
Abstract
Sertoli cells provide the structural and nutritional support for germ cell development; they actively metabolize glucose and convert it to lactate, which is an important source of energy for germ cells. Furthermore, Sertoli cells can oxidize fatty acids, a metabolic process that is assumed to fulfill their own energy requirements. Fatty acids are stored as triacylglycerides within lipid droplets. The regulation of fatty acid storage in conjunction with the regulation of lactate production may thus be relevant to seminiferous tubule physiology. Our aim is to evaluate a possible means of regulation by the PPARγ activation of lipid droplet formation and lactate production. Sertoli cell cultures obtained from 20-day-old rats were incubated with Rosiglitazone (10 μM), a PPARγ activator, for various periods of time (6, 12, 24 and 48 h). Increased triacylglycerides levels and lipid droplet content were observed, accompanied by a rise in the expression of genes for proteins involved in fatty acid storage, such as the fatty acid transporter Cd36, glycerol-3-phosphate-acyltransferases 1 and 3, diacylglycerol acyltransferase 1 and perilipins 1, 2 and 3, all proteins that participate in lipid droplet formation and stabilization. However, PPARγ activation increased lactate production, accompanied by an augmentation in glucose uptake and Glut2 expression. These results taken together suggest that PPARγ activation in Sertoli cells participates in the regulation of lipid storage and lactate production thereby ensuring simultaneously the energetic metabolism for the Sertoli and germ cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spermatogenesis is an intricate process that results from multiple and complex interactions between various testicular cells (Parvinen 1982). Sertoli cells (SC) are widely accepted to be necessary in order to provide an adequate and protected environment for germ cell development within the seminiferous tubules. Studies of SC glucose metabolism have shown that SC actively metabolize glucose but that the vast majority is converted to lactate (Robinson and Fritz 1981). Lactate produced by SC represents the preferential energy substrate for germ cells in the adluminal compartment (Boussouar and Benahmed 2004). In this metabolic scenario, palmitate was observed to be oxidized to CO2 and the oxidation of fatty acids (FA) was suggested to yield much of the energy required by SC (Jutte et al. 1985). More recently, FA have been demonstrated to be needed to sustain SC energy status (Xiong et al. 2009). The presence of numerous lipid droplets (LD) is characteristic in SC and these LD are assumed to constitute the storage of FA. Despite numerous studies that have analyzed diverse aspects of SC physiology, the possible regulation of FA storage, which would ensure FA availability for SC energetic requirements, has not been analyzed so far.
Needless to say, FA must enter into the cell in order to be utilized. One of the proteins involved in FA uptake is FAT/CD36, an integral membrane glycoprotein that has been found in a wide variety of cells (Abumrad et al. 1993; Bonen et al. 2004). Once FA are incorporated into the cells, they are activated by being covalently linked to coenzyme A forming an acyl-CoA derivative. These acyl-CoAs are involved in the first committed step in triacylglycerol (TAG) synthesis, which is the acylation of glycerol-3-phosphate by glycerol-3-phosphate acyltransferase (GPAT; Wendel et al. 2009). The last step in this pathway converts diacylglycerol to TAG through the action of diacylglycerol acyltransferase (DGAT; Shi and Cheng 2009). TAG are stored in LD and are considered the main energy source within adipocytes. They possibly have a similar role in SC. In the last decade, proteomic and genetic studies have shown that LD appear as complex dynamic organelles (Walther and Farese 2012). LD contain a core of neutral lipid surrounded by a phospholipid monolayer and coated by specific proteins, which include proteins such as perilipins (PLINs; Sztalryd and Kimmel 2014). PLINs not only participate in lipid storage but also regulate lipid metabolism through the recruitment of lipases and other regulatory proteins to the LD surface. Thus far, the regulation of molecular mechanisms participating in FA storage as TAG and those participating in LD formation in SC have not been analyzed.
Peroxisome proliferator-activated receptor (PPAR) α (NR1C1), β/δ (NR1C2) and γ (NR1C3) are ligand-activated transcription factors that belong to the nuclear-hormone receptor superfamily (Michalik et al. 2006). All PPARs act as sensors of FA and FA derivatives and thus, PPARs control metabolic pathways involved in energy metabolism (Green and Wahli 1994). In the liver, PPARα, among other biological actions, promotes FA catabolism (Jump et al. 2005). PPARβ/δ has also been shown to enhance FA catabolism in skeletal muscle and adipose tissue and its participation in the hepatic response to starvation has been observed (Sanderson et al. 2009). With regard to PPARγ, its activation has been shown to be involved in adipocyte proliferation and differentiation (Siersbaek et al. 2010). In general terms, PPARα and PPARβ/δ regulate lipid catabolism, whereas PPARγ regulates lipid anabolism. Additionally, a role for PPARγ in the induction of genes involved in glucose metabolism has been demonstrated (Picard and Auwerx 2002). Recently, we observed that pharmacological PPARα and PPARβ/δ activation regulates the expression of genes involved in FA catabolism, such as Fat/Cd36, carnitine palmitoyltransferase 1 (Cpt1) and long- and medium-chain 3-hydroxyacyl-CoA dehydrogenases (Lcad and Mcad) in SC (Regueira et al. 2014). We also observed that PPARβ/δ activation can simultaneously regulate FA oxidation and lactate production. These results have been interpreted as a coordinated mechanism that ensures the concurrent provision of energy to SC and germ cells. A possible role of PPARγ in the regulation of the energetic metabolism of the seminiferous tubule has not been examined so far.
In the present study, we investigate a role for PPARγ activation in the regulation of FA storage as TAG within LD in SC. In addition, we hypothesize that PPARγ activation simultaneously regulates FA storage and glucose metabolism in order to ensure the provision of energy to SC and germ cells, respectively.
Materials and methods
Materials
[2,6-3H]-2-deoxy-D-glucose (2-DOG) was purchased from NEN (Boston, Mass., USA). Culture media were purchased from Invitrogen (Carlsbad, Calif., USA). Rosiglitazone and all other drugs and reagents were purchased from Sigma-Aldrich (St Louis, Mo., USA).
SC isolation and culture
SC from 20-day-old Sprague-Dawley rats (Rattus norvegicus) were isolated as previously described (Meroni et al. 2002). Briefly, decapsulated testes were digested with 0.1% w/v collagenase and 0.006% w/v soybean trypsin inhibitor in Hank’s balanced salt solution for 5 min at room temperature. Seminiferous tubules were saved, cut and submitted to 1 M glycine/2 mM EDTA (pH 7.4) treatment to remove peritubular cells. The washed tubular pellet was then digested again with collagenase for 10 min at room temperature to remove germinal cells. The SC suspension, collected by sedimentation, was resuspended in culture medium that consisted in a 1:1 mixture of Ham’s F-12 and Dulbecco’s modified Eagle’s medium, supplemented with 20 mM HEPES, 100 IU/ml penicillin, 2.5 μg/ml amphotericin B, 1.2 mg/ml sodium bicarbonate, 10 μg/ml transferrin, 5 μg/ml insulin, 5 μg/ml vitamin E, and 4 ng/ml hydrocortisone. SC were cultured in 25-cm2 flasks, 6-, 24–, or 96-multiwell plates, or 8-well chamber Permanox slides (5 μg DNA/cm2) at 34 °C in a mixture of 5% CO2:95% v/v air.
The purity of the Sertoli cells reached 95% after 4 days in culture, as examined by phase-contrast microscopy. No myoid cell contamination was revealed when an immunoperoxidase technique was applied to the Sertoli cell cultures by using a specific antiserum to alpha smooth-muscle actin. The remaining cell contaminants were of germ cell origin.
Culture conditions
SC were allowed to attach for 48 h in the presence of insulin and medium was replaced at this time with fresh medium without insulin. Stimulation with Rosiglitazone (RSG) was performed with variable doses and for variable periods of time. The cells treated for 48 h with 1 and 10 μM of RSG were harvested on day five and used to evaluate TAG content, the presence of LD, glucose uptake, lactate dehydrogenase (LDH) activity and phospho-pyruvate dehydrogenase complex (P-PDC) protein levels and the 48-h–conditioned media were utilized to evaluate lactate production and glucose consumption. Cells incubated for 6, 12, 24, or 48 h with 10 μM RSG and harvested on day five were used to determine Fat/Cd36, Gpats, Dgat1, Plins, Glut1, Glut2 and LdhA mRNA levels. During those time periods in which the stimulation of gene expression was observed, incubations with a lower dose of RSG (1 μM) were also performed.
Reverse transcription plus polymerase chain reaction
Liver, skeletal muscle, kidney and testicular tissue, plus purified SC from 20-day-old rats and germ cells from 30-day-old rat (Galardo et al. 2014), were utilized to isolate total RNA by using TRI Reagent (Sigma–Aldrich) according to the manufacturer’s recommendations. The amount of RNA was estimated by spectrophotometry at 260 nm. Reverse transcription (RT) was performed as previously described (Regueira et al. 2015). The polymerase chain reaction (PCR) products were resolved by 2% w/v agarose gel and stained with ethidium bromide.
RT with real-time PCR
Total RNA was isolated from SC cultured in 6-multiwell plates with TRI Reagent (Sigma-Aldrich). The amount of RNA was estimated by spectrophotometry at 260 nm. RT was performed on 2 μg RNA at 42 °C for 50 min with a mixture containing 200 U MMLV reverse transcriptase enzyme, 125 ng random primers and 0.5 mM dNTP Mix (Invitrogen).
Real-time PCR (qPCR) was performed with a Step One Real Time PCR System (Applied Biosystems, Warrington, UK). The specific primers for RT-qPCR are shown in Table 1. Amplification was carried out as recommended by the manufacturer: 25 μl reaction mixture containing 12.5 μl SYBR Green PCR Master mix (Applied Biosystems), the appropriate primer concentration and 1 μl cDNA. The relative cDNA concentrations were established by a standard curve by using sequential dilutions of a cDNA sample. The data were normalized to hypoxanthine phosphoribosyl transferase (HPRT). The amplification program included the initial denaturation step at 95 °C for 10 min, 40 cycles of denaturation at 95 °C for 15 s and annealing and extension at 60 °C for 1 min. Fluorescence was measured at the end of each extension step. After amplification, melting curves were acquired and used to determine the specificity of PCR products. The comparative ΔΔCt method was used to calculate relative gene expression. Results were expressed as fold stimulation (means ± SD of four independent experiments).
Oil red O staining
The LD were visualized by oil red O (ORO) staining (Wang et al. 2006). Briefly, SC cultured in 8-well chamber Permanox slides were fixed with 10% v/v formalin for 1 h and stained with ORO solution (Sigma; ORO saturated solution in isopropanol:water, 3:2) for 30 min. The background was removed by washing the cells with 70% v/v alcohol for 5 s and hematoxilin staining was performed. The LD present in SC were analyzed under an Eclipse 50i microscope with a DS Fi1 digital camera and NIS Elements version 3.0 BR imaging software (Nikon Instruments, Melville, N.Y., USA). The ratio of LD to cell nucleus was calculated. Two hundred SC were analyzed for each experimental condition performed in quadruplicate. Results were expressed as the number of lipid droplets per cell (means ± SD).
TAG measurement
Cells cultured in 25 cm2 flasks were washed twice with phosphate-buffered saline (PBS) at room temperature. SC monolayers were then treated with trypsin 0.05% w/v and EDTA 0.02% w/v in PBS, pH 7.4 for 2 min at 34 °C until the cells were detached. The trypsin action was stopped by the addition of 0.3% w/v soybean trypsin inhibitor in PBS pH 7.4. Cells collected by centrifugation at 800g for 5 min were resuspended in PBS pH 7.4 and homogenized by ultrasonic irradiation. Adequate aliquots were destined for TAG and DNA determinations. TAG was measured by a routinely used method based on the colorimetric determination of the glycerol released upon the action of lipoproteinlipase on the TAGs (TG Color, GPO/PAP AA; Wiener Lab, Rosario, Argentina). Results were expressed as micrograms TAG/micrograms DNA.
Glucose consumption evaluation
Glucose levels in fresh medium and in the 48-h-conditioned media obtain from cells cultured in 24-multiwell plates were evaluated. Glucose was measured by a standard enzymatic method according to the manufacturer’s protocol (Wiener Lab). Glucose consumption was calculated as the difference between glucose levels in fresh medium and in conditioned media. Results were expressed as micrograms glucose/micrograms DNA.
Western blot analysis
Cells cultured in 6-multiwell plates were washed once with PBS at room temperature. Then, 200 μl PBS containing 2 μl protease inhibitor cocktail (P-8340; Sigma-Aldrich), 1 mM NaF, 1 mM EGTA, 1 mM EDTA, 50 nM okadaic acid and 2 mM phenylmethane sulfonylfluoride was added to each well. Cells were then placed on ice and disrupted by ultrasonic irradiation. Western blot analysis was performed as previously described (Regueira et al. 2014). Membranes were probed with specific antibodies that recognized the phosphorylated form of P-PDC (pyruvate dehydrogenase E1-α subunit [P-Ser293] antibody; Novus Biologicals, Littleton, Colo., USA) or total β-tubulin (T8328 Sigma-Aldrich). A 1:1000 dilution of primary antibodies, as indicated by the manufacturer, was used. For the chemiluminescent detection of the blots, a commercial kit from Cell Signaling Technology was used. The intensities of the autoradiographic bands were estimated by densitometry scanning by using NIH Image software (Scion). Levels of β-tubulin were used as a loading control.
Measurement of 2-deoxyglucose uptake
Glucose transport was studied by using the uptake of the labeled non-metabolizable glucose analog 2-deoxyglucose (2-DOG) on cells cultured in 24-multiwell plates as previously described (Regueira et al. 2014).
Lactate determination
Conditioned media obtained from cells cultured in 24-multiwell plates were used to determine lactate production. Lactate was measured by a standard method involving the conversion of NAD+ to NADH as described in Regueira et al. (2014).
LDH activity measurement
Cells cultured in 24-multiwell plates were disrupted by ultrasonic irradiation in 0.9% w/v NaCl and aliquots were preserved for DNA determination. The remaining material was centrifuged at 15800g for 10 min. The supernatant was used to measure total LDH activity as described in Regueira et al. (2014).
Other assays
A cell viability test was performed in cells cultured in 96-multiwell by using a commercial kit (CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay; Promega). DNA was determined by the method of Labarca and Paigen (1980). Protein content was determined by Lowry’s assay.
Statistical analysis
All experiments were run in triplicates and repeated 3–4 times. A one way analysis of variance and post hoc analysis by using Tukey-Krämer’s multiple comparisons test were performed with GraphPad InSat version 3.00 for Windows 95 (GraphPad Software, San Diego, Calif., USA). P values <0.05 were considered statistically significant.
Results
Effect of PPARγ activation on SC lipid metabolism and LD formation
In order to evaluate the effects of PPARγ activation on TAG levels and LD formation, SC were incubated with 1 or 10 μM Rosiglitazone (RSG), a pharmacological activator of PPARγ. Cell viability tests performed after a 48-h incubation period showed that RSG did not modify cell viability at either dose tested (data not shown). A 48-h treatment with 10 μM RSG increased TAG levels in SC (Fig. 1a), whereas this increase in TAG levels was accompanied by a rise in the number of LD (Fig. 1b–b’’, c).

Effect of Rosiglitazone (RSG) on triacylglyceride (TAG) levels and lipid droplet (LD) formation in Sertoli cells (SC). SC were maintained under basal conditions or incubated with 1 or 10 μM RSG for 48 h. a TAG levels were determined in cell lysates and expressed as micrograms TAG/micrograms DNA. *P < 0.05 versus basal. b–b’’ Photomicrographs of SC monolayers stained with ORO (arrows LD). c LD content in SC monolayers. Values represent means ± SD of number of LD per SC in one representative experiment out of four. *P < 0.05 versus basal
In the next set of determinations, we analyzed the molecular mechanisms that might have been involved in the previous observations. The levels of expression of the following genes participating in FA storage were evaluated: Fat/Cd36, Gpats, Dgat1 and Plins. Sertoli cell cultures were incubated for various periods of time (6, 12, 24 and 48 h) with 10 μM RSG.
First, as the facilitated transport of FA (the main substrates for TAG synthesis) is mediated by the integral transmembrane protein FAT/CD36, we evaluated whether PPARγ activation modified Fat/Cd36 expression. Fat/Cd36 mRNA levels increased when cells were incubated for 48 h with 10 μM RSG (1.78 ± 0.13*, *P < 0.05 vs basal; Fig. 2a). A 1 μM dose also increased Fat/Cd36 mRNA levels (1.56 ± 0.12*, *P < 0.05 vs basal).

Effect of RSG on Fat/Cd36, Gpats and Dgat1 mRNA levels in Sertoli cells (SC). a, c-g SC were maintained under basal conditions or incubated for various periods of time (6, 12, 24, or 48 h) with 10 μM RSG. Total RNA was extracted and analysis by reverse transcription plus real-time polymerase chain reaction (RT-qPCR) was performed. The comparative ΔΔCt method was used to calculate relative gene expression. Results are expressed as means ± SD of four independent experiments, *P < 0.05 versus basal. b–b’’’ Characterization of Gpat1–4 expression in SC. Total RNA from Sertoli cells (SC), germ cells (GC), liver (L) and kidney (K) were extracted, analyzed by RT-PCR and visualized by ethidium bromide staining (NT no template control)
Secondly, the presence of the four isoforms of glycerol-3-phosphate acyltransferase (GPAT1–4), which catalyze the first step in TAG synthesis, were evaluated. The SC expressed all isoforms of GPAT (Fig. 2b–b’’’). In addition, 12-, 24-,and 48-h treatments with 10 μM RSG increased Gpat1 mRNA levels (Fig. 2c; 1.30 ± 0.02*, 1.36 ± 0.08* and 1.53 ± 0.03*, respectively, *P < 0.05 vs basal). A 48-h treatment with 1 μM RSG also produced an increase in Gpat1 mRNA levels (1.31 ± 0.03*, *P < 0.05 vs basal). Treatments for 6 and 12 h with 10 μM RSG increased Gpat3 mRNA levels (Fig. 2e; 2.03 ± 0.30* and 2.19 ± 0.31*, *P < 0.05 vs basal). Gpat2 and Gpat4 mRNA levels were not modified by RSG treatment (Fig. 2d, f).
Thirdly, regarding the last step in TAG synthesis, two isoforms of diacylglycerol acyltransferase (DGAT1, DGAT2) have been described, although only DGAT1 is expressed in SC (Oresti et al. 2013). Therefore, we analyzed the role of PPARγ activation in the regulation of Dgat1 mRNA levels. Treatment with 10 μM RSG treatment for 12 h increased Dgat1 mRNA levels (Fig. 2g; 1.52 ± 0.07*, *P < 0.05 vs basal).
Fourthly, we analyzed whether PPARγ activation regulated the expression of PLINs, proteins involved in lipid droplet formation and stabilization. Five isoforms of PLINs (PLIN1–5) had been described. Figure 3a–a’’’’ shows that SC express Plin1, Plin2, Plin3 and Plin4, whereas Fig. 3b reveals that 12-, 24- and 48-h treatments with 10 μM RSG increased Plin1 mRNA levels (2.23 ± 0.22*, 2.67 ± 0.19* and 2.33 ± 0.80*, respectively, *P < 0.05 vs basal). In 12-, 24-,and 48-h incubations with 10 μM RSG, an increase in Plin2 mRNA levels was observed (Fig. 3c; 1.39 ± 0.20*, 1.53 ± 0.12* and 1.70 ± 0.30*, respectively, *P < 0.05 vs basal). A 48-h treatment with 1 μM RSG also increased Plin2 mRNA levels (1.72 ± 0.31*, *p < 0.05 vs basal). Treatment with 10 μM RSG increased Plin3 mRNA levels after 12-, 24- and 48-h incubations (Fig. 3d; 1.36 ± 0.10, 1.27 ± 0.13* and 1.50 ± 0.09*, respectively, *P < 0.05 vs basal). Finally, Plin4 mRNA levels were not modified by RSG treatment (Fig. 3e).

Effect of RSG on Plins mRNA levels in Sertoli cells. a–a’’’’ Characterization of Plin1–5 expression in SC. Total RNA from Sertoli cells (SC), germ cells (GC), testes (T) and muscle (M) were extracted, analyzed by RT-PCR and visualized by ethidium bromide staining (NT no template control). b-e SC were maintained under basal conditions or incubated for various periods of time (6, 12, 24, or 48 h) with 10 μM. Total RNA was extracted and RT-qPCR was performed. The comparative ΔΔCt method was used to calculate relative gene expression. Results are expressed as means ± SD of four independent experiments. *P < 0.05 versus basal
Effect of PPARγ activation on SC lactate production
The next set of experiments was performed in order to analyze a possible effect of PPARγ activation on lactate production. SC were incubated with 1 or 10 μM RSG and glucose consumption and lactate production were analyzed in 48-h conditioned media. Additionally, molecular mechanisms demonstrated to be involved in lactate production, such as glucose uptake, LDH activity, P-PDC levels and the expression of glucose transporters 1 and 2 (Glut1, Glut2) and the Ldha subunit, were evaluated.
Glucose is well known as being the predominant source of carbon atoms for lactate production. This fact prompted us to analyze whether PPARγ activation led to a modification in glucose consumption. Treatment with 10 μM RSG for 48 h increased glucose consumption (Fig. 4a; basal: 37.2 ± 2.3 μg/μDNA; RSG: 81.2 ± 4.6* μg/μDNA, *P < 0.05 versus basal). Under similar experimental conditions, an increment in lactate production was observed (Fig. 4b; basal: 6.9 ± 0.4 μg/μDNA; RSG: 15.5 ± 0.5* μg/μDNA, *P < 0.05 versus basal).

Effect of RSG on glucose consumption and lactate production in Sertoli cells (SC). SC were maintained under basal conditions or incubated with 1 or 10 μM RSG for 48 h. Levels of glucose (a) and lactate (b) were determined in the conditioned media. Values are expressed as means ± SD of triplicate incubations in one representative experiment out of three. *P < 0.05 versus basal
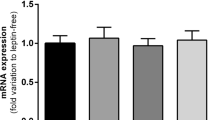
The first step in glucose metabolism is its entrance into the cells. Consequently, we decided to analyze whether PPARγ activation regulates glucose uptake in SC. Treatment for 48 h with 10 μM RSG increased 2DOG incorporation (Fig. 5a). Additionally, treatment with 10 μM RSG did not modify Glut1 expression (Fig. 5b). On the other hand, Glut2 mRNA levels increased after a 48-h incubation with 10 μM RSG (Fig. 5c; 1.87 ± 0.05*, *P < 0.05 vs basal). As increased lactate production in SC has been correlated with higher LDH activity, which is accompanied by an increase in Ldha mRNA levels (Nehar et al. 1997; Riera et al. 2001), we decided to evaluate the effect of RSG on LDH activity and Ldha expression. Treatments for 48 h with 1 or 10 μM RSG did not modify LDH activity (Fig. 6a). Ldha mRNA levels were not modified under the experimental conditions studied (Fig. 6b).

Effect of RSG on glucose uptake and on Glut1 and Glut2 mRNA levels in Sertoli cells (SC). a SC were maintained under basal conditions or incubated with 1 or 10 μM RSG for 48 h. A glucose uptake assay (2-DOG uptake) was performed after the corresponding incubation period. Results are expressed as disintegrations per minute (dpm) per microgram (μg) DNA and represent means ± SD of triplicate incubation in one representative experiment out of four. *P > 0.05 versus basal. b, c SC were maintained under basal conditions or incubated for various periods of time (6, 12, 24, or 48 h) with 10 μM RSG. Total RNA was extracted and RT-qPCR was performed. The comparative ΔΔCt method was used to calculate relative gene expression. Results are expressed as means ± SD of four independent experiments. *P < 0.05 versus basal

Effect of RSG on lactate dehydrogenase (LDH) activity, Ldha mRNA and phospho-pyruvate dehydrogenase complex (P-PDC) levels in Sertoli cells (SC). a SC were maintained under basal conditions or incubated with 1 or 10 μM RSG for 48 h. LDH activity was evaluated. Results are expressed as micro-international units per microgram DNA and represent means ± SD of triplicate incubation in one representative experiment out of four. b SC were maintained under basal conditions or incubated for various periods of time (6, 12, 24, or 48 h) with 10 μM RSG. Total RNA was extracted and RT-qPCR was performed. The comparative ΔΔCt method was used to calculate relative gene expression. Results are expressed as means ± SD of four independent experiments. c, c’ SC were maintained under basal conditions or incubated with RSG 1 or 10 μM for 48 h. Cell extracts were prepared and utilized for Western blot analysis by using antibodies specific for phosphorylated PDC (P-PDC) or β-tubulin. c Representative experiment out of four. c’ Pooled data of four independent experiments (means ± SD). Results are expressed as the fold variation in P-PDC levels (ratio of P-PDC to β-tubulin in each sample) relative to basal
Searching for additional mechanisms that might be involved in the RSG effect on lactate production, we explored the possible regulation of the availability of pyruvate, a substrate of LDH. As PDC catalyzes the conversion of pyruvate to acetyl-CoA and as PDC phosphorylation inhibits PDC activity, the state of phosphorylation of this enzyme has been correlated with pyruvate levels and consequently with lactate production (Regueira et al. 2014). Treatments for 48 h with 1 or 10 μM RSG did not modify P-PDC levels (Fig. 6c, c’).
Discussion
Energetic metabolism in the seminiferous tubule has been considered to have unique features. The most important energetic metabolite used by post meiotic germ cells is lactate, which is produced from glucose in SC (Boussouar and Benahmed 2004). Consequently, SC cannot rely on glucose for its own energy requirements and this cell type has been demonstrated to utilize FA as an energy source. SC are well known to contain abundant LD whose number varies throughout the spermatogenic cycle (Kerr and De Kretser 1975). A temporal relationship between the phagocytosis of residual bodies and LD formation in SC has been observed (Kerr et al. 1984). Despite the importance of FA in energetic metabolism and the abundance of LD in SC, the mechanisms involved in FA storage have received little attention until now.
As mentioned in the Introduction, PPAR are ligand-activated transcription factors that control metabolic pathways involved in lipid and carbohydrate metabolism. The expression of PPAR varies in different tissues. In adult SC, PPARα, PPARβ/δ and PPARγ seem to be expressed at various degrees (Braissant et al. 1996). Additionally, a developmental pattern of Ppar gene expression that occurs in SC during spermatogenesis and that probably reflects the physiological role of these nuclear receptors for germ cell development has been reported (Thomas et al. 2011). We recently demonstrated that the activation of PPARα and PPARβ/δ regulates lipid and carbohydrate metabolism (Regueira et al. 2014). However, a possible role of PPARγ in the energetic metabolism of SC has not been analyzed as yet. The present study shows that the activation of PPARγ regulates TAG levels, LD formation and lactate production in SC. These results suggest that PPARγ also has a relevant role in the regulation of the energetic metabolism of the seminiferous tubule.
LD formation has been closely associated with TAG metabolism in various cell types such as adipocytes (Ahmadian et al. 2010), cardiomyocytes (Bosma et al. 2014) and skeletal muscle cells (Bosma 2016). With regard to SC, their LD have been suggested to result from the breakdown products of phagocytosed residual bodies and apoptotic spermatogenic cells (Kerr et al. 1984; Ueno and Mori 1990). More recently, SC in culture were found to be able to use engulfed apoptotic spermatogenic cells to form LD (Wang et al. 2006). Moreover, ATP levels have been shown to increase after the phagocytosis of apoptotic spermatogenic cells by SC (Xiong et al. 2009). In the last-mentioned work, the authors concluded that LD are the source of FA, which constitute the energy substrate for SC. The results presented herein showing that PPARγ activation results in an increase in TAG levels that is accompanied by increased LD formation suggest that PPARγ activation is a mechanism that participates in lipid storage in SC.
We next wondered what molecular mechanisms might contribute to the observed increase in TAG and LD levels. As mentioned above, the facilitated transport of FA through the plasma membrane is mediated by FAT/CD36. This protein has been found near residual bodies engulfed by the SC during the spermiation process and a role of FAT/CD36 in the phagocytosis not only of residual bodies but also of apoptotic germ cells has been suggested (Gillot et al. 2005). Our results reveal that PPARγ activation increases Fat/Cd36 expression in SC. The increase in Fat/Cd36 would probably favor FA uptake, the phagocytosis of apoptotic germ cell and residual bodies and in summary, the FA availability for TAG synthesis and LD formation.
Once within the cells, FA must be activated in order to be metabolized. They can undergo oxidative degradation or can be stored as TAG according to the energy requirements of the cell. Four isoforms of GPAT catalyze the first committed step in TAG synthesis and two isoforms of DGAT are responsible for catalyzing the last step in this pathway. Thus far, no information has been available as to which isoforms of GPAT are expressed in SC. With regard to DGAT enzymes, only DGAT1 has been demonstrated as being present in SC (Oresti et al. 2013). In the present study, we evaluated the presence of the different Gpat isoforms in SC and the possible regulation of Gpats and Dgat1 by PPARγ activation. Our results show that SC express Gpat1, Gpat2, Gpat3 and Gpat4 isoforms. We observed that PPARγ activation increases Gpat1, Gpat3 and Dgat1 mRNA levels, whereas it does not modify the expression of Gpat2 and Gpat4. Many studies support the idea that GPAT1 has an important role in regulating TAG synthesis (Igal et al. 2001; Lewin et al. 2005; Linden et al. 2004). GPAT1 is an intrinsic protein of the outer mitochondrial membrane and appears to compete with carnitine palmitoyltransferase-1 (CPT1) for the same long-chain acyl-CoA pool. GPAT1 has been concluded to have an important role in channeling acyl-CoAs towards the synthetic pathway (Hammond et al. 2005). On the other hand, GPAT3 and DGAT1 are localized in the endoplasmic reticulum (Takeuchi and Reue 2009; Liu et al. 2012). Similarly to GPAT1, GPAT3 and DGAT1 mRNAs are highly expressed in white adipose tissue and are up-regulated during adipocyte differentiation (Cao et al. 2006; Cases et al. 1998). GPAT3 mRNA has been shown to increase in the white adipose tissue of mice treated with RSG and furthermore, GPAT3 overexpression in human embryonic kidney (HEK)-293 cells leads to the increased incorporation of exogenous oleic acid into TAG (Cao et al. 2006). With regard to DGAT1, the treatment of mice with RSG resulted in an increase in adipose expression of the enzyme, as does the treatment of adipocytes in vitro with the drug (Ranganathan et al. 2006). Taken together, these observations indicate that GPAT1, GPAT3 and DGAT1 are key players in the PPARγ regulation of lipid biosynthesis. The observed increase in Gpat1, Gpat3 and Dgat1 mRNA levels accompanied by an increase in TAG content after PPARγ activation in SC strongly suggests that GPAT1, GPAT3 and DGAT1 also have a relevant role in directing acyl-CoAs towards the biosynthetic pathway in this cell type.
As previously mentioned, LD have a TAG-rich core enclosed by a single layer of phospholipids and surrounded by specific proteins that drive TAG storage and regulate TAG hydrolysis. PLINs quantitatively represent the most abundant and distinctive label of the LD machinery. The mammalian genome encodes five Plin genes (Kimmel et al. 2010). Oresti et al. (2013) demonstrated that SC express Plin2. However, no additional studies have analyzed whether other PLINs isoforms are expressed in this cell type. Our results show that Plin1, Plin2, Plin3 and Plin4 are expressed in SC, while Plin5 is not. Several reports have shown an association between PPARγ activation and the regulation of Plin expression in various cell types such as adipocytes and hepatocytes (Arimura et al. 2004; Okumura 2011). Our results indicate that PPARγ activation increases Plin1, Plin2 and Plin3 mRNA levels in SC. Levels of PLINs have been correlated with the intracellular content of TAG and LD. Furthermore, the levels of PLINs increase under conditions of augmented FA flux (Brasaemle 2007). FA can act as ligands for PPAR receptors (Xu et al. 1999). In this way, the transcriptional activation of the Gpat and Plin genes by FA may provide a feed-forward mechanism, whereby the final reaction products of neutral lipid synthesis initiated by GPATs and the new synthetized PLINs provide the molecular mechanisms to sequester new TAG in LD (Bickel et al. 2009). Therefore, we are tempted to speculate that the observed augmentation in TAG and LD levels in SC are the result of the increase in Gpat1 and Plin expression following PPARγ activation.
We then hypothesized that the regulation of lipid metabolism should be related to the regulation of carbohydrate metabolism in order to sustain the energetic status of the seminiferous tubule. In view of the importance of lactate production for spermatogenesis, we decided to analyze whether PPARγ activation had a role in lactate production by SC. We observed that PPARγ activation increased the consumption of glucose and that this was accompanied by an increase in lactate production. Recently, another PPARã activator, namely pioglitazone, has also been reported to stimulate glucose consumption and lactate production in human SC (Meneses et al. 2016). The role of PPARγ activation in glucose homeostasis has been demonstrated in several tissues. For example, PPARγ agonists trigger the expression of genes related to glucose metabolism (Wu et al. 1998) and the insulin signal transduction pathway (Rieusset et al. 1999; Smith et al. 2001) in differentiated adipocytes. Additionally, direct effects of PPARγ ligands on insulin-stimulated glucose uptake in L6 myotubes and cultured human skeletal muscle cells have been reported (Cha et al. 2001; Zhang et al. 1994). The results presented herein indicate that PPARγ activation also modulates glucose metabolism in SC thereby ensuring lactate delivery to germ cells.
A role for lactate as an immune suppressive molecule has been proposed. In this respect, lactate modulates the activity of the local immune response, helping to create immune tolerance that favors uterine implantation and invasion, at the blastocyst stage (Gardner 2015). In cancer cells, lactate has been proposed to have a role in the suppression of the anticancer immune response (Choi et al. 2013). Taking into account that the mammalian testis is considered an immunoprivileged organ, we are tempted to speculate that lactate is one of the set of immunosuppressive molecules, such as activin A, TGF-β, PDL-1, Gas6, ProS and testosterone, which support this immune privilege status of the seminiferous tubules (Zhao et al. 2014). In this scenario, we envisage that the activation of PPARγ is important not only to provide energy to germ cells but also to create the immunological environment protecting germ cells from systemic immune attack.
Several biochemical mechanisms have been previously shown to be targets for the hormonal regulation of lactate production in SC (Nehar et al. 1997, 1998; Riera et al. 2001, 2002, 2007; Galardo et al. 2008, 2010; Rato et al. 2012; Oliveira et al. 2012). We wondered which mechanisms might contribute to the increase in lactate production observed after PPARγ activation. In this respect, we observed that PPARγ activation leads to an increase in glucose uptake accompanied by an increment in the expression of Glut2, a gene with a PPRE in its promoter region (Kim et al. 2000). Notably, the up-regulation of Glut2 expression by PPARγ activation is essential to achieve an improvement in glucose-stimulated insulin secretion in Zucker diabetic fatty rats (Higa et al. 1999). Liver GLUT2 may also be a direct target of PPARγ ligands and may contribute to glucose transport in type 2 diabetes or in severe obesity (Im et al. 2005; Kim and Ahn 2004). The increase in Glut2 expression in response to PPARγ activation may explain the increased glucose uptake and the subsequent augmentation in lactate production observed in SC.
Finally, LDH and PDC activities have also been shown to participate in the regulation of lactate production. Increased lactate production in response to various stimuli has been correlated with increased LDH activity accompanied by an increase in Ldha mRNA levels in SC (Riera et al. 2001, 2002, 2007). However, RSG treatment does not modify either LDH activity or Ldha mRNA levels. The latter results indicate that, similarly to the results demonstrated for the activation of PPARβ/δ, LDH activity is not a mechanism participating in the PPARγ regulation of lactate production in SC. With regard to PDC activity, we demonstrated that the regulation of the phosphorylated levels of this enzyme (P-PDC) is involved in the regulation of lactate production by PPARβ/δ activation (Regueira et al. 2014). Nevertheless, PPARγ activation does not modify P-PDC levels, suggesting that this mechanism is not relevant for the regulation of lactate production under these experimental conditions.
Taken together, our findings, which are summarized in Fig. 7, indicate that PPARγ activation in SC stimulates the expression of genes involved in FA transport and storage as TAG in LD. In addition, the results presented herein demonstrate that PPARγ activation also regulates lactate production by increasing glucose uptake and Glut2 expression. The simultaneous regulation of FA storage and glucose metabolism by PPARγ activation in SC suggests that this nuclear receptor has a relevant physiological role in the maintenance of the energetic status of the seminiferous tubule.

Representation of the mechanisms involved in LD formation and lactate production by PPARγ activation in Sertoli cells. The activation of PPARγ induces the transcription of Fat/Cd36, Gpat1, Gpat3 and Dgat1 and of Plins 1, 2 and 3, leading to an increase in FA transport and storage as LD (pink pathway). On the other hand, PPARγ activation promotes lactate increment by increasing glucose uptake and Glut2 expression (blue pathway)
References
Abumrad NA, el-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA (1993) Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem 268:17665–17668
Ahmadian M, Wang Y, Sul HS (2010) Lipolysis in adipocytes. Int J Biochem Cell Biol 42:555–559
Arimura N, Horiba T, Imagawa M, Shimizu M, Sato R (2004) The peroxisome proliferator-activated receptor gamma regulates expression of the perilipin gene in adipocytes. J Biol Chem 279:10070–10076
Bickel PE, Tansey JT, Welte MA (2009) PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta 1791:419–440
Bonen A, Campbell SE, Benton CR, Chabowski A, Coort SL, Han XX, Koonen DP, Glatz JF, Luiken JJ (2004) Regulation of fatty acid transport by fatty acid translocase/CD36. Proc Nutr Soc 63:245–249
Bosma M (2016) Lipid droplet dynamics in skeletal muscle. Exp Cell Res 340:180–186
Bosma M, Dapito DH, Drosatos-Tampakaki Z, Huiping-Son N, Huang LS, Kersten S, Drosatos K, Goldberg IJ (2014) Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim Biophys Acta 1841:1648–1655
Boussouar F, Benahmed M (2004) Lactate and energy metabolism in male germ cells. Trends Endocrinol Metab 15:345–350
Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W (1996) Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, −beta, and -gamma in the adult rat. Endocrinology 137:354–366
Brasaemle DL (2007) Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res 48:2547–2559
Cao J, Li JL, Li D, Tobin JF, Gimeno RE (2006) Molecular identification of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proc Natl Acad Sci U S A 103:19695–19700
Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, Sande E, Novak S, Collins C, Welch CB, Lusis AJ, Erickson SK, Farese RV Jr (1998) Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci U S A 95:13018–13023
Cha BS, Ciaraldi TP, Carter L, Nikoulina SE, Mudaliar S, Mukherjee R, Paterniti JR Jr, Henry RR (2001) Peroxisome proliferator-activated receptor (PPAR) gamma and retinoid X receptor (RXR) agonists have complementary effects on glucose and lipid metabolism in human skeletal muscle. Diabetologia 44:444–452
Choi SY, Collins CC, Gout PW, Wang Y (2013) Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol 230:350–355
Galardo MN, Riera MF, Pellizzari EH, Chemes HE, Venara MC, Cigorraga SB, Meroni SB (2008) Regulation of expression of Sertoli cell glucose transporters 1 and 3 by FSH, IL1 beta, and bFGF at two different time-points in pubertal development. Cell Tissue Res 334:295–304
Galardo MN, Riera MF, Pellizzari EH, Sobarzo C, Scarcelli R, Denduchis B, Lustig L, Cigorraga SB, Meroni SB (2010) Adenosine regulates Sertoli cell function by activating AMPK. Mol Cell Endocrinol 330:49–58
Galardo MN, Regueira M, Riera MF, Pellizzari EH, Cigorraga SB, Meroni SB (2014) Lactate regulates rat male germ cell function through reactive oxygen species.PLoS One 9:e88024
Gardner DK (2015) Lactate production by the mammalian blastocyst: manipulating the microenvironment for uterine implantation and invasion? BioEssays 37:364–371
Gillot I, Jehl-Pietri C, Gounon P, Luquet S, Rassoulzadegan M, Grimaldi P, Vidal F (2005) Germ cells and fatty acids induce translocation of CD36 scavenger receptor to the plasma membrane of Sertoli cells. J Cell Sci 118:3027–3035
Green S, Wahli W (1994) Peroxisome proliferator-activated receptors: finding the orphan a home. Mol Cell Endocrinol 100:149–153
Hammond LE, Neschen S, Romanelli AJ, Cline GW, Ilkayeva OR, Shulman GI, Muoio DM, Coleman RA (2005) Mitochondrial glycerol-3-phosphate acyltransferase-1 is essential in liver for the metabolism of excess acyl-CoAs. J Biol Chem 280:25629–25636
Higa M, Zhou YT, Ravazzola M, Baetens D, Orci L, Unger RH (1999) Troglitazone prevents mitochondrial alterations, beta cell destruction, and diabetes in obese prediabetic rats. Proc Natl Acad Sci U S A 96:11513–11518
Igal RA, Wang S, Gonzalez-Baro M, Coleman RA (2001) Mitochondrial glycerol phosphate acyltransferase directs the incorporation of exogenous fatty acids into triacylglycerol. J Biol Chem 276:42205–42212
Im SS, Kim JW, Kim TH, Song XL, Kim SY, Kim HI, Ahn YH (2005) Identification and characterization of peroxisome proliferator response element in the mouse GLUT2 promoter. Exp Mol Med 37:101–110
Jump DB, Botolin D, Wang Y, Xu J, Christian B, Demeure O (2005) Fatty acid regulation of hepatic gene transcription. J Nutr 135:2503–2506
Jutte NH, Eikvar L, Levy FO, Hansson V (1985) Metabolism of palmitate in cultured rat Sertoli cells. J Reprod Fertil 73:497–503
Kerr JB, De Kretser DM (1975) Cyclic variations in Sertoli cell lipid content throughout the spermatogenic cycle in the rat. J Reprod Fertil 43:1–8
Kerr JB, Mayberry RA, Irby DC (1984) Morphometric studies on lipid inclusions in Sertoli cells during the spermatogenic cycle in the rat. Cell Tissue Res 236:699–709
Kim HI, Ahn YH (2004) Role of peroxisome proliferator-activated receptor-gamma in the glucose-sensing apparatus of liver and beta-cells. Diabetes 53:S60–S65
Kim HI, Kim JW, Kim SH, Cha JY, Kim KS, Ahn YH (2000) Identification and functional characterization of the peroxisomal proliferator response element in rat GLUT2 promoter. Diabetes 49:1517–1524
Kimmel AR, Brasaemle DL, McAndrews-Hill M, Sztalryd C, Londos C (2010) Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J Lipid Res 51:468–471
Labarca C, Paigen K (1980) A simple, rapid, and sensitive DNA assay procedure. Anal Biochem 102:344–352
Lewin TM, Wang S, Nagle CA, Van Horn CG, Coleman RA (2005) Mitochondrial glycerol-3-phosphate acyltransferase-1 directs the metabolic fate of exogenous fatty acids in hepatocytes. Am J Physiol Endocrinol Metab 288:E835–E844
Linden D, William-Olsson L, Rhedin M, Asztely AK, Clapham JC, Schreyer S (2004) Overexpression of mitochondrial GPAT in rat hepatocytes leads to decreased fatty acid oxidation and increased glycerolipid biosynthesis. J Lipid Res 45:1279–1288
Liu Q, Siloto RM, Lehner R, Stone SJ, Weselake RJ (2012) Acyl-CoA:diacylglycerol acyltransferase: molecular biology, biochemistry and biotechnology. Prog Lipid Res 51:350–377
Meneses MJ, Bernardino RL, Sá R, Silva J, Barros A, Sousa M, Silva BM, Oliveira PF, Alves MG (2016)Pioglitazone increases the glycolytic efficiency of human Sertoli cells with possible implications for spermatogenesis.Int J Biochem Cell Biol 79:52–60
Meroni SB, Riera MF, Pellizzari EH, Cigorraga SB (2002) Regulation of rat Sertoli cell function by FSH: possible role of phosphatidylinositol 3-kinase/protein kinase B pathway. J Endocrinol 174:195–204
Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W (2006) International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev 58:726–741
Nehar D, Mauduit C, Boussouar F, Benahmed M (1997) Tumor necrosis factor-alpha-stimulated lactate production is linked to lactate dehydrogenase a expression and activity increase in porcine cultured Sertoli cells. Endocrinology 138:1964–1971
Nehar D, Mauduit C, Boussouar F, Benahmed M (1998) Interleukin 1alpha stimulates lactate dehydrogenase a expression and lactate production in cultured porcine Sertoli cells. Biol Reprod 59:1425–1432
Okumura T (2011) Role of lipid droplet proteins in liver steatosis. J Physiol Biochem 67:629–636
Oliveira PF, Alves MG, Rato L, Laurentino S, Silva J, Sa R, Barros A, Sousa M, Carvalho RA, Cavaco JE, Socorro S (2012) Effect of insulin deprivation on metabolism and metabolism-associated gene transcript levels of in vitro cultured human Sertoli cells. Biochim Biophys Acta 1820:84–89
Oresti GM, Garcia-Lopez J, Aveldano MI, Del Mazo J (2013) Cell-type-specific regulation of genes involved in testicular lipid metabolism: fatty acid-binding proteins, diacylglycerol acyltransferases, and perilipin 2. Reproduction 146:471–480
Parvinen M (1982) Regulation of the seminiferous epithelium. Endocr Rev 3:404–417
Picard F, Auwerx J (2002) PPAR(gamma) and glucose homeostasis. Annu Rev Nutr 22:167–197
Ranganathan G, Unal R, Pokrovskaya I, Yao-Borengasser A, Phanavanh B, Lecka-Czernik B, Rasouli N, Kern PA (2006) The lipogenic enzymes DGAT1, FAS, and LPL in adipose tissue: effects of obesity, insulin resistance, and TZD treatment. J Lipid Res 47:2444–2450
Rato L, Alves MG, Socorro S, Carvalho RA, Cavaco JE, Oliveira PF (2012) Metabolic modulation induced by oestradiol and DHT in immature rat Sertoli cells cultured in vitro. Biosci Rep 32:61–69
Regueira M, Riera MF, Galardo MN, Pellizzari EH, Cigorraga SB, Meroni SB (2014) Activation of PPAR alpha and PPAR beta/delta regulates Sertoli cell metabolism. Mol Cell Endocrinol 382:271–281
Regueira M, Artagaveytia SL, Galardo MN, Pellizzari EH, Cigorraga SB, Meroni SB, Riera MF (2015) Novel molecular mechanisms involved in hormonal regulation of lactate production in Sertoli cells. Reproduction 150:311–321
Riera MF, Meroni SB, Gomez GE, Schteingart HF, Pellizzari EH, Cigorraga SB (2001) Regulation of lactate production by FSH, iL1beta, and TNFalpha in rat Sertoli cells. Gen Comp Endocrinol 122:88–97
Riera MF, Meroni SB, Schteingart HF, Pellizzari EH, Cigorraga SB (2002) Regulation of lactate production and glucose transport as well as of glucose transporter 1 and lactate dehydrogenase a mRNA levels by basic fibroblast growth factor in rat Sertoli cells. J Endocrinol 173:335–343
Riera MF, Galardo MN, Pellizzari EH, Meroni SB, Cigorraga SB (2007) Participation of phosphatidyl inositol 3-kinase/protein kinase B and ERK1/2 pathways in interleukin-1beta stimulation of lactate production in Sertoli cells. Reproduction 133:763–773
Rieusset J, Auwerx J, Vidal H (1999) Regulation of gene expression by activation of the peroxisome proliferator-activated receptor gamma with Rosiglitazone (BRL 49653) in human adipocytes. Biochem Biophys Res Commun 265:265–271
Robinson R, Fritz IB (1981) Metabolism of glucose by Sertoli cells in culture. Biol Reprod 24:1032–1041
Sanderson LM, Degenhardt T, Koppen A, Kalkhoven E, Desvergne B, Muller M, Kersten S (2009) Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) but not PPARalpha serves as a plasma free fatty acid sensor in liver. Mol Cell Biol 29:6257–6267
Shi Y, Cheng D (2009) Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am J Physiol Endocrinol Metab 297:E10–E18
Siersbaek R, Nielsen R, Mandrup S (2010) PPARgamma in adipocyte differentiation and metabolism—novel insights from genome-wide studies. FEBS Lett 584:3242–3249
Smith U, Gogg S, Johansson A, Olausson T, Rotter V, Svalstedt B (2001) Thiazolidinediones (PPARgamma agonists) but not PPARalpha agonists increase IRS-2 gene expression in 3T3-L1 and human adipocytes. FASEB J 15:215–220
Sztalryd C, Kimmel AR (2014) Perilipins: lipid droplet coat proteins adapted for tissue-specific energy storage and utilization, and lipid cytoprotection. Biochimie 96:96–101
Takeuchi K, Reue K (2009) Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab 296:E1195–E1209
Thomas K, Sung DY, Chen X, Thompson W, Chen YE, McCarrey J, Walker W, Griswold M (2011) Developmental patterns of PPAR and RXR gene expression during spermatogenesis. Front Biosci (Elite Ed) 3:1209–1220
Ueno H, Mori H (1990) Morphometrical analysis of Sertoli cell ultrastructure during the seminiferous epithelial cycle in rats. Biol Reprod 43:769–776
Walther TC, Farese RV Jr (2012) Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 81:687–714
Wang H, Wang H, Xiong W, Chen Y, Ma Q, Ma J, Ge Y, Han D (2006) Evaluation on the phagocytosis of apoptotic spermatogenic cells by Sertoli cells in vitro through detecting lipid droplet formation by oil red O staining. Reproduction 132:485–492
Wendel AA, Lewin TM, Coleman RA (2009) Glycerol-3-phosphate acyltransferases: rate limiting enzymes of triacylglycerol biosynthesis. Biochim Biophys Acta 1791:501–506
Wu Z, Xie Y, Morrison RF, Bucher NL, Farmer SR (1998) PPARgamma induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPalpha during the conversion of 3T3 fibroblasts into adipocytes. J Clin Invest 101:22–32
Xiong W, Wang H, Wu H, Chen Y, Han D (2009) Apoptotic spermatogenic cells can be energy sources for Sertoli cells. Reproduction 137:469–479
Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, Kliewer SA, Milburn MV (1999) Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell 3:397–403
Zhang B, Szalkowski D, Diaz E, Hayes N, Smith R, Berger J (1994) Potentiation of insulin stimulation of phosphatidylinositol 3-kinase by thiazolidinedione-derived antidiabetic agents in Chinese hamster ovary cells expressing human insulin receptors and L6 myotubes. J Biol Chem 269:25735–25741
Zhao S, Zhu W, Xue S, Han D (2014) Testicular defense systems: immune privilege and innate immunity. Cell Mol Immunol 11:428–437
Acknowledgements
The technical help of Mercedes Astarloa is gratefully acknowledged. We also thank Celia Nieto for revising our English usage. This work was supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT) (PICT 2014/945) and the Consejo Nacional de Investigaciones Cientificas y Técnicas (CONICET) (PIP 2011/187). M.F. Riera, M.N. Galardo and S.B. Meroni are established investigators of CONICET. A. Gorga is a recipient of an ANPCYT fellowship. G.M. Rindone and M. Regueira are recipients of CONICET fellowships.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gorga, A., Rindone, G.M., Regueira, M. et al. PPARγ activation regulates lipid droplet formation and lactate production in rat Sertoli cells. Cell Tissue Res 369, 611–624 (2017). https://doi.org/10.1007/s00441-017-2615-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-017-2615-y