
Abstract
The vascular endothelium is a cellular interface between the blood and the interstitial space of tissue, which controls the exchange of fluid, solutes and cells by both transcellular and paracellular means. To accomplish the demands on barrier function, the regulation of the endothelium requires quick and adaptive mechanisms. This is, among others, accomplished by actin dynamics that interdependently interact with both the VE-cadherin/catenin complex, the main components of the adherens type junctions in endothelium and the membrane cytoskeleton. Actin filaments in endothelium are components of super-structured protein assemblies that control a variety of dynamic processes such as endo- and exocytosis, shape change, cell–substrate along with cell–cell adhesion and cell motion. In endothelium, actin filaments are components of: (1) contractile actin bundles appearing as stress fibers and junction-associated circumferential actin filaments, (2) actin networks accompanied by endocytotic ruffles, lamellipodia at leading edges of migrating cells and junction-associated intermittent lamellipodia (JAIL) that dynamically maintain junction integrity, (3) cortical actin and (4) the membrane cytoskeleton. All these structures, most probably interact with cell junctions and cell–substrate adhesion sites. Due to the rapid growth in information, we aim to provide a bird’s eye view focusing on actin filaments in endothelium and its functional relevance for entire cell and junction integrity, rather than discussing the detailed molecular mechanism for control of actin dynamics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Endothelial cells in vivo and in cell culture express non-muscle β-actin and γ-actin that are both members of the six different and highly conserved, actin isoforms (Rubenstein 1990). Non-muscle actin comprises about 10 % of the total endothelial protein (Patterson and Lum 2001; Schnittler et al. 1990). Actin appears as monomers (globular or G-actin) and as actin filaments (F-actin). Actin monofilaments display a diameter between 5 and 7 nm and are polymerized from G-actin under the control of actin-regulating and actin-binding proteins (Dickinson 2009; Disanza et al. 2005; Dominguez 2010; Pollard et al. 2000).
Actin filaments in endothelial cells are in most cases components of super-structured protein assemblies that include actin bundles, actin networks, cortical actin filaments and the membrane cytoskeleton. Actin-containing structures are functionally associated with specialized subcellular differentiations such as focal contacts, cell–cell contacts and the membrane cytoskeleton (Fig. 1). In this way, actin filaments are involved in controlling the dynamics of cell shape, cell polarity, cell–substrate adhesion, cell–cell adhesion, cell migration and endo- and exocytosis in both physiological and pathological conditions. One of the most pivotal roles of the endothelium is barrier function, which can be changed due to many different challenges such as oxygen radicals, bacterial endo- and exotoxins and endogenous mediators such as cytokines. However, many of those challenges target the actin-containing structures in the cells and manipulate actin dynamics in close interaction with adhesion receptors mediating cell–substrate and cell–cell adhesion (Fig. 1) and can involve components of the membrane cytoskeleton (Benz et al. 2008) and maybe its associated receptors, respectively (see below). Apart from endogenous cellular mechanisms that control actin dynamics and permeability, bacterial toxins have been shown to highjack actin-controlling targets such as Rho-GTPases or directly target actin, e.g., by ADP-ribosylation and thus significantly alter the cytoskeleton (Aktories et al. 2011; Lemichez and Aktories 2013). A special feature of actin filaments and actin filament-containing structures in non-muscle cells, including endothelium, is their ability to quickly rearrange in response to stimulations to fulfill adaptive functions, e.g., in wound healing and inflammation, situations accompanied by increased cellular dynamics such as changes in cell shape, migration and cell growth (Pollard et al. 2000). Accordingly, the appearance of actin filament-containing structures differ significantly between quiescent endothelium and endothelium that is proliferating, migrating, or challenged by mediators such as cyto- or chemokines, by growth factors, or by hemodynamic loads. Remodeling of actin filaments is frequently mediated by rapid assembly and disassembly of actin filaments, a process that depends on many actin-binding and actin-regulating proteins, as well as the ratio between filamentous and globular actin (Pollard et al. 2000). A recent paper demonstrated that β-actin balances the ratio between globular and filaments actin, particularly for controlling cell migration and cell growth (Bunnell et al. 2011). Together, in this way, actin filament dynamics are central in controlling tissue homeostasis, vascular morphogenesis and repair mechanisms in physiology and pathology. Here, we discuss the role of actin filaments and actin filament-containing structures in endothelium under both physiological and pathological conditions, particularly with respect to their importance in the dynamics of intercellular junctions and barrier function regulation.

Potential appearance of actin filament-containing structures in endothelium in confluent and resting endothelium (upper) as well as in subconfluent or activated endothelium (lower)
Organization and function of actin bundles in endothelium
Actin bundles in endothelium are closely packed actin filaments that assemble with α−actinin, tropomyosin and myosin II (Drenckhahn 1982; Drenckhahn and Wagner 1986; Schnittler et al. 1990; Tojkander et al. 2011; Wong et al. 1983). Actin bundles in endothelium are contractile structures (Drenckhahn and Wagner 1986; Schnittler et al. 1990) that are activated by myosin light chain kinase (Wysolmerski and Lagunoff 1990, 1991) in balance with myosin light chain phosphatase (Rigor et al. 2013; Shen et al. 2010). The assembly of actin bundles such as stress fibers together with myosin II was recently shown in osteosarcoma cells to depend on tropomyosin isoforms (Tojkander et al. 2011). The contractile features of actin bundles indicate them as critical filaments for cellular and junction remodeling and cell dynamics. Actin bundles in endothelium appear in two forms: (1) as junction-associated circumferential actin filaments that are predominantly expressed at mature endothelial cell junctions under resting conditions and (2) as cytoplasmic stress fibers that develop in response to certain stimulations and can terminate at the apical plasma membrane (apical stress fibers), focal contacts (basal stress fibers) and at interendothelial adherens junctions (Hoelzle and Svitkina 2012; Huveneers et al. 2012; Katoh et al. 2008; Millan et al. 2010) (Figs. 1, 2, and 4, 5, below). A third type of actin bundles, the transverse arcs, have been described in U2 osteosarcoma cells (Hotulainen and Lappalainen 2006) but, to our best knowledge, are not described for the endothelium. The association of stress fibers with focal contacts via integrins has been nicely investigated in endothelium (Napione et al. 2007; Shyy and Chien 2002; Stupack and Cheresh 2002), while the association with endothelial cell junctions is not fully understood.

a, b Abdominal aorta of a C57/Bl6 mouse labeled by phalloidin-TRITC for filamentous actin and by anti-VE-cadherin for adherens junctions. Note: prominent circumferential actin filaments (a; arrows) and the absence of stress fibers. Actin filaments colocalize with VE-cadherin (b; arrows). c–e Confluent HUVEC cultures (1.2 × 10 E 5 cells/cm2) were wounded by scratch and, after 2.5 h, fixed and triple-labeled with phalloidin-TRITC for filamentous actin and by anti-VE-cadherin and DAPI for nuclear staining. c Overview showing the wounded culture area (right side) with cells in higher magnification (d, e) that are either distant (d) or close (e) to the wound rim. Cells with a certain distance to the wound rim (d) are small in size, display a continuous VE-cadherin pattern and have circumferential actin filaments along the junctions (arrows). Note: stress fibers are absent. e Cells close to the rim are large in size, display an interrupted VE-cadherin pattern and show prominent stress fibers (arrowheads) (Schnittler and Lindeman, unpublished result). Bars (c) 20 μm, (d, e) 10 μm
Interaction of VE-cadherin with actin filament-containing bundles
There is agreement that the vascular endothelial (VE)-cadherin/catenin complex, the backbone of adherens-type junctions in endothelium can be associated with both types of actin bundles (Hoelzle and Svitkina 2012; Huveneers et al. 2012; Millan et al. 2010; Seebach et al. 2007). The regulation of these interactions has frequently been proposed to be essential in controlling endothelial barrier function.
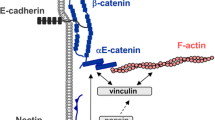
VE-cadherin (Lampugnani et al. 1992) is an endothelial-specific type II cadherin, consisting in (1) a long extracellular domain with five repeats (EC1-EC5) that mediate calcium-dependent cellular adhesion in trans, (2) a membrane-spanning domain and (3) a short cytoplasmic domain that connects VE-cadherin via connector molecules such as catenins, vinculin and EPLIN to the actin cytoskeleton (Fig. 3). The juxtamembrane domain (JMD) of the cytoplasmic VE-cadherin-tail binds p120ctn, an armadillo family protein, which in turn has been indicated to control VE-cadherin turnover, lateral clustering and junction integrity (Alcaide et al. 2008, 2012; Chiasson et al. 2009; Hatanaka et al. 2011; Iyer et al. 2004; Konstantoulaki et al. 2003; Potter et al. 2005; Vandenbroucke St Amant et al. 2012; Xiao et al. 2005). The distal part of the cytoplasmic VE-cadherin tail binds the chain proteins β- and γ-catenin that in turn are linked to α-catenin (Dejana and Vestweber 2013; Komarova and Malik 2010; Niessen et al. 2011). There is general agreement that α-catenin mediates the connection to actin bundles directly and/or indirectly via adaptor molecules. Earlier studies also showed that α-catenin binds both actin and β−catenin (Rimm et al. 1995), which led to the hypothesis that α-catenin might link the cadherin/catenin complex to the actin cytoskeleton. Many other groups have supported the perception of a direct interaction. However, this concept was challenged by the demonstration that α-catenin is an allosteric molecule (Pokutta and Weis 2007) incapable of binding to both β-catenin and actin filaments at the same time (Weis and Nelson 2006). It was shown that α-catenin exist as monomers preferentially binding to β-catenin while α-catenin dimers preferentially bind to actin filaments (Drees et al. 2005; Yamada et al. 2005). Furthermore, the actin-binding potential of α-catenin dimers was suggested to compete with ARP2/3 complex for actin binding. In this way, a highly dynamic interaction between actin filaments and the cadherin/catenin complex was proposed (Drees et al. 2005; Yamada et al. 2005). An indirect interaction of α-catenin with actin filaments can be assumed by showing that α-catenin binds α-actinin (Knudsen et al. 1995), vinculin (Weiss et al. 1998), ZO-1 (Itoh et al. 1997), afadin (AF6) (Pokutta et al. 2002), ajuba (Marie et al. 2003), formins (Kobielak et al. 2004) and EPLIN (Abe and Takeichi 2008) proteins, which in addition interact with actin or regulate actin dynamics. The morphological appearance and the entire functionality of both the VE-cadherin/catenin complex and the circumferential actin filaments indicate a structurally and functionally association but the molecular architecture and the underlying cooperative regulations are poorly defined. Recent papers have placed the locally restricted and actin-driven and ARP2/3 complex-controlled junction-associated intermittent lamellipodia (JAIL) in a central role to coordinate the VE-cadherin dynamics and VE-cadherin-mediated cell adhesion (Taha and Schnittler 2014; Taha et al. 2014). Furthermore, there are several pieces of evidence demonstrating that junction integrity is controlled by ARP2/3 complex-activating molecules (Kovacs et al. 2002; Martinelli et al. 2013; Rajput et al. 2013; Zhou et al. 2013). The current possibilities of interaction between actin and VE-cadherin-mediated cell adhesion are illustrated in Fig. 3.

Possible interactions between actin filaments and VE-cadherin
Junction-associated circumferential actin bundles
Under non-pathological conditions, the endothelium of arteries, veins and the microvascular bed display junction-associated circumferential actin bundles that are supposed to stabilize VE-cadherin-mediated cell adhesion, which is supposed to facilitate barrier function and the integrity of the entire endothelium (Gabbiani et al. 1975, 1979, 1983; White and Fujiwara 1986; White et al. 1983; Wong et al. 1983; Huttner et al. 1985). The circumferential actin bundles are regularly observed when a continuous labeling of VE-cadherin/catenin complex is observed along the junctions (Figs. 2a–c, 4b). Challenging the endothelium, e.g., under wound healing conditions (Fig. 1c) or by inflammatory mediators, induces the disassembly of the circumferential actin bundles accompanied by stress fiber development (Aepfelbacher et al. 1997; Petrache et al. 2001; Thurston and Turner 1994). Stress fiber formation is accompanied by actin/myosin II-mediated contraction and remodeling of the VE-cadherin/catenin complex from a continuous to an interrupted patterning (Essler et al. 1998; Wong et al. 1999). These processes require many signaling mechanisms such as proteins of the Rho GTPase family, kinases and phosphatases that cause intercellular gap formation and thus a breakdown of endothelial barrier function (Dejana and Vestweber 2013; Komarova and Malik 2010; Liebner et al. 2006). There are many stimuli that target the VE-cadherin/catenin complex and actin filaments, respectively, followed by increases in paracellular permeability and changes in cell dynamcis at different time scales. This includes cleavage of the protease-activated receptor 1 (PAR1) by thrombin (for review, see Beckers et al. 2010; Bogatcheva et al. 2002; Komarova and Malik 2010), treatment with the pro-inflammatory cytokine TNF-α (Wahl-Jensen et al. 2005), or treatment with endothelial growth factor VEGF (Beckers et al. 2010; Rousseau et al. 2000; Wojciak-Stothard and Ridley 2002). Those stimuli change cell migration, induce an interrupted VE-cadherin patterning and stress fiber formation (Choi et al. 2009; Huveneers et al. 2012; Mirzapoiazova et al. 2006; Nakamura et al. 2008; Rousseau et al. 2000). Irrespective of many identified signaling mechanisms required for remodeling of the VE-cadherin/catenin complex and the actin bundles, the step-by-step follow-up mechanism that, in the end, mediates the remodeling of cell junctions and causes and closes the intercellular gap formation, remains unexplained. Together, it is reasonable to assume that stress fiber formation is generally characteristic for an activated and challenged endothelium.

Immunolabeling and determination of the transendothelial electrical resistance of HUVEC under resting and shear stress conditions as indicated. Subsequently, cells were fixed and labeled with anti-VE-cadherin (a, b) and phalloidin-TRITC to label filamentous actin (c, d). Merged images (e, f). Under resting conditions, VE-cadherin colocalizes with actin filaments at only a few areas and with stress fibers (e, arrows). In contrast, application of shear stress for 5 min recruited actin bundles to the cell junctions (d, arrowheads). Circumferential actin bundles colocalize better with VE-cadherin (f, arrowheads). Determination of the transendothelial electrical resistance (TER) by impedance spectroscopy of HUVEC after transient exposure to 50 dyn/cm2 of fluid shear stress under control conditions (not infected, GFP) and after expression of either dominant negative N17rac1 or dominant negative N19rhoA. The shear stress-induced TER increase depended on Rac1 but not on RhoA. Figure is taken from Seebach et al. (2007)
Stress fibers in endothelium in vivo and in culture
Stress fibers in vivo are less observed in non-muscle cells under physiological conditions but develop under pathologies when accompanied by cell migration, proliferation and/or activation as typically occurring in wound healing and inflammation (Gabbiani et al. 1983; Gordon and Staley 1990; Gotlieb 1990; Vyalov et al. 1996). In endothelium, however, stress fibers have been demonstrated in different vessels and the heart of certain species but the different reports are not always in line with each other. This might be due to species, ages and vascular segments investigated. In particular, endothelial stress fibers have been demonstrated to occur only in some areas of the heart and in particular areas of large arteries, veins and some parts of the microvascular beds (Thurston and Baldwin 1994; White and Fujiwara 1986; White et al. 1983; Fraccaroli et al. 2012; Jinguji 2003; Katoh and Noda 2012; Yu et al. 1997). There are arteriosclerotic prone sites, distal to arterial branches that display increased apoptosis and increased cell turnover. Those endothelial cells also exhibit large amounts of stress fibers (Kano et al. 2000; Katoh et al. 2008), while the endothelium of other parts of the arteries do not show them, particularly segments of the arteries that are exposed to unidirectional laminar shear stress (Gabbiani et al. 1983; Gordon and Staley 1990; Gotlieb 1990; Vyalov et al. 1996). However, there is agreement that stress fibers develop in endothelium under pathological conditions, e.g., under wound healing or experimental hypertension (Gabbiani et al. 1975, 1979, 1983; White and Fujiwara 1986; White et al. 1983; Wong et al. 1983; Huttner et al. 1985). Furthermore, irrespective of the carefully performed studies, a systematic evaluation of stress fibers in endothelium in vivo throughout the vascular bed and during vascular development under non-pathological conditions is still incomplete, particularly with respect to species, organs, ages and gender.
Appearance of stress fibers under pathological conditions and during cell growth might have different functional meanings. Firstly, stress fibers are of critical importance for cell migration, as a force of retraction at the rear end of migrating cells is provided (Tojkander et al. 2012). Rho-GTPases-mediated stress fiber development in endothelium, due to proinflamatory stimulations, such as thrombin, histamine, TNF-α and VEGF and under wound healing conditions and its contraction potential, raised the idea that this is an important mechanism in breaking down the endothelial barrier function (Beckers et al. 2010; Birukova et al. 2013; Wojciak-Stothard et al. 1998; Wojciak-Stothard and Ridley 2002). These stimulations and conditions create a challenging environment for the cells and it can be proposed that compensation mechanisms have been developed. Following this path, the formation of stress fibers might indeed have a protective function, as they increase the bending strength and stiffness of the entire cell and might also facilitate transient cell adhesion to the substrate by interacting with focal adhesion complexes and focal contacts (Tojkander et al. 2012). Stress fibers characteristically develop in growing endothelial cell cultures and frequently persist during proliferation and cell migration (Figs. 1, 2c, 5), even after reaching high cell density (1–1.2 ×105 for HUVEC) (Fig. 4a).

Life cell imaging of HUVEC expressing Lifeact-fluorescent-protein (LifeAct-EGFP) at different time points as indicated. LifeAct-EGFP binds to actin filaments and allows dynamic analyses of actin dynamics. Labeling actin filaments with LifeAct-EGFP most probably do not disturb either the actin dynamics or the polymerization/depolymerization features (Riedl et al. 2008). HUVEC cultures display cell density-dependent actin patterns and dynamics. In sparce cultures (a, b), migrating cells display lamellipodia that appear at leading edges (arrows), while in subconfluent cultures (c, d), JAIL appear between adjacent cells (arrows). In highly confluent cultures (e, f), the circumferential actin bundles are predominant and stress fibers appear much fewer. Arrows indicate small JAIL. Taken from Taha et al. (2014)
Mechanical force induced stress fibers; challenging a paradigm
Application of experimental fluid shear stress and cyclic stretch, two mechanical stimuli generated in vivo by blood flow and blood pressure significantly increased stress fiber formation as first demonstrated by Franke et al. (1984) after shear stress application and by Shirinsky et al. (1989) after cyclic stretch. These phenomena have been frequently confirmed in many studies using different types of cultured endothelium isolated from various species and organs. The background of the fluid shear stress-provoked stress fiber formation in cultured endothelial is not quite clear, as certain studies have shown that the endothelium of the arterial system in vivo display stress fibers in a few locations only (Kano et al. 2000; Katoh et al. 2008) (compare Figs. 2a and 4b). It is reasonable to assume that the background of shear stress-induced stress fiber formation in cell culture models is related to cell culture conditions, particularly with respect to cell density and the cell culture substrate. Furthermore, development of stress fiber is usually accompanied by remodeling of the VE-cadherin/catenin complex, resulting in an interrupted VE-cadherin patterning (Fig. 2c), a phenomenon that is accompanied by a breakdown of endothelial barrier function. In contrast, by optimization of endothelial cell culture conditions, it was demonstrated that laminar shear stress applied to primary cultured, highly confluent human umbilical vein endothelial cells (about 105 cells/cm2) transiently up-regulated the paraendothelial barrier function dose-dependently (DePaola et al. 2001; Katoh et al. 2008; Seebach et al. 2000, 2007), increased VE-cadherin clustering and recruited actin filament bundles to the cell junctions, forming circumferential actin bundles and ultimately the disappearance of stress fibers (Seebach et al. 2007) (Fig. 4). This mechanism was shown to depend on Rac1 activation (Fig. 4c) and occurs within a few minutes. Furthermore, the shear stress-induced circumferential actin bundles persist within the investigated time of 24 h even during cell alignment and cell elongation (Seebach et al. 2007), indicating that junction dynamics and remodeling can take place without disturbing the junction integrity and without stress fiber formation. The experimental shear stress-induced circumferential actin bundles and the continuous VE-cadherin pattern are consistent with laminar shear stress-exposed arterial endothelium in vivo out of arteriosklerosis prone sites (Fig. 2a, b). A comparable phenomenon can be observed upon treatment of the endothelium with peroxyvanadate, a phosphatase inhibitor, which reorganizes actin filaments (Ayalon and Geiger 1997; Seebach et al. 2005). Application of peroxyvanadate transiently increased barrier function at certain concentrations, with recruitment of actin filament bundles to the junctions but prolonged treatment with a high concentration (50 μM) ends up with barrier function breakdown (Seebach et al. 2005). This indicates that the actin and junction patterning and in turn barrier function depend on a highly quantitative balance between activation and silencing. Thus, small variations in this balance might close or open endothelial cell junctions.
Culture conditions significantly influence stress fiber formation
In our opinion, there is an essential need for objective and standardized parameters of endothelial cell culture conditions for analyzing cell and molecular dynamics. This would better allow comparing results from different published experimentations, specifically in terms of quantification. The three parameters that might be helpful include definition of (1) cell density, (2) the VE-cadherin patterning at the junctions together with actin filament distribution and (3) the cell culture substrate. The first two parameters are directly dependent on each other as described in human umbilical cord vein endothelium (Taha et al. 2014). The cell density should be precisely defined by, e.g., cells/cm2 rather than using blurred terms. VE-cadherin is one of the most critical cell adhesion complexes in endothelium and is involved in controlling barrier function and the cell cycle (Caveda et al. 1996; Nelson and Chen 2003; Taddei et al. 2008). The total amount of VE-cadherin in HUVEC cultures is independent of the cell density (Lampugnani et al. 1995; Taha et al. 2014) and thus makes the VE-cadherin patterning suitable for cell density evaluation, as a given amount of VE-cadherin is distributed along the junctions. As a result, subconfluent cultures (<<1 × 105 cells/cm2) that are large in size with long cell perimeters display an interrupted VE-cadherin patterning, while highly confluent endothelial cultures (≤1 × 105 cells/cm2) with short cell perimeters exhibit a continuous VE-cadherin line along the junctions (Taha and Schnittler 2014). The continuous line of VE-cadherin patterning is also characteristic of quiescent, shear stress-aligned arterial and polygonal venous endothelium in vivo. Thus, the VE-cadherin patterning is a usable parameter for defining cell confluence. The different VE-cadherin patterning is accompanied by a respective distribution of actin filaments. For example, highly confluent endothelial cultures display fewer stress fibers with a continuous VE-cadherin patterning, while subconfluent cultures exhibit many stress fibers accompanied by an interrupted VE-cadherin patterning (Lampugnani et al. 1995; Taha et al. 2014) (compare Fig. 3). The differences in cell density-dependent VE-cadherin and actin patterning significantly modulate the dynamics of both VE-cadherin (Huveneers et al. 2012; Taha et al. 2014) and actin (Taha et al. 2014). Thirdly, the cell culture substrate is also of critical importance for the expression of actin filaments. For example, the extracellular matrix protein fibronectin is frequently used as a culture substrate. Interestingly, fibronectin is up-regulated in atherosclerotic lesions (Magnusson and Mosher 1998) and was found to be deposited at atherosclerosis-prone sites without signs of atherosclerotic lesions (Orr et al. 2005). This is consistent with stress fiber formation at these sites, as fibronectin is required for Rho-A activation, a signal that is well known to promote stress fibers (Bourdoulous et al. 1998). In our laboratory, we use cross-linked gelatin (Smeets et al. 1992), which acts as a scaffold for deposition of the endothelial-produced endogenous extracellular matrix proteins. As a result, cells maintain endothelial barrier function during shear stress-induced dynamic alignment and develop a continuous VE-cadherin patterning and actin patterning, with the development of circumferential actin filament bundles and the nearly complete disappearance of stress fibers, a patterning characteristic for arterial endothelium in vivo (Schnittler et al. 1997; Seebach et al. 2000, 2007). Furthermore, the appearance and the dynamics of actin filaments strongly depend on cell density, as recently demonstrated (Taha and Schnittler 2014; Taha, et al. 2014) (Fig. 5). Stress fibers mostly disappear with increasing cell density and in the end can develop a circumferential junction-associated actin filament pattern exhibiting great variation in actin dynamics (Fig. 5), (compare: http://www.molbiolcell.org/content/suppl/2013/11/11/mbc.E13-07-0404v1.DC1/mc-E13-07-0404-s07.mp4). Life cell imaging of endothelium, particularly the dynamics of fluorescence-tagged proteins in endothelial cells has been limited for a long time, as the endothelium is less susceptible to genetic manipulation by classical transfection protocols (Lindemann and Schnittler 2009). Considerable progress has been made in recent years, as genetic manipulation of primary cultures of endothelial cells using virus-based gene transduction protocols such as adenovirus and lentivirus gave sufficient results (Kuldo et al. 2013; Lindemann and Schnittler 2009; Mannell et al. 2012; Taflin et al. 2013; Witting et al. 2013). In 2008, the group of Wedlich-Söldner introduced a novel and beautiful tool for studying actin dynamics (Riedl et al. 2008). This is a 17-amino-acid-long peptide, named Lifeact, which binds highly specifically to actin filaments and can be fused to fluorescent proteins such as EGFP or mCherry without significant disturbance of actin dynamics (Riedl et al. 2008). Cloning of Lifeact-fluorescent-protein (LifeAct-EGFP or LifeAct-mChery) into the lentiviral vector allowed the study of actin dynamics in endothelial cells by fluorescent life cell imaging over periods of hours (Taha et al. 2014). In summary, it is reasonable to propose that endothelial stress fibers are typical structures appearing in endothelium in regeneration, cell proliferation and after activation due to inflammatory stimulation and thus seem to compensate for increased cellular vulnerability and also contribute the enforced cell dynamics needed under those conditions.
Actin networks and endothelial cell junctions
Junction formation, remodeling and maintenance in endothelium depend on the formation of lamellipodia/filopodia and the junction-associated intermittent lammellipodia (JAIL) that are actin-driven membrane protrusions developed at the leading edges of migrating cells and at established cell junctions (JAIL), respectively (Lecuit 2008; Nelson et al. 2013; Niessen et al. 2011; Taha and Schnittler 2014).
Actin-driven initial cell contact formation in endothelium
Initial cell contact formation of both epithelium and endothelium is mediated by plasma membrane protrusions, lamellipodia, which develop at the leading edge of migrating cells. In general, lamellipodia formation is initiated by the activated actin-related protein (ARP) 2/3 complex that controls actin network formation due to initiating actin nucleation and branching. Filament formation occurs in a polarized manner from which a slow growing pointed end and a quick growing barbed end can be distinguished (Goley and Welch 2006; Padrick and Rosen 2010; Pollard and Borisy 2003; Rottner et al. 2010). Physical interaction of the plasma membrane of adjacent cells allows the formation of a first adhesion complex most likely by the homophilic engagement of cadherins (Lecuit 2008; Nelson et al. 2013; Niessen et al. 2011; Taha and Schnittler 2014; Hoelzle and Svitkina 2012) that exist as free-floating mono- or multimers in the plasma membrane (Iino et al. 2001). The details about the molecular mechanisms of actin driven lamellipodia and filopodia formation have been thoroughly discussed in great detail in recent reviews (Goley and Welch 2006; Mattila and Lappalainen 2008; Padrick and Rosen 2010; Pollard and Borisy 2003; Rottner et al. 2010) and are not within the scope of this paragraph. Apart from lamellipodia, filopodia can also contribute to the initial formation of intercellular cell adhesion. Filopodia are finger-like plasma membrane protrusions with a diameter of up to 0.3 μm, which display actin filaments aligned in parallel (Mattila and Lappalainen 2008). Filopodia develop from the plasma membrane of the entire cell body or even from the leading edge of lamellipodia. Filopodia function as antennae to probe the local environment, as they express receptors for diverse surface receptors for molecules such as cadherins, integrins and extracellular matrix proteins and might also transmit a variety of signals to the cytoplasm (Mattila and Lappalainen 2008). Further details related to filopodia formation and function have been described in a recent review (Mattila and Lappalainen 2008). Initial cell contact formation in endothelial cell cultures was shown to be mediated by lamellipodia that overlap plasma membranes of adjacent cells, a process that causes VE-cadherin engagement in trans followed by the formation of filopodia-like structures involving fascin and the vasodilatator-stimulated phosphoprotein (VASP) (Hoelzle and Svitkina 2012). This led to individual VE-cadherin-based endothelial cell contacts that are associated with myosin II-containing stress fibers (Hoelzle and Svitkina 2012). Those VE-cadherin clusters alternate with VE-cadherin-free gaps, forming an interrupted VE-cadherin patterning (Lampugnani et al. 1995). These immature junctions display high dynamic remodeling when compared to cell junctions in confluent cells (Huveneers et al. 2012; Taha et al. 2014). However, VE-cadherin-mediated cell adhesion is organized by individual VE-clusters of various sizes, as demonstrated by stimulated emission depletion (STED) microscopy with a lateral resolution of about 30–40 nm (Seebach et al. 2007). Individual clusters still exist, even if a continuous VE-cadherin patterning is seen by confocal laser microscopy with a maximal lateral resolution of about 300 nm (Seebach et al. 2007). These clusters can fuse together under certain stimulations, a phenomenon that was correlated with increased endothelial barrier function (Seebach et al. 2007). VE-cadherin clusters have been described as associating with stress fibers (Hoelzle and Svitkina 2012; Huveneers et al. 2012; Kronstein et al. 2012; Millan et al. 2010), but many of the VE-cadherin are not obviously associated with stress fibers (Schnittler, own observation). Furthermore, there are polygonal organized VE-cadherin superstructures (Geyer et al. 1999) that appear at areas of overlapping plasma membranes. Whether and how individual and superstructured VE-cadherin clusters connect to actin filaments is not entirely clear.
Junction-associated intermittent lamellipodia (JAIL)
Recent work now demonstrates that actin-driven lamellipodia formation is not only required for initial cell junction formation but also essential for cell junction dynamics and the maintenance of adherens junction integrity and barrier function in mature junctions (Rajput et al. 2013; Taha and Schnittler 2014; Taha et al. 2014; Tang and Brieher 2012; Zhou et al. 2013). The background of this mechanism is based on the observation that lamellipodia-like structures appear at endothelial cell junctions, particularly in subconfluent cells with a cell density <<105 cells/cm2 (Taha and Schnittler 2014; Taha et al. 2014). Subconfluent cultures display an interrupted VE-cadherin patterning (Lampugnani et al. 1995) with intercellular gaps in-between; a location where actin-driven and spatio-temporally restricted lamellipodia-like structures typically occur. Accordingly, these structures were designated as junction-associated intermittent lamellipodia (JAIL) (Taha et al. 2014). JAIL are driven by actin polymerization under the control of the ARP2/3 complex and nucleation promoting factors (NPFs) such as N-WASP (for review, see Taha and Schnittler 2014). JAIL develop at gaps between individual VE-cadherin clusters (Figs. 6 and 7) and cause a spatio-temporally restricted overlap of the respective parts of the plasma membranes (Taha and Schnittler 2014; Taha et al. 2014). This process facilitates VE-cadherin engagement and leads to the formation of lamellar-like VE-cadherin adhesion sites that are subsequently incorporated into the cell junctions during JAIL retraction. These dynamics change the VE-cadherin patterning and drive the dynamic remodeling of VE-cadherin (compare movie at http://www.molbiolcell.org/content/suppl/2013/11/11/mbc.E13-07-0404v1.DC1/mc-E13-07-0404-s11.mp4). According to this mechanism, actin-driven JAIL are large and appear frequently at junctions of subconfluent cell cultures displaying large gaps between individual VE-cadherin clusters. In addition, the JAIL size (up to 48 μm2), duration (up to 5 min) and frequency all decreased with increasing cell density due to increased VE-cadherin cluster formation (Taha et al. 2014). JAIL formation explains the recently described high VE-cadherin dynamic remodeling of the VE-cadherin in subconfluent versus confluent endothelial cell cultures (Huveneers et al. 2012; Taha et al. 2014). The coordination between VE-cadherin-mediated cell adhesion and ARP2/3-controlled and actin-driven junction-associated intermittent lamellipodia (JAIL) formation appears to be an interdependent mechanism that permits concurrent junction dynamics and cell adhesion at the same time. This also explains the ability of cell junctions to respond to stimulations at different spatiotemporal scales.

Scheme illustrating JAIL formation at endothelial cell junctions lacking VE-cadherin

Actin-driven JAIL formation. JAIL are driven by the N-WASP-activated ARP2/3 complex. The time point of maximal JAIL extension (about 5 min) in HUVEC is shown. JAIL induces formation of VE-cadherin adhesion plaque. Taken from Taha et al. (2014)
Cortical actin and the membrane cytoskeleton
Another type of actin filaments are the cortical actin filaments, which are also termed cortical actin rim and are located below the plasma membrane and seem to be connected to the classical membrane cytoskeleton that connects to integral membrane proteins and membrane lipids. For detailed reviews, we refer readers to Kapus and Janmey (2013), Pesen and Hoh (2005) and Prasain and Stevens (2009). The membrane cytoskeleton functions as a stabilizing protein network that connects the semi-fluidic plasma membrane with the cortical actin filaments and the classical cytoskeleton (Pesen and Hoh 2005). Thus, corical actin filaments are, firstly, involved in determination of the cell shape and, secondly, might provide a physical link for cell signaling (Kapus and Janmey 2013). The organization of the membrane cytoskeleton of non-erythrocytes including endothelium (Kapus and Janmey 2013; Pesen and Hoh 2005; Prasain and Stevens 2009) seems to be comparable to the membrane cytoskeleton of erythrocytes (Bennett 1982). It consists in spectrin and actin, which are further organized and regulated by adaptor proteins such as ankyrin, proteins 4.1 and 4.2, ezrin-radixin-moesin (ERM) proteins and adducin (Kapus and Janmey 2013; Prasain and Stevens 2009). The endothelial membrane cytoskeleton (Pesen and Hoh 2005), together with cortical actin filaments, have been indicated to be critical in the mechanically induced release of nitric oxide (Fels et al. 2012). It can be assumed that both cortical actin and the membrane cytoskeleton dynamically interact with actin filament bundles and most likely also hold influence with actin networks. In this way, cortical actin and the membrane cytoskeleton might be involved in controlling interendothelial and cell substrate adhesion (Kapus and Janmey 2013; Prasain and Stevens 2009). However, as outlined above, the appearance and dynamics of actin-containing bundles and the actin networks critically depend on cell density (Taha et al. 2014). It is currently unclear if the organization and the functional behavior of the cortical actin and the membrane cytoskeleton also depend on cell density. Of note, it has been demonstrated that (1) spectrin and (2) MYADM, a membrane micro-domain organizing protein, are critical for endothelial barrier function regulation (Aranda et al. 2011; Benz et al. 2008). However, the molecular architecture of how the cortical actin and the membrane cytoskeleton interact with cell junctions and whether or how this interaction is regulated, are still matters remaining to be investigated.
Future directions and outlook
Actin dynamics plays a central role in establishing and maintaining the integrity of the entire endothelium and, at the same time, allowing plasticity due to physiological and pathological demands. Direct and/or adaptor molecule-mediated indirect interactions between actin filaments and surface receptors such as integrins, cadherins, or other receptor types are required to fulfill these tasks. Detailed studies in the past have unraveled a significant number of signaling cascades and mechanisms that target and modulate actin dynamics at the attachment sites of the plasma membrane, such as cell–cell junctions, focal adhesion sites, or the membrane cytoskeleton. However, the molecular architecture that mediates and regulates the close interaction between these structures needs to be further clarified. This is particularly the case for the interaction between actin filaments and endothelial cell junctions, as well as actin filaments and the membrane cytoskeleton. Furthermore, there is an essential need to understand how different actin filament-containing structures, such as actin bundles, actin networks and the membrane cytsoskeleton, interact with each other, particularly at sites where focal adhesion and intercellular adhesion take place. Novel papers have demonstrated the regulatory role of actin nucleators such as the ARP2/3 complex and its activators, N-WASP and cortactin, in interendothelial adhesion dynamics and in turn endothelial barrier function. Since actin regulation and VE-cadherin regulation depend on a significant number of structural and regulatory molecules, these interactions need to be further analyzed.
References
Abe K, Takeichi M (2008) EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci USA 105:13–19
Aepfelbacher M, Essler M, Huber E, Sugai M, Weber PC (1997) Bacterial toxins block endothelial wound repair. Evidence that Rho GTPases control cytoskeletal rearrangements in migrating endothelial cells. Arterioscler Thromb Vasc Biol 17:1623–1629
Aktories K, Lang AE, Schwan C, Mannherz HG (2011) Actin as target for modification by bacterial protein toxins. FEBS J 278:4526–4543
Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, Luscinskas FW (2008) p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood 112:2770–2779
Alcaide P, Martinelli R, Newton G, Williams MR, Adam A, Vincent PA, Luscinskas FW (2012) p120-Catenin prevents neutrophil transmigration independently of RhoA inhibition by impairing Src dependent VE-cadherin phosphorylation. Am J Physiol Cell Physiol 303:C385–C395
Aranda JF, Reglero-Real N, Kremer L, Marcos-Ramiro B, Ruiz-Saenz A, Calvo M, Enrich C, Correas I, Millan J, Alonso MA (2011) MYADM regulates Rac1 targeting to ordered membranes required for cell spreading and migration. Mol Biol Cell 22:1252–1262
Ayalon O, Geiger B (1997) Cyclic changes in the organization of cell adhesions and the associated cytoskeleton, induced by stimulation of tyrosine phosphorylation in bovine aortic endothelial cells. J Cell Sci 110(Pt 5):547–556
Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP (2010) Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost 103:40–55
Bennett V (1982) The molecular basis for membrane - cytoskeleton association in human erythrocytes. J Cell Biochem 18:49–65
Benz PM, Blume C, Moebius J, Oschatz C, Schuh K, Sickmann A, Walter U, Feller SM, Renne T (2008) Cytoskeleton assembly at endothelial cell-cell contacts is regulated by alphaII-spectrin-VASP complexes. J Cell Biol 180:205–219
Birukova AA, Tian X, Tian Y, Higginbotham K, Birukov KG (2013) Rap-afadin axis in control of Rho signaling and endothelial barrier recovery. Mol Biol Cell 24:2678–2688
Bogatcheva NV, Garcia JG, Verin AD (2002) Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry (Mosc) 67:75–84
Bourdoulous S, Orend G, MacKenna DA, Pasqualini R, Ruoslahti E (1998) Fibronectin matrix regulates activation of RHO and CDC42 GTPases and cell cycle progression. J Cell Biol 143:267–276
Bunnell TM, Burbach BJ, Shimizu Y, Ervasti JM (2011) beta-Actin specifically controls cell growth, migration, and the G-actin pool. Mol Biol Cell 22:4047–4058
Caveda L, Martin-Padura I, Navarro P, Breviario F, Corada M, Gulino D, Lampugnani MG, Dejana E (1996) Inhibition of cultured cell growth by vascular endothelial cadherin (cadherin-5/VE-cadherin). J Clin Invest 98:886–893
Chiasson CM, Wittich KB, Vincent PA, Faundez V, Kowalczyk AP (2009) p120-catenin inhibits VE-cadherin internalization through a Rho-independent mechanism. Mol Biol Cell 20:1970–1980
Choi YS, Choi HJ, Min JK, Pyun BJ, Maeng YS, Park H, Kim J, Kim YM, Kwon YG (2009) Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood 114:3117–3126
Dejana E, Vestweber D (2013) The Role VE-Cadherin in Vascular Morphogenesis and Permeability Control. In: VanRoy F (ed) Molecular Biology of Cadherins, vol 116. Progress in Molecular Biology and Translational Science, pp 119-144
DePaola N, Phelps JE, Florez L, Keese CR, Minnear FL, Giaever I, Vincent P (2001) Electrical impedance of cultured endothelium under fluid flow. Ann Biomed Eng 29:648–656
Dickinson RB (2009) Models for actin polymerization motors. J Math Biol 58:81–103
Disanza A, Steffen A, Hertzog M, Frittoli E, Rottner K, Scita G (2005) Actin polymerization machinery: the finish line of signaling networks, the starting point of cellular movement. Cell Mol Life Sci 62:955–970
Dominguez R (2010) Structural insights into de novo actin polymerization. Curr Opin Struct Biol 20:217–225
Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI (2005) Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell 123:903–915
Drenckhahn D (1982) Cell motility and cytoüplasmic filaments in vascular endothelium. In: Hammersen MK (ed) Bodensee Symposium on microcirculation. Karger, Basel, pp 60–79
Drenckhahn D, Wagner J (1986) Stress fibers in the splenic sinus endothelium in situ: molecular structure, relationship to the extracellular matrix, and contractility. J Cell Biol 102:1738–1747
Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M (1998) Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem 273:21867–21874
Fels J, Jeggle P, Kusche-Vihrog K, Oberleithner H (2012) Cortical actin nanodynamics determines nitric oxide release in vascular endothelium. PLoS ONE 7:e41520
Fraccaroli A, Franco CA, Rognoni E, Neto F, Rehberg M, Aszodi A, Wedlich-Soldner R, Pohl U, Gerhardt H, Montanez E (2012) Visualization of endothelial actin cytoskeleton in the mouse retina. PLoS ONE 7:e47488
Franke RP, Grafe M, Schnittler H, Seiffge D, Mittermayer C, Drenckhahn D (1984) Induction of human vascular endothelial stress fibres by fluid shear stress. Nature 307:648–649
Gabbiani G, Badonnel MC, Rona G (1975) Cytoplasmic contractile apparatus in aortic endothelial cells of hypertensive rats. Lab Invest 32:227–234
Gabbiani G, Elemer G, Guelpa C, Vallotton MB, Badonnel MC, Huttner I (1979) Morphologic and functional changes of the aortic intima during experimental hypertension. Am J Pathol 96:399–422
Gabbiani G, Gabbiani F, Lombardi D, Schwartz SM (1983) Organization of actin cytoskeleton in normal and regenerating arterial endothelial cells. Proc Natl Acad Sci USA 80:2361–2364
Geyer H, Geyer R, Odenthal-Schnittler M, Schnittler HJ (1999) Characterization of human vascular endothelial cadherin glycans [In Process Citation]. Glycobiology 9:915–925
Goley ED, Welch MD (2006) The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol 7:713–726
Gordon SR, Staley CA (1990) Role of the cytoskeleton during injury-induced cell migration in corneal endothelium. Cell Motil Cytoskeleton 16:47–57
Gotlieb AI (1990) The endothelial cytoskeleton: organization in normal and regenerating endothelium. Toxicol Pathol 18:603–617
Hatanaka K, Simons M, Murakami M (2011) Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. Am J Physiol Heart Circ Physiol 300:H162–H172
Hoelzle MK, Svitkina T (2012) The cytoskeletal mechanisms of cell-cell junction formation in endothelial cells. Mol Biol Cell 23:310–323
Hotulainen P, Lappalainen P (2006) Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J Cell Biol 173:383–394
Huttner I, Walker C, Gabbiani G (1985) Aortic endothelial cell during regeneration. Remodeling of cell junctions, stress fibers, and stress fiber-membrane attachment domains. Lab Invest 53:287–302
Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, Akhmanova A, Rehmann H, de Rooij J (2012) Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol 196:641–652
Iino R, Koyama I, Kusumi A (2001) Single molecule imaging of green fluorescent proteins in living cells: E-cadherin forms oligomers on the free cell surface. Biophys J 80:2667–2677
Itoh M, Nagafuchi A, Moroi S, Tsukita S (1997) Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments. J Cell Biol 138:181–192
Iyer S, Ferreri DM, DeCocco NC, Minnear FL, Vincent PA (2004) VE-cadherin-p120 interaction is required for maintenance of endothelial barrier function. Am J Physiol Lung Cell Mol Physiol 286:L1143–L1153
Jinguji Y (2003) Developmental stage dependent expression of the endothelial stress fibers and organization of fibronectin fibrils in the aorta of chick embryos. Zool Sci 20:1359–1366
Kano Y, Katoh K, Fujiwara K (2000) Lateral zone of cell-cell adhesion as the major fluid shear stress- related signal transduction site. Circ Res 86:425–433
Kapus A, Janmey P (2013) Plasma membrane–cortical cytoskeleton interactions: a cell biology approach with biophysical considerations. Compr Physiol 3:1231–1281
Katoh K, Noda Y (2012) Distribution of cytoskeletal components in endothelial cells in the Guinea pig renal artery. Int J Biol 2012:439349
Katoh K, Kano Y, Ookawara S (2008) Role of stress fibers and focal adhesions as a mediator for mechano-signal transduction in endothelial cells in situ. Vasc Health Risk Manag 4:1273–1282
Knudsen KA, Soler AP, Johnson KR, Wheelock MJ (1995) Interaction of alpha-actinin with the cadherin//catenin cell-cell adhesion complex via alpha-catenin. J Cell Biol 130:67–77
Kobielak A, Pasolli HA, Fuchs E (2004) Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat Cell Biol 6:21–30
Komarova Y, Malik AB (2010) Regulation of endothelial permeability via paracellular and transcellular transport pathways. Ann Rev Physiol 72:463–493
Konstantoulaki M, Kouklis P, Malik AB (2003) Protein kinase C modifications of VE-cadherin, p120, and beta-catenin contribute to endothelial barrier dysregulation induced by thrombin. Am J Physiol Lung Cell Mol Physiol 285:L434–L442
Kovacs EM, Goodwin M, Ali RG, Paterson AD, Yap AS (2002) Cadherin-directed actin assembly: E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr Biol 12:379–382
Kronstein R, Seebach J, Grossklaus S, Minten C, Engelhardt B, Drab M, Liebner S, Arsenijevic Y, Taha AA, Afanasieva T, Schnittler HJ (2012) Caveolin-1 opens endothelial cell junctions by targeting catenins. Cardiovasc Res 93:130–140
Kuldo JM, Asgeirsdottir SA, Zwiers PJ, Bellu AR, Rots MG, Schalk JA, Ogawara KI, Trautwein C, Banas B, Haisma HJ, Molema G, Kamps JA (2013) Targeted adenovirus mediated inhibition of NF-kappaB-dependent inflammatory gene expression in endothelial cells in vitro and in vivo. J Control Release 166:57–65
Lampugnani MG, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, Ruco LP, Dejana E (1992) A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol 118:1511–1522
Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B, Dejana E (1995) The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta-catenin, and alpha- catenin with vascular endothelial cadherin (VE-cadherin). J Cell Biol 129:203–217
Lecuit T (2008) “Developmental mechanics”: cellular patterns controlled by adhesion, cortical tension and cell division. HFSP J 2:72–78
Lemichez E, Aktories K (2013) Hijacking of Rho GTPases during bacterial infection. Exp Cell Res 319:2329–2336
Liebner S, Cavallaro U, Dejana E (2006) The multiple languages of endothelial cell-to-cell communication. Arterioscler Thromb Vasc Biol 26:1431–1438
Lindemann D, Schnittler H (2009) Genetic manipulation of endothelial cells by viral vectors. Thromb Haemost 102:1135–1143
Magnusson MK, Mosher DF (1998) Fibronectin: structure, assembly, and cardiovascular implications. Arterioscler Thromb Vasc Biol 18:1363–1370
Mannell H, Pircher J, Rathel T, Schilberg K, Zimmermann K, Pfeifer A, Mykhaylyk O, Gleich B, Pohl U, Krotz F (2012) Targeted endothelial gene delivery by ultrasonic destruction of magnetic microbubbles carrying lentiviral vectors. Pharm Res 29:1282–1294
Marie H, Pratt SJ, Betson M, Epple H, Kittler JT, Meek L, Moss SJ, Troyanovsky S, Attwell D, Longmore GD, Braga VM (2003) The LIM protein Ajuba is recruited to cadherin-dependent cell junctions through an association with alpha-catenin. J Biol Chem 278:1220–1228
Martinelli R, Kamei M, Sage PT, Massol R, Varghese L, Sciuto T, Toporsian M, Dvorak AM, Kirchhausen T, Springer TA, Carman CV (2013) Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. J Cell Biol 201:449–465
Mattila PK, Lappalainen P (2008) Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol 9:446–454
Millan J, Cain RJ, Reglero-Real N, Bigarella C, Marcos-Ramiro B, Fernandez-Martin L, Correas I, Ridley AJ (2010) Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol 8:11
Mirzapoiazova T, Kolosova I, Usatyuk PV, Natarajan V, Verin AD (2006) Diverse effects of vascular endothelial growth factor on human pulmonary endothelial barrier and migration. Am J Physiol Lung Cell Mol Physiol 291:L718–L724
Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, Razvi M, Kini V, Mahadev K, Goldstein BJ, McKinney R, Fukai T, Ushio-Fukai M (2008) Role of protein tyrosine phosphatase 1B in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res 102:1182–1191
Napione L, Cascone I, Mitola S, Serini G, Bussolino F (2007) Integrins: a flexible platform for endothelial vascular tyrosine kinase receptors. Autoimmun Rev 7:18–22
Nelson CM, Chen CS (2003) VE-cadherin simultaneously stimulates and inhibits cell proliferation by altering cytoskeletal structure and tension. J Cell Sci 116:3571–3581
Nelson WJ, Dickinson DJ, Weis WI (2013) Roles of cadherins and catenins in cell-cell adhesion and epithelial cell polarity. Prog Mol Biol Transl Sci 116:3–23
Niessen CM, Leckband D, Yap AS (2011) Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol Rev 91:691–731
Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA (2005) The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol 169:191–202
Padrick SB, Rosen MK (2010) Physical mechanisms of signal integration by WASP family proteins. Annu Rev Biochem 79:707–735
Patterson CE, Lum H (2001) Update on pulmonary edema: the role and regulation of endothelial barrier function. Endothelium 8:75–105
Pesen D, Hoh JH (2005) Micromechanical architecture of the endothelial cell cortex. Biophys J 88:670–679
Petrache I, Verin AD, Crow MT, Birukova A, Liu F, Garcia JG (2001) Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 280:L1168–L1178
Pokutta S, Weis WI (2007) Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol 23:237–261
Pokutta S, Drees F, Takai Y, Nelson WJ, Weis WI (2002) Biochemical and structural definition of the l-afadin- and actin-binding sites of alpha-catenin. J Biol Chem 277:18868–18874
Pollard TD, Borisy GG (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112:453–465
Pollard TD, Blanchoin L, Mullins RD (2000) Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct 29:545–576
Potter MD, Barbero S, Cheresh DA (2005) Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem 280:31906–31912
Prasain N, Stevens T (2009) The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res 77:53–63
Rajput C, Kini V, Smith M, Yazbeck P, Chavez A, Schmidt T, Zhang W, Knezevic N, Komarova Y, Mehta D (2013) Neural Wiskott-Aldrich syndrome protein (N-WASP)-mediated p120-catenin interaction with Arp2-Actin complex stabilizes endothelial adherens junctions. J Biol Chem 288:4241–4250
Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R (2008) Lifeact: a versatile marker to visualize F-actin. Nat Methods 5:605–607
Rigor RR, Shen Q, Pivetti CD, Wu MH, Yuan SY (2013) Myosin light chain kinase signaling in endothelial barrier dysfunction. Med Res Rev 33:911–933
Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS (1995) Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA 92:8813–8817
Rottner K, Hänisch J, Campellone KG (2010) WASH, WHAMM and JMY: regulation of Arp2/3 complex and beyond. Trends Cell Biol 20:650–661
Rousseau S, Houle F, Huot J (2000) Integrating the VEGF signals leading to actin-based motility in vascular endothelial cells. Trends Cardiovasc Med 10:321–327
Rubenstein PA (1990) The functional importance of multiple actin isoforms. BioEssays 12:309–315
Schnittler HJ, Wilke A, Gress T, Suttorp N, Drenckhahn D (1990) Role of actin and myosin in the control of paracellular permeability in pig, rat and human vascular endothelium. J Physiol 431:379–401
Schnittler HJ, Puschel B, Drenckhahn D (1997) Role of cadherins and plakoglobin in interendothelial adhesion under resting conditions and shear stress. Am J Physiol 273:H2396–H2405
Seebach J, Dieterich P, Luo F, Schillers H, Vestweber D, Oberleithner H, Galla HJ, Schnittler HJ (2000) Endothelial barrier function under laminar fluid shear stress. Lab Invest 80:1819–1831
Seebach J, Madler HJ, Wojciak-Stothard B, Schnittler HJ (2005) Tyrosine phosphorylation and the small GTPase rac cross-talk in regulation of endothelial barrier function. Thromb Haemost 94:620–629
Seebach J, Donnert G, Kronstein R, Werth S, Wojciak-Stothard B, Falzarano D, Mrowietz C, Hell SW, Schnittler HJ (2007) Regulation of endothelial barrier function during flow-induced conversion to an arterial phenotype. Cardiovasc Res 75:596–607
Shen Q, Rigor RR, Pivetti CD, Wu MH, Yuan SY (2010) Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res 87:272–280
Shirinsky VP, Antonov AS, Birukov KG, Sobolevsky AV, Romanov YA, Kabaeva NV, Antonova GN, Smirnov VN (1989) Mechano-chemical control of human endothelium orientation and size. J Cell Biol 109:331–339
Shyy JY, Chien S (2002) Role of integrins in endothelial mechanosensing of shear stress. Circ Res 91:769–775
Smeets EF, von Asmuth EJ, van der Linden CJ, Leeuwenberg JF, Buurman WA (1992) A comparison of substrates for human umbilical vein endothelial cell culture. Biotech Histochem 67:241–250
Stupack DG, Cheresh DA (2002) ECM remodeling regulates angiogenesis: endothelial integrins look for new ligands. Science’s STKE 2002:pe7
Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E (2008) Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 10:923–934
Taflin C, Favier B, Charron D, Glotz D, Mooney N (2013) Study of the allogeneic response induced by endothelial cells expressing HLA class II after lentiviral transduction. Methods Mol Biol 960:461–472
Taha AA, Schnittler H (2014) Dynamics between actin and the VE-cadherin/catenin complex-novel aspects of the ARP2/3 complex in regulation of endothelial junctions. Cell Adhesion Migration (in press)
Taha AA, Taha M, Seebach J, Schnittler HJ (2014) ARP2/3-mediated junction-associated lamellipodia control VE-cadherin-based cell junction dynamics and maintain monolayer integrity. Mol Biol Cell 25:245–256
Tang VW, Brieher WM (2012) alpha-Actinin-4/FSGS1 is required for Arp2/3-dependent actin assembly at the adherens junction. J Cell Biol 196:115–130
Thurston G, Baldwin AL (1994) Endothelial actin cytoskeleton in rat mesentery microvasculature. Am J Physiol 266:H1896–H1909
Thurston G, Turner D (1994) Thrombin-induced increase of F-actin in human umbilical vein endothelial cells. MicrovascRes 47:1–20
Tojkander S, Gateva G, Schevzov G, Hotulainen P, Naumanen P, Martin C, Gunning PW, Lappalainen P (2011) A molecular pathway for myosin II recruitment to stress fibers. Curr Biol 21:539–550
Tojkander S, Gateva G, Lappalainen P (2012) Actin stress fibers–assembly, dynamics and biological roles. J Cell Sci 125:1855–1864
Vandenbroucke St Amant E, Tauseef M, Vogel SM, Gao XP, Mehta D, Komarova YA, Malik AB (2012) PKCalpha activation of p120-catenin serine 879 phospho-switch disassembles VE-cadherin junctions and disrupts vascular integrity. Circ Res 111:739–749
Vyalov S, Langille BL, Gotlieb AI (1996) Decreased blood flow rate disrupts endothelial repair in vivo. Am J Pathol 149:2107–2118
Wahl-Jensen VM, Afanasieva TA, Seebach J, Stroher U, Feldmann H, Schnittler HJ (2005) Effects of Ebola Virus Glycoproteins on Endothelial Cell Activation and Barrier Function. J Virol 79:10442–10450
Weis WI, Nelson WJ (2006) Re-solving the cadherin-catenin-actin conundrum. J Biol Chem 281:35593–35597
Weiss EE, Kroemker M, Rudiger AH, Jockusch BM, Rudiger M (1998) Vinculin is part of the cadherin-catenin junctional complex: complex formation between alpha-catenin and vinculin. J Cell Biol 141:755–764
White GE, Fujiwara K (1986) Expression and intracellular distribution of stress fibers in aortic endothelium. J Cell Biol 103:63–70
White GE, Gimbrone MA Jr, Fujiwara K (1983) Factors influencing the expression of stress fibers in vascular endothelial cells in situ. J Cell Biol 97:416–424
Witting SR, Vallanda P, Gamble AL (2013) Characterization of a third generation lentiviral vector pseudotyped with Nipah virus envelope proteins for endothelial cell transduction. Gene Ther 20:997–1005
Wojciak-Stothard B, Ridley AJ (2002) Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol 39:187–199
Wojciak-Stothard B, Entwistle A, Garg R, Ridley AJ (1998) Regulation of TNF-alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. J Cell Physiol 176:150–165
Wong AJ, Pollard TD, Herman IM (1983) Actin filament stress fibers in vascular endothelial cells in vivo. Science 219:867–869
Wong RK, Baldwin AL, Heimark RL (1999) Cadherin-5 redistribution at sites of TNF-alpha and IFN-gamma-induced permeability in mesenteric venules. Am J Physiol 276:H736–H748
Wysolmerski RB, Lagunoff D (1990) Involvement of myosin light-chain kinase in endothelial cell retraction. Proc Natl Acad Sci USA 87:16–20
Wysolmerski RB, Lagunoff D (1991) Regulation of permeabilized endothelial cell retraction by myosin phosphorylation. Am J Physiol 261:C32–C40
Xiao K, Garner J, Buckley KM, Vincent PA, Chiasson CM, Dejana E, Faundez V, Kowalczyk AP (2005) p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell 16:5141–5151
Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ (2005) Deconstructing the cadherin-catenin-actin complex. Cell 123:889–901
Yu PK, Yu D, Alder VA, Seydel U, Su E, Cringle SJ (1997) Heterogeneous endothelial cell structure along the porcine retinal microvasculature. Exp Eye Res 65:379–389
Zhou K, Muroyama A, Underwood J, Leylek R, Ray S, Soderling SH, Lechler T (2013) Actin-related protein2/3 complex regulates tight junctions and terminal differentiation to promote epidermal barrier formation. Proc Natl Acad Sci USA 110:E3820–E3829
Acknowledgement
The German Research Council, DFG INST 2105/24-1 and SCHN 430/6-1 to H.S. supported this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schnittler, H., Taha, M., Schnittler, M.O. et al. Actin filament dynamics and endothelial cell junctions: the Ying and Yang between stabilization and motion. Cell Tissue Res 355, 529–543 (2014). https://doi.org/10.1007/s00441-014-1856-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-014-1856-2