
Abstract
Pluripotent stem cells are still generally accepted not to exist in adult human ovaries, although increasing studies confirm the presence of pluripotent/multipotent stem cells in adult mammalian ovaries, including those of humans. The aim of this study is to isolate, characterize and differentiate in vitro stem cells that originate from the adult human ovarian cortex and that express markers of pluripotency/multipotency. After enzymatic degradation of small ovarian cortex biopsies retrieved from 18 women, ovarian cell cultures were successfully established from 17 and the formation of cell colonies was observed. The presence of cells/colonies expressing some markers of pluripotency (alkaline phosphatase, surface antigen SSEA-4, OCT4, SOX-2, NANOG, LIN28, STELLA), germinal lineage (DDX4/VASA) and multipotency (M-CAM/CD146, Thy-1/CD90, STRO-1) was confirmed by various methods. Stem cells from the cultures, including small round SSEA-4-positive cells with diameters of up to 4 μm, showed a relatively high degree of plasticity. We were able to differentiate them in vitro into various types of somatic cells of all three germ layers. However, these cells did not form teratoma when injected into immunodeficient mice. Our results thus show that ovarian tissue is a potential source of stem cells with a pluripotent/multipotent character for safe application in regenerative medicine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The existence of stem/progenitor cells in adult mammalian ovaries has recently been confirmed, although significant confusion remains about the types of stem/progenitor cells present and their properties. The latest studies have shown that stem cells occur in most of the compartments of adult ovaries, from the ovarian surface epithelium (OSE; Bukovsky et al. 2005; Szotek et al. 2008; Virant-Klun et al. 2008, 2009, 2011, 2013) and cortex (Zou et al. 2009; Pacchiarotti et al. 2010; Gong et al. 2010; White et al. 2012) to the follicles (Honda et al. 2007; Kossowska-Tomaszczuk et al. 2009).
The OSE has been studied by using various approaches. Gene expression profiling of OSE that was not cultured but only removed from the surface of healthy and malignant human ovaries and analysed has shown a high expression of genes related to adult stem cell maintenance and multipotency (Bowen et al. 2009). Similarly, Szotek et al. (2008) have identified a population of label-retaining cells exhibiting stem/progenitor cell characteristics in the OSE layer. In cell cultures from OSE scrapings, the presence of putative pluripotent stem cells has been confirmed by using more detailed gene expression analysis. These putative stem cells are able to develop into oocyte-like, embryoid body-like and parthenogenetic embryo-like structures and express some markers of pluripotency, such as SOX-2, OCT-4 and NANOG (Virant-Klun et al. 2008, 2009, 2011). These findings have also been extended to other mammalian species: rabbit, sheep and monkey (Parte et al. 2011). All these studies have demonstrated that probably two distinct populations of stem/progenitor cells exist in the OSE: a population of small cells resembling very small embryonic-like stem cells (VSELs) and a population of larger putative stem cells. The potential presence of VSELs in adult human ovaries is in accordance with the findings of Ratajczak and his co-workers who first discovered VSELs in human bone marrow (Kucia et al. 2006) and some other adult tissues and organs (Ratajczak et al. 2008; Paczkowska et al. 2009).
Although the existence of stem/progenitor cells located in the OSE is no longer questionable, the existence of stem/progenitors cells in adult ovarian cortical tissue has not been confirmed until now. Approximately 40 years ago, specific cells from the ovarian stroma were demonstrated to be involved in the development of follicles but they were not identified in detail (Peters and Pedersen 1967; Byskov and Lintern-Moore 1973). From today’s perspective, they can be named “stem” or “progenitor” cells. The recent report by White et al. (2012) is in accordance with some previous studies in the mouse model. Zou et al. (2009) used a similar isolation protocol to that of White et al. (2012) and successfully established the long-term culture of Mvh (human VASA)-positive cells from adult and neonatal mouse ovaries. Pacchiarotti et al. (2010) established a multipotent germ stem cell line by using the transgenic Oct4-GFP mouse model. Gong et al. (2010) described the isolation of colony-forming cells, which expressed some markers specific for embryonic stem cells, formed embryoid bodies and teratomas but did not express germline markers MVH and Fragilis. All these studies support the existence of stem/progenitor cells in the adult ovarian cortex but these cells might be of different types. White et al. (2012) claimed to have isolated mitotically active germ cells (germline stem cells) from adult human ovarian cortical tissue by using fluorescence-activated cell sorting (FACS) of cells expressing DDX4/VASA. Although this work is important, some doubts remain about the effectiveness of the isolation protocol. Zhang et al. (2012) used the Rosa26rbw/+;Ddx4-Cre germline reporter mouse model but did not find any mitotically active Ddx4-expressing cells in postnatal mouse ovaries. Whereas the presence of two different types of pluripotent stem/progenitor cells is indicated in the cortical tissue of adult mouse ovaries, only germline stem cells have been reported to be present in the adult ovarian cortex in humans. Therefore, the aims of this study were (1) to create ovarian stem cell lines from adult human ovarian cortex biopsies (after enzymatic degradation of tissue to retain most cell types of the ovarian niche), (2) to confirm and characterize stem cells expressing some markers of pluripotency/multipotency, (3) to isolate a relatively homogeneous population of stem cells from the cell cultures and (4) to differentiate stem cells in vitro into the somatic cells of all three germ layers.
Materials and methods
Ovarian cortex biopsies were collected from 18 patients who had a mean age of 50 years (range: 35–73 years) and who were surgically treated at the University Medical Centre Ljubljana for various gynaecological reasons (non-ovarian cancer, removal of ovaries to prevent breast cancer, removal of myoma). This study was approved by the Slovenian National Medical Ethics Committee (no. 135/09/09). Each patient was included in the study only after written consent was obtained confirming their voluntary participation.
Enzymatic degradation of ovarian tissue
Collected ovarian biopsies were first cut into small pieces by using a scalpel and then incubated for 10 min in 0.6 mg/ml collagenase type XI (Sigma-Aldrich) in basal medium consisting of DMEM/F-12 culture medium (Sigma-Aldrich) with 3.7 g/l NaHCO3, 1 % penicillin/streptomycin (Sigma-Aldrich), pH adjusted to 7.4 with 1 M NaOH. Following centrifugation at 1400 rpm for 8 min, the supernatant was discarded and the pellet was re-suspended in an enzyme mixture of collagenase type XI (0.6 mg/ml) and commercially prepared hyaluronidase (SynVitro Hydase, Origio). After 10 min, the enzymes were inactivated by using 10 % FBS (fetal bovine serum; Gibco) and the suspension of cells was centrifuged again. The supernatant was discarded and the pellet was re-suspended in basal medium. The suspension of cells was passed through a 70-μm cell strainer (BD Falcon), divided into three centrifuge tubes and then centrifuged again.
Culture of ovarian cells
After centrifugation, the pellets were re-suspended in 4 ml of the following culture media: (1) basal medium with 20 % (v/v) FBS, (2) basal medium with 20 % follicular fluid (FF) from the in vitro fertilization program (prepared as blood serum) or (3) a medium usually used to culture human embryonic stem cells (hESCs) and composed of basal medium, 20 % KnockOut Serum Replacement (Gibco), 1 mM L-glutamine (PAA), 1 % non-essential amino acids (PAA), 0.1 mM 2-mercaptoethanol (Invitrogen), 13 mM HEPES, 8 ng/ml human basic fibroblast growth factor (bFGF; Sigma-Aldrich). The suspensions of cells were then transferred into 0.2 % gelatine-coated 12-well culture plates (TPP) in a volume of 1 ml/well. Cells were passaged approximately every 12–18 days by using 0.15 % trypsin-EDTA (Sigma-Aldrich). In the optimal cell cultures, approximately 20 passages were performed during the 8-month cell culturing period. Cells were monitored daily under a heat-staged inverted microscope (Nikon ECLIPSE TE-2000S).
Analyses of cells/colonies for pluripotency or multipotency
Alkaline phosphatase staining
An Alkaline Phosphatase Detection Kit (Millipore) was used to detect alkaline phosphatase (AP) activity. Cells were fixed in 4 % paraformaldehyde (PFA) for 1 min and incubated in a working solution of reagents that consisted of Fast Red Violet, Naphtol AS-BI phosphate solution and water in a 2:1:1 ratio. After 15 min, the cells were rinsed with phosphate-buffered saline (PBS) and observed under an inverted microscope (Hoffman illumination). The cells or cell colonies expressing AP activity were stained pink to red.
Immunocytochemistry
Pluripotent and mesenchymal stem cells markers were used to analyse cultured cells and to determine their stemness. Immunocytochemical staining was performed by using two different indirect methods: in the first, the secondary antibodies were conjugated with biotin and, in the second, they were conjugated with fluorophores.
When using biotin-conjugated secondary antibodies, the staining was performed as follows. The cells were fixed in 4 % PFA, permeabilized with 0.2 % Triton (when intracellular protein was analysed), incubated with 3 % H2O2 to block the endogenous peroxidase activity and with 10 % FBS to block the non-specific binding sites and then incubated with primary antibodies for 1 h at room temperature. The following primary antibodies were used to determine the presence of pluripotent stem cell markers: a mouse anti-stage-specific embryonic antigen-4 (SSEA-4) monoclonal antibody (clone MC-813-70, diluted 1:100, Millipore) and a mouse anti-SOX2 monoclonal antibody (diluted 1:100, Abcam). To determine the presence of mesenchymal stem cell markers, the primary antibodies included in the Human Mesenchymal Stem Cell Characterization Kit (Millipore) were used, namely a mouse anti-CD14 monoclonal antibody (clone 2D-15C, diluted 1:500), a mouse anti-CD19 monoclonal antibody (clone FMC63, diluted 1:500), a mouse anti-STRO-1 monoclonal antibody (clone STRO-1, diluted 1:500), a mouse anti-Thy-1 monoclonal antibody (clone F15-42-1, diluted 1:500) and a mouse anti-M-CAM monoclonal antibody (clone P1H12, diluted 1:500). After incubation with the primary antibodies, the cells were incubated for 30 min with biotinylated polyclonal rabbit anti-mouse immunoglobulins (diluted 1:400, DakoCytomation) and then with ABC reagent (Vectastain Elite ABC Kit, Vector Laboratories). Following thorough washes, the cells were incubated in diaminobenzidine substrate (Sigma-Aldrich) until brown staining appeared (but no longer than 5 min) and observed under an inverted microscope (Hoffman illumination). As a negative control, the primary antibodies were omitted from the procedure and replaced with 1 % FBS. Positive cells or cell colonies were stained brown.
When using fluorophore-conjugated secondary antibodies, immunostaining was performed as follows. The cells were fixed in 4 % PFA, permeabilized with 0.2 % Triton (when intracellular protein was analysed), incubated for 20 min with 10 % FBS and then incubated with primary antibodies for 1 h at room temperature. The following primary antibodies were used: mouse anti-stage-specific embryonic antigen-4 (SSEA-4) monoclonal antibody (clone MC-813-70, diluted 1:100, Millipore), mouse anti-SOX2 monoclonal antibody (diluted 1:100, Millipore), mouse anti-OCT4 monoclonal antibody (clone 7 F9.2, diluted 1:100, Millipore), rabbit anti-OCT4 monoclonal antibody (diluted 1:200, Stemgent), mouse anti-NANOG monoclonal antibody (diluted 1:100, Abgent), rabbit anti-LIN28 antibody (diluted 1:100, Millipore) and mouse anti-STELLA monoclonal antibody (1:200, Millipore). To determine the presence of germ cells, rabbit anti-VASA/DDX4 polyclonal antibodies (1:400, Millipore) were used. After incubation with primary antibodies, the cells were visualized with goat anti-mouse immunoglobulins conjugated to Alexa Fluor 488 (1:200, Molecular Probes) or with goat anti-rabbit immunoglobulins conjugated to Cy3 (1:200, Molecular Probes). Following a 30-min incubation, the cells were washed and counterstained with 4,6-diamidino-2-phenylindole (DAPI). As a negative control, the primary antibodies were omitted from the procedure and replaced with 1 % FBS.
Flow cytometry
The cells were analysed by using anti-SSEA-4-phycoerythrin (PE) antibodies (BD Pharmingen). Mouse IgG3-PE were used as an isotype control. Cells were harvested from cultures by using trypsin to achieve single cell suspension, except for one sample analysed immediately after thawing. The samples were analysed with a FACSCalibur (BD). Data were analysed with BD CellQuest Pro Software.
FACS of SSEA-positive cells
SSEA-4-positive cells were isolated from the cell suspensions of four samples by using FACS (FACSAria, BD Biosciences, San Jose, Calif., USA). Briefly, 106 cells were resuspended in PBS with 1 % penicillin/streptomycin (Sigma) and stained with anti-SSEA-4-PE antibodies (BD Pharmingen, clone MC813-70). Mouse IgG3 κ-PE (BD Pharmingen) was used as isotype control. Suspension of cells with added antibodies were incubated for 30 min in the dark, then washed and re-suspended for sorting in PBS with penicillin/streptomycin at a concentration of 2 × 106 cells/ml. Sorted cells were collected in prepared basal medium with 20 % FBS and observed under an inverted microscope.
Gene expression analyses of single cell colonies
Gene expression analyses of putative stem cells (single cell colonies) cultured in various media were performed by using the Biomark real-time quantitative polymerase chain reaction (qPCR) system (Fluidigm). In all samples, the expressions of 18 genes, namely LIN28, SOX-2, OCT4A, TDGF1, UTF1, NANOS, NANOG, STELLA, MYC, TERT, KLF4, LIN28B, CD9, STAT3, DNMT1, REX01 and DNMT3B, mostly related to pluripotency and of the housekeeping gene for glyceraldehyde 3-phoshate dehydrogenase (GAPDH), which was used for normalization, were analysed in comparisons with hESCs (positive control) and fibroblasts (negative control). The sample handling and data analysis were performed as described previously (Stimpfel et al. 2012).
Telomerase assay
Three samples of ovarian stem cell culture and positive controls (hESCs and human breast adenocarcinoma cells MCF7) were analysed. Total RNA was isolated from 105 or 106 cells by using a RNAqueous-4PCR Kit (Ambion) according to the manufacturer’s instructions. RNA was quantified by using the NanoDrop 1000 spectrophotometer (Thermo Scientific) and cDNA was synthesized from 500 ng total RNA by using a High-Capacity cDNA Reverse Transcription Kit with an RNase Inhibitor (Applied Biosystems) according to the manufacturer’s protocol. The expression of human telomerase reverse transcriptase (hTERT) in tested samples was performed by using the TaqMan Gene Expression Assay Hs00972646_m1 (Applied Biosystems) according to the manufacturer’s instructions. The housekeeping gene GAPDH was used as an endogenous control. Briefly, qPCR was performed by using an ABI 7900 instrument (Applied Biosystems). Individual qPCRs were carried out in 10 μl reaction mix with 2× TaqMan Universal PCR Master Mix (Applied Biosystems), 1× TaqMan Gene Expression Assay (Applied Biosystems) and 200 ng cDNA. Each sample was analysed in triplicate. RNA isolated from MCF7 cells was used as a positive control for hTERT expression. The data were analysed by SDS2.4 software and Ct values were extracted. Fold-differences in hTERT expression were calculated by using the comparative Ct method as described previously (Livak and Schmittgen 2001).
TaqMan protein expression assay
The reagents of the TaqMan Protein Assay enable the detection and relative quantitation of proteins in cultured cells and tissue lysates. The reagents use an adapted form of proximity ligation assay technology (Fredriksson et al. 2002; Gullberg et al. 2004) in combination with real-time qPCR. We used this technology to analyse the cells from ovarian stem cell cultures. hESCs were used as a positive control, with human adult dermal fibroblasts as a negative control; a non-protein control (NPC) was also employed (double-distilled H2O added instead of the sample to the reagent mixture). The sample cells were collected and counted by using a Neubauer counting chamber. The sample lysis solutions were prepared with Cell Lysis Reagent (part of the Protein Expression Sample Prep Kit; Life Technologies), Calbiochem Protease Inhibitor Cocktail Set I and Calbiochem Phosphatase Inhibitor Cocktail Set II (EMD Chemicals) as specified by the manufacturer. The cell lysate dilutions representing 250, 125 and 62 cells per reaction were then prepared. A TaqMan Protein Assay Kit (hOct3/4; Life Technologies), in combination with a TaqMan Protein Assays Core Reagents Base Kit and 2× TaqMan Protein Assays Fast Master Mix, was used following the manufacturer’s recommendations. All qPCRs were carried out on an ABI PRISM 7500 Fast instrument (Life Technologies). The fluorescence in the amplification reactions was analysed by using Sequence Detection Software, v2.1 (Life Technologies). Final analysis was performed by ProteinAssist Software (Life Technologies). In data analysis, the cycle thresholds (Ct) for each dilution from the amplification plot by using a threshold of 2 were obtained. Protein expression fold changes between NPC and tested samples were determined by first calculating ΔCt values (Ct for cell input minus Ct for NPC) for each lysate dilution point and for each protein target. Then, ΔCt values were plotted against cell input per assay reaction. The slopes from the resulting plots were used to determine linear dynamic range and fold changes. All relative fold changes (up and down) were calculated against the positive or negative controls.
Western blot assay for protein expression
Five different cultures of putative ovarian stem cells were analysed by Western blot assay for the expression of OCT4A and LIN28 proteins in comparison with a positive control (hESCs) and negative control (human adult fibroblasts). The proteins were extracted by M-PER Mammalian Protein Extraction reagent (Thermo Scientific), to which protease and phosphatase inhibitor cocktail sets were added (EMD Chemicals). Cell lysates were centrifuged at 14,000 g for 15 min at 4 °C. Supernatants were solubilized in 2× Laemmli buffer, boiled for 2 min and separated by 12 % SDS-polyacrylamide gel electrophoresis on mini gels (1.5 mm thick) in a Mini-Protean Tetra Cell (Bio-Rad); 20–40 μg of hESC lysate and 100 μg ovarian and fibroblast cell lysate were loaded per lane. The proteins were transferred to a 0.2 μm polyvinylidene difluoride membrane (Immobilon-PSQ, Millipore) overnight at a constant voltage (20 V), followed by a high-intensity transfer at a constant 1 A for 30–60 min, both under cooling in a Criterion Blotter (Bio-Rad). Protein standards from 10 kDa to 250 kDa (Precision Plus Protein Western C standards) were from Bio-Rad. Membranes were blocked in 5 % non-fat dried milk (Bio-Rad) in PBS-Tween for 1 h and then incubated with mouse monoclonal antibodies against OCT-4 (clone 7 F9.2, diluted 1:1000, Millipore) for 3 h at room temperature and rabbit polyclonal antibodies against LIN28 (diluted 1:500, Millipore) overnight at 4 °C. After being washed in PBS-Tween, the blots were incubated with secondary antibodies: anti-mouse horseradish-peroxidase-conjugated IgGs from sheep (diluted 1:10,000, GE Healthcare, Amersham) for 2 h at room temperature or goat anti-rabbit horseradish-peroxidase-conjugated IgGs (H + L; diluted 1:5000, Thermo Scientific, Pierce Biotechnology) for 1 h at room temperature. Immunoreactive proteins were visualized by using chemiluminescent substrate ECL Prime Western Blotting Detection Reagent (GE Healthcare). Specificity of the immune-reaction was tested by probing the membrane with the secondary antibody only. The used anti-OCT-4 and anti-LIN28 antibodies detect intracelullar proteins with molecular weights ∼39 kDa and ∼28 kDa according to manufacturer’s declaration.
In vitro differentiation of ovarian stem cells
Adipogenic differentiation was induced by using adipogenic induction medium. The medium consisted of DMEM/F-12 high glucose (Gibco), GlutaMax (Gibco), recombinant human insulin (Sigma), dexamethasone (Sigma), indomethacin (Fluka), 3-isobutyl-1-methylxanthine (Sigma), human serum and gentamycin (Gibco). After 3 weeks of culture in induction medium, the cells were fixed in 4 % PFA, incubated for 10 min in Oil Red O working solution, rinsed with PBS three times and observed under an inverted microscope (Hoffman illumination). Lipid droplets were stained red.
To induce osteogenic differentiation, the cells were cultured in osteogenic induction medium consisting of DMEM low glucose, L-glutamin, FBS, dexamethasone (Sigma-Aldrich), L-ascorbic acid 2-phosphate (Sigma-Aldrich), β-glycerophosphate (Sigma-Aldrich) and penicillin/streptomycin. After 4 weeks, the cells were analysed by using von Kossa staining. The cells were fixed in 4 % PFA, incubated for 10 min in the dark in 2 % silver nitrate, washed with distilled water and exposed to UV-light for 25 min. After being washed, the cultures were observed by light microscopy to detect deposited silver as metallic silver.
Neural differentiation was initially performed as described previously (Jang et al. 2010). Briefly, the cells were cultured for 7 days in DMEM/F-12 supplemented with 1 % FBS and 80 ng/ml human bFGF and then for 7–10 days in DMEM/F-12 supplemented with 10 μM forskolin. Then, the cells were analysed by using immunocytochemistry. Following a few attempts of successful differentiation, we modified the protocol to increase the number of differentiated cells. The cells from various passages (from primary culture to the 10th passage) were cultured in DMEM/F-12 supplemented with 80 ng/ml human bFGF, 30 μM forskolin, 2 % FBS, 1 % non-essential amino acids, 0.1 mM 2-mercaptoethanol and 1 % ITS (insulin-transferrin-selenium). Following 1 week of culture, the cells were analysed by using immunocytochemistry.
To initiate pancreatic differentiation, the cells were cultured for 7 days on gelatine or on Matrigel in pancreatic proliferation medium (DMEM/F-12 supplemented with 1 % N2, 2 % B27, 1 % penicillin/streptomycin and 25 ng/ml bFGF) and then for 15 days in pancreatic differentiation medium (DMEM/F-12 supplemented with 1 % N2, 2 % B27, 1 % penicillin/streptomycin and 10 mM nicotinamide; Lumelsky et al. 2001; Segev et al. 2004). The cells were then analysed by using dithizon and immunocytochemistry.
Immunocytochemistry for expression of neuronal and pancreatic markers
The cells were stained by using the protocol described in Immunocytochemisty with biotin-conjugated secondary antibodies. Biotinylated polyclonal rabbit anti-mouse immunoglobulins (diluted 1:400, DakoCytomation) and biotinylated polyclonal goat anti-rabbit immunoglobulins (diluted 1:600, DakoCytomation) were employed. To determine the presence of neuronal markers in neuronal-like cells, the following primary antibodies were used: a mouse anti-nestin monoclonal antibody (clone 10C2, diluted 1:200, Millipore), a mouse anti-β-tubulin III monoclonal antibody (clone SDL.3D10, diluted 1:500, Sigma-Aldrich) and a rabbit anti-S100 polyclonal antibody (diluted 1:500, DakoCytomation). To determine the presence of insulin and C-peptide in pancreatic-like cells, the following primary antibodies were used: a rabbit anti-insulin (H-86) polyclonal antibody (diluted 1:200, SantaCruz) and a mouse anti-C-peptide monoclonal antibody (diluted 1:100, Biovendor).
Teratoma formation test
Some of the cell cultures were examined for their ability to form teratomas in vivo. In selected cell cultures, trypsin was used to detach the cells from the surface. The cells were then centrifuged for 6 min at 1200 rpm. The supernatant was discarded and the pellet was re-suspended in basal medium. The suspension of cells was placed on ice and delivered to the Department of Animal Science (Biotechnical Faculty, University of Ljubljana, Slovenia) where approximately 1-2 × 106 cells were mixed with Matrigel and injected subcutaneously into NOD-SCID (non-obese diabetic/severe combined immunodeficiency) mice. hESCs, IGROV-1 cells and melanoma cells were used as positive controls.
Results
Ovarian cortex biopsies were collected from 18 patients with a mean age of 50 years (range: 35–73 years old). Five women were in the reproductive period of life (age 35–45), seven women in perimenopause (age 47–50) and six women in menopause/postmenopause (age 52–73). We were able to establish ovarian cell cultures with cells from 17 patients (ovarian biopsies). We did not observe any differences in the number or dimensions of cell colonies and the stem cell characteristics according to the age of the patients. The only exception was a 73-year-old patient for whom the ovarian cell culture was established but no cell colonies formed.
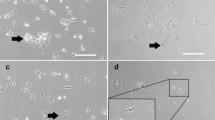
The ovarian cell cultures consisted of various types of cells, including fibroblasts forming a natural feeder layer and cell colonies (Fig. 1a), which resembled early ESC colonies (Fig. 1b). The best cell cultures providing the highest number of cell colonies (approximately 10 colonies with diameters of up to 200 μm per well in a 12-well plate) were cultured in basal medium with FBS. After prolonged culture, embryoid-body-like structures (Fig. 1c) resembling those of ESCs (Fig. 1d) also formed. The cell cultures were successfully established on autologous ovarian fibroblasts without the use of mouse embryonic fibroblasts or any other animal cells as a feeder layer. Only gelatine was used as a supportive layer to improve the attachment of cells; it was also needed in cultures after further passages, except when cells were cultured in basal medium with FBS. We were able to maintain the cell cultures as viable and in good condition for more than 6 months but, after 6 months, the natural feeder-layer consisting of autologous ovarian fibroblasts underwent senescence and, consequently, the cell colonies did not develop or grow further.

Colonies of various sizes and embryoid-body-like structures formed in ovarian and human embryonic stem cell (hESC) cultures and markers of pluripotency expressed in ovarian cell cultures. a Colony in the ovarian cell culture. b hESC culture. c Embryoid-body-like structures in ovarian cell cultures. d hESC-derived embryoid bodies. e, f Alkaline-phosphatase-positive cluster in 1-month-old culture and in 5 month-old culture, respectively. g SSEA-4-positive colony. h SOX-2-positive cell colony. Bars 100 μm
Characterization of stem cells from the cortex of adult human ovaries
The formed cell clusters were tested for the expression of various markers of pluripotency: AP, SSEA-4, SOX-2, OCT-4, NANOG, LIN28, STELLA. Stainings were performed at various times in several ovarian stem cell cultures. Positive staining of cells/colonies for AP was observed in various culture media but AP was most expressed in hESC culture medium and in basal medium with FBS (Fig. 1e, f). The initial AP activity was found on the eighth day of cell culturing but otherwise was found regardless of the time of culturing, even after 5 months. Additionally, if the cell cultures were not passaged on time, the cell colonies either no longer stained for AP activity or stained weakly 14 days after passage. Immunocytochemistry showed that cells/colonies from ovarian stem cell cultures were also SSEA-4-positive (Fig. 1g) and SOX-2-positive (Fig. 1h) when cultured in basal medium with FBS. At the same time, markers of mesenchymal stem cells were applied in the same ovarian cell cultures and the presence of cells/colonies expressing these markers was also confirmed (Electronic Supplementary Material, Fig. S1). For staining of markers of pluripotency and multipotency, similar staining patterns of cell colonies were observed: cell colonies were not homogeneously stained and were only partially stained for various markers (Fig. 1e-h, Electronic Supplementary Material, Fig. S1), whereas negative controls were not stained. Markers M-CAM (CD146) and THY-1 (CD90) were strongly expressed, STRO-1 was weakly expressed and CD14 and CD19 markers were not expressed (Electronic Supplementary Material, Fig. S1).
Moreover, immunofluorescence revealed that a proportion of cells or cell colonies was positively stained for the markers of pluripotency: LIN28, NANOG, OCT-4, SOX-2 (Fig. 2), SSEA-4 and STELLA (Fig. 3a-f). The cell colonies did not stain on germinal-lineage-related marker DDX4/VASA but we noticed some rare individual cells that had diameters of 15–20 μm and that were VASA-positive (Fig. 3g-i). A proportion of single cells with mesenchymal-like morphology were present that expressed markers of pluripotency, namely OCT-4, SSEA-4, SOX-2 and NANOG (Electronic Supplementary Material, Fig. S2); these cells expressed cytoplasmic positivity for NANOG, as was recently published for mesenchymal stem cells by Gu et al. (2012).

Cells/cell colonies in ovarian cell cultures showing positive immunofluorescence staining for markers of pluripotency, namely OCT-4, LIN28, NANOG and SOX-2. All cells/cell colonies were from the same 3-month-old cell culture and were cultured in basic medium with 20 % FBS. a–d Cells/cell colonies were double-stained for (a) OCT-4 and (b) NANOG. c Nuclei stained with DAPI. d Merged image of a–c. e–h Cells/cell colonies were double-stained for (e) OCT-4 and (f) SOX-2. g Nuclei stained with DAPI. h Merged image of e–g. i–l Cells/cell colonies were double-stained for (i) OCT-4 and (j) LIN28. k Nuclei stained with DAPI. l Merged image of i–k. m–p Cells/cell colonies were double-stained for (m) NANOG and (n) LIN28. o Nuclei stained with DAPI. p Merged image of m–o. Bars 100 μm

Cells/cell colonies in ovarian cell cultures showing positive immunofluorescence staining for markers SSEA-4, STELLA and VASA. Cells/cell colonies were from three different cell cultures and cultured in basic medium with 20 % FBS. Cells/colonies in a–c were from a single 8.5-month-old cell culture. a SSEA-4-positive cells (arrows). b Nuclei stained with DAPI. c Merged image of a, b. Cells/cell colonies in d–f were from a 1-month-old cell culture. d STELLA-positive cells. e Nuclei stained with DAPI. f Merged image of d, e. g–i Cells/cell colonies were from a different 1-month-old cell culture from that with the STELLA-positive cells. g Some rare individual cells were VASA-positive (arrow). h Nuclei stained with DAPI. i Merged image of g, h. j–l Negative control (primary antibodies were omitted). j Non-stained cells. k Nuclei stained with DAPI. l Merged image of j, k. Bars 100 μm
Flow-cytometry
Cell samples from three different ovarian cell cultures in DMEM/F-12 culture medium with FBS were analysed. In all analysed cell samples, which contained the entire amount of cells from the wells, the presence of cells expressing marker SSEA-4 was confirmed. The highest proportion of SSEA-4-positive cells in one cell culture was 3.4 %. In the two other cell cultures, the proportions of SSEA-4-positive cells were lower: 3.1 % and 0.8 % of all cells.
SSEA-4-positive cells sorted by FACS
Four ovarian stem cell cultures were collected in order to sort the SSEA-4-positive cells. All of them contained a similar homogeneous population of SSEA-4-positive cells. These cells represented up to 6 % of all cells in the cell culture (Fig. 4c, d) and were small round yellow cells with diameters of 2–4 μm (Fig. 4e). The expression of SSEA-4-surface antigen made them putative stem cells, which appeared as single cells or cells attached to larger round cells (epithelial cells, possibly). These cells were in a “dormant” state after sorting. They were alive but did not proliferate or grow. When exposed to temperature stress (2 h in a refrigerator at 4 °C) and placed back into the CO2-incubator (37 °C, 6 % CO2 in air), they started to attach to the dish bottom and subsequently began to grow or form small cell clusters, which were mostly SSEA-4-positive, as revealed by the ABC method (Fig. 4f). Similar small round yellow cells were observed during culture in all ovarian stem cell cultures. In some cultures, they were abundant (Fig. 4g) and were OCT-4-positive, as revealed by immunofluorescence (Fig. 4h–j).

Ovarian cell cultures analysed by flow-cytometry and fluorescence-activated cell sorting (FACS) for the expression of SSEA-4. a Subpopulation of SSEA-4-positive cells. b Isotype control. c Subpopulation of SSEA-4-positive cells sorted with FACS by using SSEA-4-phycoerythrin (PE) antibodies. d Isotype control. e Sorted SSEA-4-positive cells: small round yellow cells with diameters of up to 4 μm. f Sorted SSEA-4-positive cells additionally confirmed by immunocytochemistry (brown). g Similar population of small round yellow cells among ovarian cell culture. h Positive immunoflouorescence staining for the expression of marker OCT-4 reveals small putative stem cells with diameters of up to 4 μm. i DAPI staining. j Merged image of h, i. Bars 10 μm (e–g), 100 μm (h–j)
Expression of genes related to pluripotency
Gene expression analysis of four single ovarian stem cell colonies (OSC1, OSC4, OSC5, OSC6) was performed by using the Biomark real-time qPCR system, Fluidigm. The samples were taken from three different cultures (samples OSC4 and OSC5 were from the same culture). Samples OSC4 and OSC5 were sampled after 5 months of culture in basic medium with 20 % FBS, OSC6 was cultured in hESC medium and sampled in the first week of establishing the culture (the first colony that appeared in the culture) and OSC1 was cultured in basic medium with 20 % follicular fluid and sampled after the first week of culture. In all samples, the expressions of 17 genes LIN28, SOX-2, OCT4A, TDGF1, UTF1, NANOS, NANOG, STELLA, MYC, TERT, KLF4, LIN28B, CD9, STAT3, DNMT1, REX01 and DNMT3B, mostly related to pluripotency and of the housekeeping gene GAPDH, which was used for normalization, were analysed in comparison with hESCs (positive control) and human fibroblasts (F161 line; negative control). As can be seen in Fig. 5a, ovarian stem cells expressed most of the analysed genes and clustered together with hESCs (150 or 200 cells); human fibroblasts (150 or 200 cells) represented a completely different group of cells (Fig. 5). Fibroblasts did not express genes of pluripotency or expressed them to a low extent. Three ovarian stem cell cultures were separately analysed for telomerase activity by qPCR, with two of them expressing activity weakly when compared with hESCs and the MCF-7 cell line (breast cancer cells) as positive controls (Electronic Supplementary Material, Fig. S3). One of the positive ovarian stem cell cultures had been cultured for 7 months at the time of analysis.

Analyses of single ovarian stem cell colonies for the expression of genes related to pluripotency by the Fluidigm system. a Heat map comparing the expression of the genes in ovarian stem cell colonies, hESCs and fibroblasts. The scale on the left represents the level of expression (red high gene expression, green low or no gene expression). b Dendrogram from hierarchical clustering comparing ovarian stem cell colonies, hESCs and fibroblasts. c Principal component analysis (green hESCs, red ovarian stem cells, blue fibroblasts)
Expression of proteins related to pluripotency by TaqMan and Western blot protein assays
Five ovarian stem cell cultures were analysed for the expression of OCT3/4 protein by TaqMan protein assay. Two of the ovarian stem cell cultures significantly expressed the protein, when compared with hESCs as the positive control and adult human dermal fibroblasts as the negative control (Fig. 6a). In both ovarian stem cell cultures, the expression level was significantly lower than that in hESCs. Similarly, the Western blot analysis confirmed a strong double-band expression of protein in hESCs (positive control), corresponding to the OCT-4A isomere with a molecular weight of ∼39 kDa (manufacturer’s declaration); in one ovarian cell culture (K27), a weak double-band appeared corresponding to OCT-4A, whereas in two other ovarian cell cultures (K14 and K19) and adult fibroblasts (negative control), it did not (Fig. 6b, c). The LIN28 protein with an appropriate molecular weight of ∼28 kDa was weakly expressed in both analysed samples of ovarian cell cultures (K28 and K39) but it was not expressed in fibroblasts (Fig. 6d). In ovarian cell cultures, the protein LIN28 was also expressed to a lower extent than in the positive control (hESCs).

TaqMan protein assay and Western blot analyses of ovarian stem cell samples for the proteins related to pluripotency in comparison with hESCs and fibroblasts. a Expression of protein OCT-4 shows the fold change chart for OCT-4 protein (K14, K16, K19, K20, K21 ovarian stem cells, REF hESC as positive control). b Western blot analysis of protein OCT-4A in hESCs (positive control) and in three different ovarian cell cultures (K14, K19, K27) revealed strong expression of protein OCT-4A (∼39 kDa) in hESCs and weak expression of this protein in the K27 ovarian cell culture. c Western blot analysis of protein OCT-4A in hESCs (positive control) and adult human fibroblasts (negative control) revealed strong OCT-4A expression in hESCs but no expression in fibroblasts. d Two ovarian cell samples (K28, K39) showed weak expression of LIN28 in comparison with hESCs, whereas this protein was not expressed in fibroblasts
Transmission electron microscopy
Three ovarian stem cell colonies resembling embryoid-body-like structures that formed in 3- and 6-week cell cultures and that were grown in hESC medium or in basal medium with FF were sectioned and analysed by transmission electron microscopy. In the centre of the cell colonies, we found a population of relatively small round cells with diameters of approximately 5 μm; these were non-differentiated and had nuclei filling almost the entire cell volume, each nucleus being only surrounded by a thin layer of cytoplasm (Fig. 7a, b). From the centre to the margins of colonies, the cells were differentiating into various types of somatic cells (Fig. 7c, d). Lipid droplets were spread among the outer layers of cells, thus confirming the possible presence of adipocytes.

Sections of embryoid-body-like structures developed in ovarian cell cultures and analysed by transmission electron microscopy. a, b Small round non-differentiated cells with diameters of approximately 5 μm in the centre of the colony (arrows). c, d Differentiated cells in the margins (arrow in c lipid droplets acumulated in differentiated cells, arrow in d margin of colony). Bars 5 μm (a, b, d), 2 μm (c)
In vitro differentiation of ovarian stem cells
Ovarian stem cells were successfully differentiated into cells of the three primary germ layers.
-
Adipogenic differentiation of ovarian stem cells (mesoderm): in three different cultures, spontaneous differentiation of ovarian stem cells into adipocytes was observed when these stem cells in primary culture or 2-month-old culture were cultured in hESC culture medium with or without FF and on matrigel (once also on gelatine; Fig. 8a). The differentiation of the ovarian stem cells into adipogenic tissue was also induced by using adipose-differentiation medium on a 2.5-month-old culture grown in DMEM/F-12 culture medium (Fig. 8b). The accumulation of lipids in the cell cultures was confirmed by Oil Red O staining (red).
Fig. 8 
In vitro differentiation of ovarian stem cells into adipogenic, osteogenic cells, neuronal-like cells and insulin-releasing pancreatic-like cells by using differentiation media. a, b Adipocytes, which release lipid droplets, stained with Oil Red O (red). a Spontaneous differentiation. b Differentiation by using differentiation medium. c Osteogenic tissue, confirmed by von Kossa staining (black) with metallic silver deposits in the cell culture. d Nets of neuronal-like cells. Small round yellow cells (arrows) were present around differentiating cells and might be involved in the differentiation process (negative control after immunocytochemistry staining). e Nestin-positive neuronal-like cells. f β-Tubulin-III-positive neuronal-like cells. g S-100-positive neuronal-like cells. h, i Pancreatic-like cells positively stained for the expression of insulin and C-peptide, respectively. j Positive cells after dithizone staining. Bars 50 μm (a, b, d–g, j), 100 μm (c, h, i)
-
Osteogenic differentiation of ovarian stem cells (mesoderm): in two cultures of ovarian stem cells, grown in basal medium with FBS and on natural autologous ovarian fibroblasts, osteogenic differentiation was induced with an osteogenic differentiation medium. The released calcium phosphate and carbonate in the cell cultures were confirmed by von Kossa staining (Fig. 8c).
-
Neural differentiation (ectoderm): stem cells in six different ovarian cell cultures, i.e., one primary culture just after the enzymatic degradation of the ovarian biopsy and five cultures aged from 1 month to 5 months, were initially cultured in basal medium with FBS and were differentiated into neuronal-like cells by being cultured in neural differentiation medium. Neural differentiation was optimal when cells were cultured on Matrigel. Neuronal-like cells were characterized by several protrusions in different directions and formed nets of cells (Fig. 8d). In all cell cultures, the presence of small round yellow cells (putative stem cells) was observed around neuronal-like cells and were possibly involved in the differentiation process. Neuronal-like cells were positively stained for various markers: nestin (Fig. 8e), β-tubulin III (Fig. 8f) and S-100 (Fig. 8g), as revealed by immunocytochemistry.
-
Pancreatic differentiation of ovarian stem cells (endoderm): in 1-month-old and 3-month-old ovarian cell cultures in basal medium with FBS and on gelatine, the differentiation of ovarian stem cells into pancreatic-like cells was successfully induced by using pancreas differentiation medium. After overnight exposure of differentiated cells to an increased concentration of sugar (glucose), insulin-positive (Fig. 8h) and C-peptide-positive cells (Fig. 8i) were confirmed by immunocytochemistry and dithizone staining (Fig. 8j).

Teratoma formation test
We established a system for the transplantation of ovarian stem cells into NOD-SCID mice in order to test their pluripotency in vivo. After transplantation of positive controls (hESCs of the H1 line), IGROV-1 ovarian cancer cells and melanoma cancer cells, a teratoma developed (Electronic Supplementary Material, Fig. S5). Ovarian stem cells from our cultures were transplanted six times into a total of 17 NOD-SCID recipients but no teratoma formed, despite the expression of several markers of pluripotency, as confirmed by various methods.
Discussion
This report confirms that the isolation and in vitro culture of pluripotent/multipotent stem cells from small biopsies of adult human ovarian cortex are possible. These putative pluripotent/multipotent stem cells, which are probably not of germline origin, have been successfully differentiated into various types of somatic cells of all three germ layers: mesoderm, ectoderm and endoderm.
The most important observation in our ovarian cell cultures was the formation of cell colonies, which resembled early hESC colonies and were comparable with colonies developed from ovarian stem cells in a mouse model (Gong et al. 2010). As in our experiment, the cells in these colonies expressed markers of pluripotency. We also found rare individual VASA-positive cells in ovarian cell cultures. These individual VASA-positive cells were not oocytes, because they were too small (diameters of 15–20 μm) and the much larger oocytes (diameters of more than 100 μm) were removed when the suspension of cells after the enzymatic digestion of ovarian tissue was filtered through a cell strainer. Therefore, the presence of stem cells related to the germinal lineage is not excluded. In some other studies, the VASA-positive cells were much more abundant and the isolation protocols were designed to isolate cells expressing germ cell markers such as VASA and OCT-4 (Zou et al. 2009; Pacchiarotti et al. 2010; White et al. 2012) and to expand them in vitro. Zou et al. (2009) found that juvenile and adult mouse ovaries possessed mitotically active germline stem cells. They isolated them and cultured them in vitro for more than 6 months. Germline stem cells were infected with the green-fluorescent-protein-labelled (GFP) virus and transplanted into the ovaries of infertile mice in which they underwent oogenesis; the recipient mice produced GFP-transgene offspring. Further, Pacchiarotti et al. (2010) confirmed the existence of ovarian germline stem cells and their ability to be cultured and expanded in vitro in mouse postnatal ovaries. In spite of prolonged culture and many passages, ovarian germline stem cells maintained telomerase activity, expressed germ cell and stem cell markers and revealed a normal karyotype. Moreover, White et al. (2012) have published a similar observation in humans. They found that the ovaries of women in the reproductive period of life possess a population of rare mitotically active cells that have a gene expression profile consistent with primitive germ cells, including pluripotency and can be isolated from the ovarian cortical tissue by FACS, propagated and developed in vitro into haploid primitive oocytes with diameters of up to 50 μm. Although these findings contribute to basic research and to an understanding of oogenesis and ovarian stem cell self-renewal, the studies by Zou et al. (2009) and White et al. (2012), in particular, have been called into question, because an investigation by Zhang et al. (2012) has shown opposing results (no mitotically active germline progenitors in mouse ovaries) as mentioned above.
In our study, the expression of multipotent/mesenchymal stem cells was also observed. The expression of markers related to both pluripotent and multipotent stem cells indicated that, in the ovarian cortex tissue, a population of cells expressed both types of markers or that different types of stem cells were present in the tissue, including pluripotent and mesenchymal stem cells. Other reports have shown that mesenchymal stem cells from adult human tissues and organs, including testicles, can also express some markers of pluripotency (Gonzalez et al. 2009; Riekstina et al. 2009; Kuroda et al. 2010; Trubiani et al. 2010). One of the possible mechanisms generating mesenchymal stem cells in adult human ovaries might be epithelial-mesenchymal transition (Ahmed et al. 2007; Okamoto et al. 2009; Zhu et al. 2010). The study by Kossowska-Tomaszczuk et al. (2009) demonstrated the multipotency of luteinizing granulosa cells (GCs) collected from mature human ovarian follicles. Cells were isolated from the ovarian follicles of infertile patients, were maintained in culture over prolonged periods and were shown to express the typical stem cell marker OCT4 (POU5F1) and markers of the mesenchymal lineage (CD29, CD44, CD90, CD105, CD117 and CD166) but no germ-line cell markers. In contrast, stem cells in our study morphologically differ from Kossowska-Tomaszczuk’s cells; they express the markers of both pluripotency and multipotency. Therefore, the co-existence or even some kind of symbiosis between the different types of stem cells is not excluded. Some reports on the successful isolation, characterization and in vitro and in vivo differentiation of putative thecal stem cells from mouse ovaries have been presented (Honda et al. 2007). All these data confirm the potential stemness of follicular cells in the ovaries.
Our results also confirm the presence of VSEL-like cells, which have been studied previously in adult human ovaries. Former studies in humans and other mammalian species have demonstrated the natural presence of similar populations of small cells comparable with VSELs in the OSE in adult human ovaries (Virant-Klun et al. 2008, 2009, 2011; Parte et al. 2011). VSELs have also been confirmed to be present in other adult human tissues and organs (Kucia et al. 2006; Ratajczak et al. 2008; Paczkowska et al. 2009). In this study, SSEA-4-positive small round cells have been found in ovarian cell cultures just after enzymatic tissue degradation and also after prolonged culturing of ovarian cells up to 6 months or even more. The small putative stem cells sorted from ovarian cell cultures of ovarian cortex biopsies are comparable with those sorted from human adult OSE scrapings (Virant-Klun et al. 2013). The small puatative stem cells from the OSE have been shown to express some primordial germ-cell-related genes, such as PRDM1/BLIMP1, PRDM14 and STELLA. Further research will show if these small round yellow cells are really VSELs or something else.
Since the main observation of this work was the formation of cell colonies expressing some markers of pluripotent/multipotent stem cells, the cell cultures were exposed to differentiation protocols and the results confirmed the robust plasticity of these stem/progenitor cells. We were able to differentiate them into somatic cells of all three germ layers, namely adipogenic and osteogenic cells (mesoderm), neuronal-like cells (ectoderm) and pancreatic-like cells (endoderm) but they did not form a teratoma after transplantation into immunodeficient mice despite their expression of markers of pluripotency. Several possible explanations can be provided for this finding. We might not have reached the critical number of stem cells to form a teratoma in vivo or their proliferation was too slow. The different methods used in this study revealed that the proportion of cells expressing markers of pluripotency was relatively low because of the heterogeneity of the ovarian cells present in the cell cultures. In addition, the cells that expressed some markers of mesenchymal stem cells in our investigation might have prevented teratoma formation. According to some studies, multipotent mesenchymal stem cells have the ability to restrain the growth of tumours (Sun et al. 2011). Moreover, if small putative stem cells expressing the SSEA-4 marker in this study were really VSELs, then this phenomenon might also be related to their specific epigenetic status. As has previously been demonstrated, VSELs are epigenetically highly controlled to prevent the formation of tumours in adult human tissues and organs (Shin et al. 2009, 2010, 2012). Even though the formation of a teratoma in vivo is a prerequisite of pluripotency in stem cell research, the inability of ovarian stem cells to form a teratoma is actually an advantage for the potential application of ovarian stem cells in regenerative medicine.
The ovary is a highly dynamic structure with intense cell proliferation and growth. Any developing germ cell must lie in an appropriate niche (e.g., granulosa cells) in order to fulfil its function in terms of cell maintenance, renewal or differentiation. The putative stem cells with either pluripotent or multipotent characteristics described in this study might well be involved in establishing such a dynamic niche.
We can conclude that the cortex of adult human ovaries is an important source of stem cells that express various markers of pluripotency/multipotency. These cells show a high degree of plasticity and can be stored, for example, for patients with ovariectomy to prevent breast cancer and safely used in regenerative medicine because of their low potential for teratoma formation.
References
Ahmed N, Thompsona EW, Quinn MA (2007) Epithelial-mesenchymal interconversion in normal ovarian surface epithelium and ovarian carcinomas: an exception to the norm. J Cell Physiol 213:581–588
Bowen NJ, Walker LD, Matyunina LV, Logani S, Totten KA, Benigno BB, McDonald JF (2009) Gene expression profiling supports the hypothesis that human ovarian surface epithelia are multipotent and capable of serving as ovarian cancer initiating cells. BMC Med Genomics 2:71
Bukovsky A, Svetlikova M, Caudle MR (2005) Oogenesis in cultures derived from adult human ovaries. Reprod Biol Endocrinol 3:17
Byskov AG, Lintern-Moore S (1973) Follicle formation in the immature mouse ovary: the role of the rete ovarii. J Anat 116:207–217
Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gústafsdóttir SM, Ostman A, Landegren U (2002) Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol 20:473–477
Gong SP, Lee ST, Lee EJ, Kim DY, Lee G, Chi SG, Ryu BK, Lee CH, Yum KE, Lee HJ, Han JY, Tilly JL, Lim JM (2010) Embryonic stem cell-like cells established by culture of adult ovarian cells in mice. Fertil Steril 93:2594–2601
Gonzalez R, Griparic L, Vargas V, Burgee K, Santacruz P, Anderson R, Schiewe M, Silva F, Patel A (2009) A putative mesenchymal stem cells population isolated from adult human testes. Biochem Biophys Res Commun 385:570–575
Gu TT, Liu SY, Zheng PS (2012) Cytoplasmic NANOG-positive stromal cells promote human cervical cancer progression. Am J Pathol 181:652–661
Gullberg M, Gústafsdóttir SM, Schallmeiner E, Jarvius J, Bjarnegård M, Betsholtz C, Landegren U, Fredriksson S (2004) Cytokine detection by antibody-based proximity ligation. Proc Natl Acad Sci USA 101:8420–8424
Honda A, Hirose M, Hara K, Matoba S, Inoue K, Miki H, Hiura H, Kanatsu-Shinohara M, Kanai Y, Kono T, Shinohara T, Ogura A (2007) Isolation, characterization, and in vitro and in vivo differentiation of putative thecal stem cells. Proc Natl Acad Sci USA 104:12389–12394
Jang S, Cho HH, Cho YB, Park JS, Jeong HS (2010) Functional neural differentiation of human adipose tissue-derived stem cells using bFGF and forskolin. BMC Cell Biol 11:25
Kossowska-Tomaszczuk K, De Geyter C, De Geyter M, Martin I, Holzgreve W, Scherberich A, Zhang H (2009) The multipotency of luteinizing granulosa cells collected from mature ovarian follicles. Stem Cells 27:210–219
Kucia M, Reca R, Campbell FR, Zuba-Surma E, Majka M, Ratajczak J, Ratajczak MZ (2006) A population of very small embryonic-like (VSEL) CXCR4(+)SSEA-1(+)Oct-4(+) stem cells identified in adult bone marrow. Leukemia 20:857–869
Kuroda Y, Kitada M, Wakao S, Nishikawa K, Tanimura Y, Makinoshima H, Goda M, Akashi H, Inutsuka A, Niwa A, Shigemoto T, Nabeshima Y, Nakahata T, Nabeshima Y, Fujiyoshi Y, Dezawa M (2010) Unique multipotent cells in adult human mesenchymal cell populations. Proc Natl Acad Sci USA 107:8639–8643
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408
Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R (2001) Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 292:1389–1394
Okamoto S, Okamoto A, Nikaido T, Saito M, Takao M, Yanaihara N, Takakura S, Ochiai K, Tanaka T (2009) Mesenchymal to epithelial transition in the human ovarian surface epithelium focusing on inclusion cysts. Oncol Rep 21:1209–1214
Pacchiarotti J, Maki C, Ramos T, Marh J, Howerton K, Wong J, Pham J, Anorve S, Chow YC, Izadyar F (2010) Differentiation potential of germ line stem cells derived from the postnatal mouse ovary. Differentiation 79:159–170
Paczkowska E, Kucia M, Koziarska D, Halasa M, Safranow K, Masiuk M, Karbicka A, Nowik M, Nowacki P, Ratajczak MZ, Machalinski B (2009) Clinical evidence that very small embryonic-like stem cells are mobilized into peripheral blood in patients after stroke. Stroke 40:1237–1244
Parte S, Bhartiya D, Telang J, Daithankar V, Salvi V, Zaveri K, Hinduja I (2011) Detection, characterization, and spontaneous differentiation in vitro of very small embryonic-like putative stem cells in adult mammalian ovary. Stem Cells Dev 20:1451–1464
Peters H, Pedersen T (1967) Origin of follicle cells in the infant mouse ovary. Fertil Steril 18:309–313
Ratajczak MZ, Zuba-Surma EK, Shin DM, Ratajczak J, Kucia M (2008) Very small embryonic-like (VSEL) stem cells in adult organs and their potential role in rejuvenation of tissues and longevity. Exp Gerontol 43:1009–1017
Riekstina U, Cakstina I, Parfejevs V, Hoogduijn M, Jankovskis G, Muiznieks I, Muceniece R, Ancans J (2009) Embryonic stem cell marker expression pattern in human mesenchymal stem cells derived from bone marrow, adipose tissue, heart and dermis. Stem Cell Rev Rep 5:378–386
Segev H, Fishman B, Ziskind A, Shulman M, Itskovitz-Eldor J (2004) Differentiation of human embryonic stem cells into insulin-producing clusters. Stem Cells 22:265–274
Shin DM, Zuba-Surma EK, Wu W, Ratajczak J, Wysoczynski M, Ratajczak MZ, Kucia M (2009) Novel epigenetic mechanisms that control pluripotency and quiescence of adult bone marrow-derived Oct4(+) very small embryonic-like stem cells. Leukemia 23:2042–2051
Shin DM, Liu R, Klich I, Wu W, Ratajczak J, Kucia M, Ratajczak MZ (2010) Molecular signature of adult bone marrow-purified very small embryonic-like stem cells supports their developmental epiblast/germ line origin. Leukemia 24:1450–1461
Shin DM, Liu R, Wu W, Waigel SJ, Zacharias W, Ratajczak MZ, Kucia M (2012) Global gene expression analysis of very small embryonic-like stem cells reveals that the Ezh2-dependent bivalent domain mechanism contributes to their pluripotent state. Stem Cells Dev 21:1639–1652
Stimpfel M, Skutella T, Kubista M, Malicev E, Conrad S, Virant-Klun I (2012) Potential stemness of frozen-thawed testicular biopsies without sperm in infertile men included into the in vitro fertilization programme. J Biomed Biotechnol 2012:291038
Sun XY, Nong J, Qin K, Warnock GL, Dai LJ (2011) Mesenchymal stem cell-mediated cancer therapy: a dual-targeted strategy of personalized medicine. World J Stem Cells 3:96–103
Szotek PP, Chang HL, Brennand K, Fujino A, Pieretti-Vanmarcke R, Lo Celso C, Dombkowski D, Preffer F, Cohen KS, Teixeira J, Donahoe PK (2008) Normal ovarian surface epithelial label-retaining cells exhibit stem/progenitor cell characteristics. Proc Natl Acad Sci USA 105:12469–12473
Trubiani O, Zalzal SF, Paganelli R, Marchisio M, Giancola R, Pizzicannella J, Bühring HJ, Piattelli M, Caputi S, Nanci A (2010) Expression profile of the embryonic markers nanog, OCT-4, SSEA-1, SSEA-4, and Frizzled-9 receptor in human periodontal ligament mesenchymal stem cells. J Cell Physiol 225:123–131
Virant-Klun I, Zech N, Rozman P, Vogler A, Cvjeticanin B, Klemenc P, Malicev E, Meden-Vrtovec H (2008) Putative stem cells with an embryonic character isolated from the ovarian surface epithelium of women with no naturally present follicles and oocytes. Differentiation 76:843–856
Virant-Klun I, Rozman P, Cvjeticanin B, Vrtacnik-Bokal E, Novakovic S, Rülicke T, Dovc P, Meden-Vrtovec H (2009) Parthenogenetic embryo-like structures in the human ovarian surface epithelium cell culture in postmenopausal women with no naturally present follicles and oocytes. Stem Cells Dev 18:137–149, Erratum in: Stem Cells Dev 18:1109
Virant-Klun I, Skutella T, Stimpfel M, Sinkovec J (2011) Ovarian surface epithelium in patients with severe ovarian infertility: a potential source of cells expressing markers of pluripotent/multipotent stem cells. J Biomed Biotechnol 2011:381928
Virant-Klun I, Skutella T, Hren M, Gruden K, Cvjeticanin B, Vogler A, Sinkovec J (2013) Isolation of small SSEA-4-positive putative stem cells from the ovarian surface epithelium of adult human ovaries by two different methods. Biomed Res Int 2013:690415
White YA, Woods DC, Takai Y, Ishihara O, Seki H, Tilly JL (2012) Oocyte formation by mitotically active germ cells purified from ovaries of reproductive-age women. Nat Med 18:413–421
Zhang H, Zheng W, Shen Y, Adhikari D, Ueno H, Liu K (2012) Experimental evidence showing that no mitotically active female germline progenitors exist in postnatal mouse ovaries. Proc Natl Acad Sci USA 109:12580–12585
Zhu Y, Nilsson M, Sundfeldt K (2010) Phenotypic plasticity of the ovarian surface epithelium: TGF-beta 1 induction of epithelial to mesenchymal transition (EMT) in vitro. Endocrinology 151:5497–5505
Zou K, Yuan Z, Yang Z, Luo H, Sun K, Zhou L, Xiang J, Shi L, Yu Q, Zhang Y, Hou R, Wu J (2009) Production of offspring from a germline stem cell line derived from neonatal ovaries. Nat Cell Biol 11:631–636
Acknowledgments
The authors thank all patients who kindly donated their ovarian tissue for this research and are also grateful to Dr. Elvira Malicev and Prof. Primoz Rozman from the Blood Transfusion Center Ljubljana for flow-cytometry analyses and the FACS service, to Prof. Gregor Sersa from the Institute of Oncology Ljubljana for providing IGROV-1 and the melanoma cell line, to Prof. Rok Romih from the Institute of Cell Biology, Medical Faculty, University of Ljubljana for transmission electron microscopy of cell colonies, to Dr. Natasa Toplak from Omega for the TaqMan Protein Expression Assays, to Sabine Conrad, MTA from the University of Tübingen, Germany for technical assistance with the Fluidigm analyses, to Dr. Lenart Girandon from Educell for providing adipogenic induction medium, Oil Red O solution and silver nitrate solution and to all the other people and institutions supporting this research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Martin Stimpfel and Irma Virant-Klun contributed equally to this work.
The authors state that they have no competing financial interests.
This study was supported by the Slovenian Research Agency (grant J3-0415 to I.V.-K.) and by the German Federal Ministry of Education and Research (grant 01GN1001 to T.S.).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. S1
Ovarian stem cell cultures positively stained on mesenchymal stem cell markers. a M-CAM. b Thy-1. c STRO-1. d Negative control (CD 14). e Negative control (CD 19). Bar 100 μm (JPEG 21 kb)
Supplementary Fig. S2
Positive staining of mesenchymal-like stem cells for the expression of markers of pluripotency, namely OCT-4, SSEA-4, SOX-2 and NANOG, as revealed by immunofluorescence. a OCT-4-positive cells. b Nuclei stained with DAPI. c Merged image of a, b. d SSEA-4-positive cells. e Nuclei stained with DAPI. f Merged image of d, e. g SOX-2-positive cells. h Nuclei stained with DAPI. i Merged image of g, h. j NANOG-positive cells. k Nuclei stained with DAPI. l Merged image of j, k. Bar 100 μm (JPEG 32 kb)
Supplementary Fig. S3
Amplification plot showing expression of telomerase (TERT telomerase reverse transcriptase) in samples of ovarian stem cells (T1 ovarian stem cells, T2 putative ovarian stem cells in 2-week-old culture, T3 human embryonic stem cells [H1 line], T4 putative ovarian stem cells in 7-month-old culture, gapdh glyceraldehyde 3-phoshate dehydrogenase) (JPEG 53 kb)
Supplementary Fig. S4
Western blot analysis in ovarian cell cultures developed without primary antibody on hESCs. Membrane was probed with secondary antibody only. Immunoreactive protein corresponded to upper band at around 75 kDa and was non-specific (JPEG 7 kb)
Supplementary Fig. S5
Teratoma formation in SCID (severe combined immunodeficiency) mice after transplantation of ovarian cancer and melanoma cells (positive controls) (JPEG 27 kb)
Rights and permissions
About this article
Cite this article
Stimpfel, M., Skutella, T., Cvjeticanin, B. et al. Isolation, characterization and differentiation of cells expressing pluripotent/multipotent markers from adult human ovaries. Cell Tissue Res 354, 593–607 (2013). https://doi.org/10.1007/s00441-013-1677-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-013-1677-8