the primary iNKT cell population. However, several challenges were faced in obtaining a highly pure population of primary iNKT cells and, as a result, an alternative system needed to be found. First, primary iNKT cell preparations are likely to be contaminated with APC and T cells. Although these cells might be present in small numbers, their contamination may lead to misleading results, as these contaminating cells are known to have abundant TLR expression. Second, the use of CD1d tetramers for the sorting of iNKT cells will probably activate iNKT cells, at least in part, during staining and thus will affect the primary aim of the present study, which is to examine TLR9/CpG-mediated effects on iNKT cell activation. Lastly, cell sorting generates a low yield of iNKT cells, which poses limitations for our investigations. Therefore, we used DN32.D3 cells that have been extensively used by many groups, as in our previous studies (Kulkarni et al. 2012; Stuart et al. 2010; Villanueva et al. 2014; Zanin-Zhorov and Cohen 2013). DN32.D3 are murine iNKT cells with the Vα14-Jα18 rearranged TCRα (Lantz and Bendelac 1994). These cells can be activated with α-GalCer in the absence of APC or FcR-expressing accessory cells to produce several cytokines including IL-2, IL-4 and IFN-γ. DN32.D3 cells express their own CD1d, as determined by flow cytometry; this enables the cross-presentation of α-GalCer leading to cellular activation (Stuart et al. 2010). Since this system provides a pure iNKT cell population, we chose to address TLR9 expression and CpG-mediated functions in the activation of iNKT cells as a useful model in our subsequent experiments, thus avoiding potential contamination by other cell types.
DN32.D3 iNKT cells were co-stimulated with CpG in the presence of α-GalCer for up to 72 h and the supernatants were analyzed to quantitate the amounts of IL-2, IL-4 and IFN-γ as indicators of iNKT cell activation. A significant reduction occurred in IL-2 levels by about three- to four-fold at 24 h post-stimulation in the group that received CpG together with α-GalCer (Fig. 2a). Importantly, this reduction was markedly prolonged for up to 48 h; however, no significant differences were observed at 72 h post-stimulation, although the trend of reduction in IL-2 was evident in the CpG-treated group (data not shown). A similar reduction in the iNKT cell production of IL-4 and IFN-γ was also observed in groups receiving CpG (Fig. 2b, c). Furthermore, we confirmed a significant transcriptional up-regulation of the TLR9 gene in DN32.D3 cells receiving CpG together with α-GalCer at 24 h post-stimulation (Fig. 2d). This observation was in agreement with the aforementioned finding that CpG treatment of TCR-activated mouse hepatic and splenic iNKT cells results in the up-regulation of TLR9 protein. Together, these findings indicate that the co-stimulation of α-GalCer-activated iNKT cells with CpG results in a significant reduction in their activation status concomitantly with the transcriptional up-regulation of TLR9 genes.

CpG-mediated co-stimulatory regulation of iNKT cell activation in the presence of α-GalCer. DN32.D3 iNKT cells were stimulated with α-GalCer alone or in the presence of CpG or CpG control. iNKT cell activation was determined by the production of interleukin-2 (IL-2; a), IL-4 (b) and IFN-γ (c) in the culture supernatants as determined by enzyme-linked immunosorbent assay (ELISA) at various time points post-stimulation, as indicated. The values in the group treated with α-GalCer + CpG were statistically different compared with those in the α-GalCer alone or the α-GalCer + CpG control groups (*P ≤ 0.05, **P ≤ 0.01). These experiments were repeated at least three times independently and the results were found to be consistently reproducible. The representative data from one independent experiment are shown here as the mean IL-2, IL-4, or IFN-γ concentration ± SD in triplicate cultures. In d, DN32.D3 cells were stimulated with α-GalCer alone or in the presence of CpG or CpG control and the cells were collected at 24, 48 and 72 h post-stimulation. Total RNA was extracted followed by cDNA synthesis and quantitative real-time polymerase chain reaction (PCR) was performed to determine the expression of the TLR9 gene in these cells. The results are shown as the expression of the TLR9 gene relative to the expression of the housekeeping gene, β-actin. Experimental cell cultures were performed in triplicates and sets of triplicate data in the goup treated with α-GalCer + CpG were statistically different compared with those in the α-GalCer alone or the α-GalCer + CpG control groups (aGC α-GalCer, CpGC non-CpG control)
CpG treatment of iNKT cells in presence of anti-CD3 mAb leads to down-regulation of iNKT activation
DN32.D3 cells can also be efficiently activated through anti-CD3 ligation (Stuart et al. 2010). In order to assess the regulatory effects of CpG in TCR-activated iNKT cells, in addition to α-GalCer, we used anti-CD3 mAb to activate iNKT cells. DN32.D3 cells were co-stimulated with CpG in the presence of anti-CD3 mAb for up to 72 h and the supernatants were analyzed to quantitate IL-2, IL-4 and IFN-γ as readouts for iNKT cell activation. A significant reduction in IL-2 by about five-fold at 24 h post-stimulation was seen in the group that received CpG together with anti-CD3 mAb (Fig. 3a). Interestingly, this reduction was significantly prolonged for up to 72 h with about a four-fold difference between the CpG-treated and untreated groups. Furthermore, a similar reduction in the iNKT cell production of IL-4 and IFN-γ was observed in groups receiving CpG treatment for up to 72 h post-stimulation (Fig. 3b, c). Taken together, our findings indicate that the CpG-mediated co-stimulation of TCR (α-GalCer or anti-CD3 mAb)-activated iNKT cells results in a significant down-regulation of iNKT cellular activation status.

CpG-mediated co-stimulatory regulation of iNKT cell activation in the presence of anti-CD3 antibodies. DN32.D3 iNKT cells were stimulated with α-GalCer alone or in the presence of anti-CD3 antibodies. iNKT cell activation was determined by the production of IL-2 (a), IL-4 (b) and IFN-γ (c) in the culture supernatants by ELISA at various time points post-stimulation, as indicated. The values in the group treated with anti-CD3 antibodies + CpG were statistically different compared with those in the α-GalCer alone group (*P ≤ 0.05). These experiments were repeated at least three times independently and the results were found to be consistently reproducible. The representative data from one independent experiment are shown here as the mean IL-2, IL-4, or IFN-γ concentration ± SD in triplicate cultures (CD3 anti-CD3 antibodies)
CpG-mediated down-regulation of TCR-induced iNKT cell activation is dependent on MyD88 signaling
To determine whether the CpG-mediated suppression of iNKT cell production of cytokines was mediated by TLR signaling, we used an inhibitor of MyD88 (PepinhMYD), which is a 26-amino-acid polypeptide that blocks MyD88 signaling by inhibiting its homodimerization through binding. As was evident from our observations, MyD88 inhibition in DN32.D3 cells significantly restored the TCR (α-GalCer or anti-CD3 mAb)-induced production of IFN-γ (Fig. 4). Since the restoration of CpG-mediated suppression was only partial (although statistically significant), particularly in cells activated in the presence of anti-CD3 mAb, other DNA-sensing PRR mechanisms might be capable of engaging CpGs, in addition to TLR9/MyD88 signaling.

MyD88-dependent CpG-mediated down-regulation of TCR-induced iNKT cell activation. DN32.D3 iNKT cells were pretreated with a peptide inhibitor of MyD88 (PepinhMYD) at a concentration of 25 μM followed by stimulation with TCR agonists (α-GalCer or anti-CD3 antibodies) with or without CpG as indicated. Culture supernatants were collected 24 h post-stimulation and the IFN-γ concentrations were quantified by ELISA. Control groups received non-CpG control and the values in MyD88 inhibitor-treated groups were statistically different compared with those that did not receive the inhibitor (*P ≤ 0.05). These experiments were repeated three times independently and the representative data from one independent experiment are shown here as the mean IFN-γ concentration ± SD in triplicate cultures (CD3 mAb anti-CD3 antibodies)
CpG treatment of iNKT cells in presence of TCR agonists affects MAPK signaling
Stimulation of T cells through TCR activates the MAPK signal transduction pathway leading to T cell activation (Gorjestani et al. 2008). We hypothesized that CpG-mediated suppression of cytokine production in iNKT cells was related to the regulation of the MAPK signaling pathway. Extracellular signal-regulated kinase (ERK), p38 and Jun N-terminal kinases (JNKs) are important members of the MAPK pathway (Dong et al. 2002). These kinases are activated upon the phosphorylation of the TxY motif in response to various cellular and environmental stimuli and play crucial roles in a variety of biological processes, including growth, survival, differentiation and cytokine secretion. Notably, the MAPK pathway involving ERK1/2 and p38 MAPKs plays an important role in the iNKT cell production of cytokines (Stuart et al. 2010). Therefore, we sought to quantitate the amounts of phosphorylated MAPKs (ERK1/2, p38 MAPKK, and JNK1/2/3) in the iNKT cells stimulated with CpG and α-GalCer, together with appropriate controls. As shown in Fig. 5a, the levels of ERK1/2 and p38 MAPKK were significantly reduced in cells treated with CpG compared with controls and indeed, these values were comparable with those observed in the unstimulated iNKT cells.

CpG treatment of iNKT cells in the presence of TCR agonists affects mitogen-activated protein kinase (MAPK) signaling. To determine whether CpG treatment of α-Galcer-activated DN32.D3 iNKT cells affected MAPK signaling, the amounts of phosphorylated MAPKs (ERK1/2, p38 MAPKK and JNK1/2/3) were quantitated by using a commercial ELISA kit (a). Whereas the lysate provided by the manufacturer was used as the positive control, the unstimulated cells were considered as a negative control. The values in the group treated with α-GalCer + CpG were statistically different compared with those in the α-GalCer alone or the α-GalCer + CpG control groups (*P ≤ 0.05). In b, c, additive effects of CpG in blocking MAPK signaling in TCR-activated iNKT cells with PD98059 (ERK1/2 inhibitor) and SB203580 (p38 MAPK inhibitor) used at 20 μM were investigated. Amounts of IL-2 were quantified in the culture supernatants as a measure of iNKT cell activation. Cytokine levels in the group treated with α-GalCer + CpG were statistically different compared with those in the α-GalCer alone or the α-GalCer + inhibitor groups (*P ≤ 0.05, **P ≤ 0.01). Experiments were repeated three times independently and the results from one independent experiment are shown as means ± SD (aGC α-GalCer, CpGC non-CpG control, PD-inh PD98059, SB-inh SB203580, CD3 anti-CD3 antibodies, Unstim unstimulated)
Since the decrease in cytokine production by cells treated with CpG and α-GalCer coincided with inhibition of ERK1/2 and p38 MAPK phosphorylation, we questioned whether these observations were linked. To examine this further, we used two MAPK inhibitors in an attempt to replicate the inhibitory effects of CpG. We also aimed to determine whether CpG had any additive effects when combined with MAPK inhibition in iNKT cells. PD98059, a potent inhibitor of MEK1 kinase, which prevents the phosphorylation of ERK1/2 leading to the suppression of cytokine secretion, and SB203580, which is an inhibitor of p38 MAPK, were used in the present study. DN32.D3 cells treated with PD98059 or SB203580 in the presence of α-GalCer exhibited a marked reduction in IL-2 production at levels comparable with that seen in cells that received α-GalCer and CpG (Fig. 5b). The addition of CpG to the cells receiving α-GalCer and the inhibitor led to a further decrease in the production of IL-2. Although the anti-CD3-activated iNKT cells receiving the inhibitor had diminished IL-2 production, the addition of CpG to the cells led to a further decrease in IL-2 production (Fig. 5c). These findings suggest that the CpG-mediated effects were additive to the actions of the MAPK inhibitors used in our study. Collectively, these observations indicate that the CpG treatment of iNKT cells in the presence of TCR agonists induces inhibitory molecular signals upstream of the MAPK signaling pathway that affects the iNKT cell production of cytokines and, thus, their activation status.
CpG-mediated signaling in iNKT cells regulates TCR signaling through induction of phosphatases
Next, we investigated the molecular mechanisms that contribute to the reduced levels of MAPKs in the CpG-treated iNKT cells. MAPK phosphatases are key regulators of MAPK signaling and dephosphorylate MAPKs into their inactive forms (Jeffrey et al. 2007). Notably, TLR stimulation has been shown to induce the expression of MAPK phosphatases, also referred to as dual-specificity phosphatases (DUSPs), in conventional CD4+ T cells (Gonzalez-Navajas et al. 2010). Another important molecule upstream of the MAPK signaling pathway is SHP-1, a tyrosine phosphatase, which is a negative regulator of TCR signaling (Hebeisen et al. 2013; Johnson et al. 2013). Therefore, we first determined the transcriptional changes in the DUSP1, DUSP2, DUSP10, SHP-1, SHP-2 and TRAF6 genes. We found that DN32.D3 cells receiving CpG exhibited a significant up-regulation in DUSP1 gene expression at 1 h post-stimulation (Fig. 6a). In addition, significant transcriptional up-regulation of SHP-1 in CpG-treated iNKT cells was also observed at 15 min and 1 h post-stimulation (Fig. 6b). An increase in SHP-1 expression at 30 min in CpG-receiving cells was also observed; however, the difference between the groups was not statistically significant. No significant transcriptional changes in the expression of DUSP2, DUSP10, SHP-2, or TRAF6 were observed (data not shown).

CpG-mediated signaling in iNKT cells regulates TCR signaling through the induction of phosphatases. DN32.D3 iNKT cells were stimulated with anti-CD3 antibodies alone or in the presence of CpG and the cells were collected at the indicated time points post-stimulation. Total RNA was extracted followed by cDNA synthesis and quantitative real-time PCR was performed to determine the expression of the DUSP1 (a) and SHP-1 (b) genes in these cells. The results are shown as the expression of the target gene relative to that of the housekeeping gene (TBP). Experimental cell cultures were performed in triplicate and sets of triplicate data in the group treated with anti-CD3 + CpG were statistically different compared with those treated with anti-CD3 + PBS (phosphate-buffered saline) control (*P ≤ 0.05). In c, d, DN32.D3 cells were stimulated with TCR agonists (anti-CD3 antibodies in c, α-GalCer in d) alone or in the presence of CpG and the cells were collected at the indicated time points post-stimulation. Cell lysate was prepared and Western blot was performed to determine the protein expression of DUSP1 (c) and SHP-1 (d) in these cells. The bar graphs (c‘, d‘) represent the fold changes in protein expression compared with cells treated with PBS alone, as determined by densitometric quantification
We further investigated the expression of DUSP1 and SHP-1 in DN32.D3 iNKT cells at the protein level. The expression of DUSP1 in the cells receiving CpG in the presence of TCR-agonist (anti-CD3 mAb) tended to increase by as early as 1 h post-stimulation (Fig. 6c). This elevation continued for up to 24 h post-stimulation in cells treated with CpGs compared with cells stimulated with anti-CD3 mAb without CpG. Using densitometry to quantify DUSP1 expression relative to β-tubulin, we found that DUSP1 expression in cells treated with anti-CD3 and CpG remained up to 5.5 times higher than in cells treated only with anti-CD3 and PBS (Fig. 6c). In addition to DUSP1 expression, we also found a two-fold increase in SHP-1 expression in cells that received CpG compared with those treated with TCR agonist alone (Fig. 6d). Taken together, these observations indicate that the CpG treatment of iNKT cells concomitantly with TCR agonists leads to the induction of certain phosphatases that interfere with MAPK and TCR signaling, which in turn negatively regulates iNKT cell activation.
Discussion
The host recognition of microbial components is a critical event in the initiation of innate responses and TLRs expressed by the cells of the immune system facilitate this process of recognition. Although only APCs were previously thought to express TLRs, various T cell subsets can clearly also express TLRs, some of which are functional (Kabelitz 2007; Kulkarni et al. 2011). Invariant NKT cells that express both NK cell recptors and TCRs are positioned strategically at the interface of innate and adaptive immune system components. Recently, we and others have shown that co-stimulation with certain TLR agonists in iNKT cells receiving suboptimal TCR signals results in the enhancement of their activation (Gardner et al. 2010; Kulkarni et al. 2012; Villanueva et al. 2014). HMGB1 (high mobility group box 1) protein, which interacts with certain TLRs, has also been suggested to induce pro-inflammatory cytokine production in T cells (Kudo et al. 2013). Interestingly, certain TLR agonists such as CpG-ODN have additionally been shown to serve a regulatory function mediated by B cells in controlling DC maturation (Maddur et al. 2012, 2014). To this end, an important observation from our previous study indicated that certain TLR ligands such as CpG can negatively regulate iNKT cell activation in the presence of higher concentrations of α-GalCer. In the present study of mouse primary iNKT cells and DN32.D3 cells, we show that CpG co-stimulation of iNKT cells in the presence of higher concentrations of TCR agonists (α-GalCer or anti-CD3 mAb) results in decreased iNKT cell activation. These regulatory effects are dependent, at least in part, on signaling by the MyD88 adapter moiety and CpG co-stimulation leads to diminished MAPK and TCR signaling through the induction of DUSP1 and SHP-1 phosphatases.
From a biological perspective, the regulation of T cell responses is essential for immune homeostasis in order to prevent unwanted T cell induced pathology (Hou et al. 2016). Recently, Juno et al. (2012) have underscored the important aspects of iNKT cell regulation and their function in the context of viral infections. Reports have been presented showing that TLRs, in addition to modulating APC responses, can also regulate T cell activation (Johnson et al. 2007; Shepherd et al. 2004). For example, Gonzalez-Navajas et al. (2010) found that mice deficient in TLR4 are more prone to develop colitis and the direct TLR4 co-stimulation of antigen-experienced CD4+ T cells with LPS leads to decreased IFN-γ production (Gonzalez-Navajas et al. 2010). Similarly, the TLR2- and TLR4-mediated modulation of natural T regulatory cells has also been documented (Zanin-Zhorov and Cohen 2013). In this context, we found that the CpG treatment of LMNC and splenocytes and of DN32.D3 iNKT cells in the presence of α-GalCer or anti-CD3 mAb results in the decreased production of IL-2, IL-4 and IFN-γ. Importantly, the down-regulation of iNKT cell activation is prolonged for up to 72 h post-stimulation in vitro (Figs. 2, 3). In the context of bacterial infections in which iNKT cells receive more than adequate activation signals (Suzuki et al. 2004; Tupin et al. 2007), our observations suggest that CpG-like ligands can modulate the cellular activation status to avoid cytokine-mediated tissue damage. Notably, the liver NKT cell population can also contain a newly characterized mucosal-associated invariant T cell (MAIT) that possesses semi-invariant TCR (Vα19-Jα33) and recognizes molecules presented on the non-polymorphic MHC-related protein 1 (Heymann and Tacke 2016; Hinks 2016). Although our ex vivo experiment involves the use of glycolipid loaded CD1d tetramers specific for Vα14-Jα18 iNKT TCR, there still exists the rare possibility of our gated iNKT cell population containing MAIT cells.
CpG is a potent TLR9 agonist, and the signaling by TLR9 requires the MyD88 adapter protein (Marshall et al. 2005). In the present study, we observed a significant up-regulation of TLR9 expression in iNKT cells at both transcript and protein levels when primary LMNC, splenocytes and DN32.D3 cells are treated with CpG and TCR agonists. Using a MyD88 inhibitor, we found that the CpG-mediated suppressive effect on iNKT cell activation is significantly abrogated (Fig. 4). However, the restoration of CpG-mediated suppression is only partial (although statistically significant), particularly in cells activated in the presence of the anti-CD3 mAb. Therefore, in addition to TLR9/MyD88 signaling, other DNA-sensing PRR mechanisms might be capable of engaging CpGs. Indeed, enough evidence exists to show that CpGs can also be recognized by other cytosolic DNA-sensing receptors, such as Z-DNA-binding protein 1 (ZBP1) and activation of IFN regulatory factors absent in melanoma 2 (AIM2) in a TLR9/MyD88-independent mechanism (Landrigan et al. 2011; Vilaysane and Muruve 2009).
Stimulation of T cells through the TLR or TCR pathway results in the activation of MAPK (ERK, p38 MAPK and JNK) and MAPK signaling in these cells is necessary for their effector functions. For example, all three MAPKs are involved in CD8+ T cell cytotoxicity (Dong et al. 2002) and whereas ERK or p38 MAPK are critical for CD4+ T cell polarization and for the production of IFN-γ (Agrawal et al. 2006; Pages et al. 1999), JNK activation is necessary for IL-4 production (Rincon et al. 1997). Notably, certain PRR ligands such as LPS can negatively regulate CD4+ T cell responses by affecting intraceullar MAPK signaling (Gonzalez-Navajas et al. 2010). Recently, Stuart et al. (2010) showed that TCR stimulation in iNKT cells leads to the activation of ERK and p38 MAPK but not JNK (Stuart et al. 2010). In the present study, we also have shown that the inhibition of ERK1/2 and p38 MAPK activation can abrogate iNKT cell activation induced by α-GalCer or anti-CD3 mAb. Moreover, the amounts of IL-2 production in iNKT cells that received α-GalCer + MAPK inhibitors were very similar to those receiving α-GalCer + CpG (Fig. 5b). This observation suggests that CpG treatment results in the downstream inhibition of MAPK activity. In support of this suggestion, we indeed observed that the addition of CpG to iNKT cells receiving the TCR agonist together with MAPK inhibitors leads to a further sizable reduction in iNKT cell activation. These additive inhibitory effects of CpG clearly indicate events that impede downstream MAPK signaling. Furthermore, we confirmed that the CpG-treated iNKT cells have substantially reduced levels of phosphorylated ERK1/2 and p38 MAPK proteins indicating an interference in MAPK signaling in these cells. Along similar lines, an earlier report also suggsested that CpG can suppress regulatory T cell functions through an affect on ERK phosphorylation and, thus, MAPK signaling (Johnson et al. 2007).
An important conjecture from our findings is that the regulatory nature of CpGs depends on the level of TCR reactivity. In our previous study, when iNKT cells were activated by using suboptimal concentrations of TCR agonists, CpG co-stimulation in these cells led to an enhancement of their activation (Kulkarni et al. 2012). Contrastingly, in the present study, CpG co-stimulation of iNKT cells in the presence of higher concentrations of TCR agonists resulted in diminished activation. This “conditional” regulation by the CpG in iNKT cells appears to operate through the fine-tuning of the control mechanisms that regulate TCR signaling. Since both the TLR and TCR signaling pathways cross-talk and utilize MAPKs to exert their actions, one of the “regulatory check-points” for the CpG-mediated effects observed in the present study is presumably at the level of MAPK signaling.
Signaling by MAPKs can be modulated at the molecular level by various mechanisms. One such mechanism is the dephosphorylation of MAPKs by DUSPs (Xu et al. 2014). DUSPs are a family of protein tyrosine phosphatases that are expressed either in the nuclear or cytosol compartment and that chiefly dephoshorylate threonine and tyrosine residues on MAPKs (Jeffrey et al. 2007). Of this family, DUSP1 has been implicated as the crucial negative regulator of the innate immune system (Liu et al. 2007). For example, Chen et al. (2002) found that LPS stimulation of a macrophage cell line leads to increased DUSP1 expression afftecting p38 MAPK signaling and similarly, others also observed DUSP1 up-regulation in APCs stimulated with CpG and peptidoglycan resulting in diminished ERK or p38 MAPK signaling (Chi et al. 2006; Salojin et al. 2006). DUSPs are also induced in T cells in response to TCR and TLR co-stimulation (Gonzalez-Navajas et al. 2010; Liu et al. 2007). In this context, we assessed the induction of DUSP1 (nuclear), DUSP2 (cytosolic) and DUSP10 (nuclear and cytosolic) in iNKT cells co-stimulated with CpG in the presence of TCR agonists. We observed an increase in DUSP1 gene transcription in CpG co-stimulated cells early on at 6 h with a corresponding increase in DUSP1 protein expression by 24 h post-stimulation. We further examined the expression of SHP-1, a well-characterized negative regulator of TCR reactivity in T cells and found that SHP-1 is also markedly increased at both the transcriptional and protein levels in CpG co-stimulated iNKT cells. Taken together, our observations suggest that CpG co-stimulation in TCR-activated iNKT cells induces the expression of certain phosphatases that affect both upstream TCR and downstream MAPK signaling to dampen cellular activation. Placing our observations in a biological perspective, we can reasonably infer that DUSP1 and SHP-1 phosphatases provide a crucial negative feedback mechanism to attenuate the activity of ERK and p38 MAPKs during innate responses against various microbial components.
In summary, our results show that CpG co-stimulation in iNKT cells can modulate TCR-induced activation independently of APC-derived signals and that the effects mediated by CpG depend, at least in part, on MyD88 signaling. These effects seem to operate through the blocking of MAPK and TCR pathway signaling via the induction of certain phosphatases such as DUSP1 and SHP-1. This may have some biological value in that excessive iNKT cell activation in response to microbial insults needs a regulatory apparatus to avoid unwanted cytokine-mediated immunopathology. Notably, mouse Vα14 NKT cells are functionally very similar to human Vα24 NKT cells in many ways including their cytotoxic activity against tumor/infected cells and their importance in autoimmunity (Takahashi et al. 2002). Furthermore, α-GalCer is a potent TCR agonist for both mouse and human iNKT cells whose engagement induces the robust production of IFN-γ and IL-4 cytokines. Therefore, our findings may have clinical implications in the control of overactive NKT-cell-mediated complications such as arthritis, liver damage, atherosclerosis and contact hypersensitivity (Chiba et al. 2008).
References
Agrawal A, Dillon S, Denning TL, Pulendran B (2006) ERK1−/− mice exhibit Th1 cell polarization and increased susceptibility to experimental autoimmune encephalomyelitis. J Immunol 176:5788–5796
Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H, Delneste Y (2005) Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol 175:1551–1557
Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y (2002) Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol 169:6408–6416
Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA (2006) Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci U S A 103:2274–2279
Chiba A, Dascher CC, Besra GS, Brenner MB (2008) Rapid NKT cell responses are self-terminating during the course of microbial infection. J Immunol 181:2292–2302
Cottalorda A, Verschelde C, Marcais A, Tomkowiak M, Musette P, Uematsu S, Akira S, Marvel J, Bonnefoy-Berard N (2006) TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol 36:1684–1693
Dong C, Davis RJ, Flavell RA (2002) MAP kinases in the immune response. Annu Rev Immunol 20:55–72
Gardner TR, Chen Q, Jin Y, Ajuebor MN (2010) Toll-like receptor 3 ligand dampens liver inflammation by stimulating Valpha 14 invariant natural killer T cells to negatively regulate gammadeltaT cells. Am J Pathol 176:1779–1789
Gelman AE, Zhang J, Choi Y, Turka LA (2004) Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol 172:6065–6073
Gong J, Yu H, Liu T, Gill JJ, Chambers JR, Wheatcroft R, Sabour PM (2008) Effects of zinc bacitracin, bird age and access to range on bacterial microbiota in the ileum and caeca of broiler chickens. Am J Pathol 104:1372–1382
Gonzalez-Navajas JM, Fine S, Law J, Datta SK, Nguyen KP, Yu M, Corr M, Katakura K, Eckman L, Lee J, Raz E (2010) TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest 120:570–581
Gorjestani S, Rider V, Kimler BF, Greenwell C, Abdou NI (2008) Extracellular signal-regulated kinase 1/2 signalling in SLE T cells is influenced by oestrogen and disease activity. Lupus 17:548–554
Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, Speiser DE, Rufer N (2013) SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest 123:1044–1056
Heymann F, Tacke F (2016) Immunology in the liver—from homeostasis to disease. Nat Rev Gastroenterol Hepatol 13:88–110
Hinks TS (2016) Mucosal-associated invariant T cells in autoimmunity, immune-mediated diseases and airway disease. Immunology 148:1–12
Hou X, Hao X, Zheng M, Xu C, Wang J, Zhou R, Tian Z (2016) CD205-TLR9-IL-12 axis contributes to CpG-induced oversensitive liver injury in HBsAg transgenic mice by promoting the interaction of NKT cells with Kupffer cells. Cell Mol Immunol 13:1–10
Jeffrey KL, Camps M, Rommel C, Mackay CR (2007) Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov 6:391–403
Johnson TV, Camras CB, Kipnis J (2007) Bacterial DNA confers neuroprotection after optic nerve injury by suppressing CD4+CD25+ regulatory T-cell activity. Invest Ophthalmol Vis Sci 48:3441–3449
Johnson DJ, Pao LI, Dhanji S, Murakami K, Ohashi PS, Neel BG (2013) Shp1 regulates T cell homeostasis by limiting IL-4 signals. J Exp Med 210:1419–1431
Juno JA, Keynan Y, Fowke KR (2012) Invariant NKT cells: regulation and function during viral infection. PLoS Pathog 8:e1002838
Kabelitz D (2007) Expression and function of toll-like receptors in T lymphocytes. Curr Opin Immunol 19:39–45
Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY (2004) TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A 101:3029–3034
Kudo D, Toyama M, Aoyagi T, Akahori Y, Yamamoto H, Ishii K, Kanno E, Maruyama R, Kaku M, Kushimoto S, Kawakami K (2013) Involvement of high mobility group box 1 and the therapeutic effect of recombinant thrombomodulin in a mouse model of severe acute respiratory distress syndrome. Clin Exp Immunol 173:276–287
Kulkarni RR, Haeryfar SM, Sharif S (2010) The invariant NKT cell subset in anti-viral defenses: a dark horse in anti-influenza immunity? J Leukoc Biol 88:635–643
Kulkarni R, Behboudi S, Sharif S (2011) Insights into the role of toll-like receptors in modulation of T cell responses. Cell Tissue Res 343:141–152
Kulkarni RR, Villanueva AI, Elawadli I, Jayanth P, Read LR, Haeryfar SM, Sharif S (2012) Costimulatory activation of murine invariant natural killer T cells by toll-like receptor agonists. Cell Immunol 277:33–43
Landrigan A, Wong MT, Utz PJ (2011) CpG and non-CpG oligodeoxynucleotides directly costimulate mouse and human CD4+ T cells through a TLR9- and MyD88-independent mechanism. J Immunol 187:3033–3043
Lantz O, Bendelac A (1994) An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J Exp Med 180:1097–1106
Liu Y, Shepherd EG, Nelin LD (2007) MAPK phosphatases—regulating the immune response. Nat Rev Immunol 7:202–212
Maddur MS, Kaveri SV, Bayry J (2012) Regulation of human dendritic cells by B cells depends on the signals they receive. Blood 119:3863–3864
Maddur MS, Sharma M, Hegde P, Stephen-Victor E, Pulendran B, Kaveri SV, Bayry J (2014) Human B cells induce dendritic cell maturation and favour Th2 polarization by inducing OX-40 ligand. Nat Commun 5:4092
Marshall JD, Fearon KL, Higgins D, Hessel EM, Kanzler H, Abbate C, Yee P, Gregorio J, Cruz TD, Lizcano JO, Zolotorev A, McClure HM, Brasky KM, Murthy KK, Coffman RL, Nest GV (2005) Superior activity of the type C class of ISS in vitro and in vivo across multiple species. DNA Cell Biol 24:63–72
Nagarajan NA, Kronenberg M (2007) Invariant NKT cells amplify the innate immune response to lipopolysaccharide. J Immunol 178:2706–2713
Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J (1999) Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 286:1374–1377
Paget C, Mallevaey T, Speak AO, Torres D, Fontaine J, Sheehan KC, Capron M, Ryffel B, Faveeuw C, Leite de Moraes M, Platt F, Trottein F (2007) Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity 27:597–609
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Rincon M, Derijard B, Chow CW, Davis RJ, Flavell RA (1997) Reprogramming the signalling requirement for AP-1 (activator protein-1) activation during differentiation of precursor CD4+ T-cells into effector Th1 and Th2 cells. Genes Funct 1:51–68
Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T (2006) Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol 176:1899–1907
Selvanantham T, Escalante NK, Cruz Tleugabulova M, Fieve S, Girardin SE, Philpott DJ, Mallevaey T (2013) Nod1 and Nod2 enhance TLR-mediated invariant NKT cell activation during bacterial infection. J Immunol 191:5646–5654
Sharif S, Arreaza GA, Zucker P, Mi QS, Delovitch TL (2002) Regulation of autoimmune disease by natural killer T cells. J Mol Med 80:290–300
Shepherd EG, Zhao Q, Welty SE, Hansen TN, Smith CV, Liu Y (2004) The function of mitogen-activated protein kinase phosphatase-1 in peptidoglycan-stimulated macrophages. J Biol Chem 279:54023–54031
Stuart JK, Bisch SP, Leon-Ponte M, Hayatsu J, Mazzuca DM, Maleki Vareki S, Haeryfar SM (2010) Negative modulation of invariant natural killer T cell responses to glycolipid antigens by p38 MAP kinase. Int Immunopharmacol 10:1068–1076
Suzuki Y, Wakita D, Chamoto K, Narita Y, Tsuji T, Takeshima T, Gyobu H, Kawarada Y, Kondo S, Akira S, Katoh H, Ikeda H, Nishimura T (2004) Liposome-encapsulated CpG oligodeoxynucleotides as a potent adjuvant for inducing type 1 innate immunity. Cancer Res 64:8754–8760
Takahashi T, Chiba S, Nieda M, Azuma T, Ishihara S, Shibata Y, Juji T, Hirai H (2002) Cutting edge: analysis of human V alpha 24+CD8+ NK T cells activated by alpha-galactosylceramide-pulsed monocyte-derived dendritic cells. J Immunol 168:3140–3144
Tupin E, Kinjo Y, Kronenberg M (2007) The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol 5:405–417
Tyznik AJ, Tupin E, Nagarajan NA, Her MJ, Benedict CA, Kronenberg M (2008) Cutting edge: the mechanism of invariant NKT cell responses to viral danger signals. J Immunol 181:4452–4456
Vilaysane A, Muruve DA (2009) The innate immune response to DNA. Semin Immunol 21:208–214
Villanueva AI, Haeryfar SM, Mallard BA, Kulkarni RR, Sharif S (2014) Functions of invariant NK T cells are modulated by TLR ligands and IFN-alpha. Innate Immun 21:275–288
Wu L, Van Kaer L (2009) Natural killer T cells and autoimmune disease. Curr Mol Med 9:4–14
Xu T, Wu X, Chen Q, Zhu S, Liu Y, Pan D, Chen X, Li D (2014) The anti-apoptotic and cardioprotective effects of salvianolic acid a on rat cardiomyocytes following ischemia/reperfusion by DUSP-mediated regulation of the ERK1/2/JNK pathway. PLoS One 9:e102292
Zanin-Zhorov A, Cohen IR (2013) Signaling via TLR2 and TLR4 directly down-regulates T cell effector functions: the regulatory face of danger signals. Front Immunol 4:211
Acknowledgements
The authors thank the NIH core tetramer facility at Emory University, Atlanta, Ga., USA for providing CD1d tetramers. They are also grateful to Emory University and University of Guelph Flow Cytometry Core Facility for help with flow cytometry. This work was partially supported by the National Institutes of Health (NIH) grants R01AI086133 and U19AI083019 to K.M.-K.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors of this manuscript declare that they have no conflict of interest.
Statement on the welfare of animals
The authors state that all applicable international, national, and/or institutional guidelines for the care and use of animals were followed, and all procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Additional information
Alexander Ian Villanueva should be considered as the co-first author
Rights and permissions
About this article

Cite this article
Kulkarni, R.R., Villanueva, A.I., Read, L.R. et al. CpG oligonucleotide-mediated co-stimulation of mouse invariant natural killer T cells negatively regulates their activation status. Cell Tissue Res 369, 541–554 (2017). https://doi.org/10.1007/s00441-017-2631-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-017-2631-y