
Abstract
Innate immune responses to lung pathogens involve the coordinated expression of myriad affector and effector molecules of innate immunity, which must be induced and appropriately regulated in response to diverse stimuli generated by microbes or the infected host. Many intercellular and intracellular signaling pathways are involved, but we propose NF-κB and STAT3 transcription factors to be especially important signaling hubs for integrating these pathways to orchestrate effective host defense without excessive inflammatory injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute lower respiratory tract infection is a major public health concern. Amongst the poorest populations, it causes a greater burden of disease than cancer, heart attack, stroke, or any other type of infection (Mizgerd 2006). In wealthy countries such as the U.S., acute lower respiratory infection causes more deaths than any other infection, and the death rate has not substantively improved since antibiotics became of widespread use in the middle of the last century (Armstrong et al. 1999). New approaches for this persistent and pervasive disease are needed.
If microbes landing in the lungs are few and avirulent, then they can be eliminated by resident defenses such as the mucociliary escalator and alveolar macrophages. Larger numbers or more virulent microbes overwhelm or circumvent resident defenses and elicit an inflammatory response consisting of neutrophils and extravascular plasma (Mizgerd 2008). These neutrophils and plasma proteins are innate immune responses essential to host defense. However, neutrophils generate products that can kill cells and digest tissue, and the extravasation of plasma into alveoli leads to the pulmonary edema that is a defining feature of acute lung injury (Ware and Matthay 2000). Thus, these processes must be exquisitely controlled during lung infection in order to allow host defense while limiting inflammatory injury.
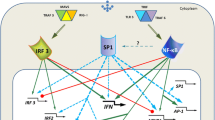
A tremendous number of molecules contribute to this control, including an ever-growing list of ligands, receptors, adaptor molecules, enzymes (including kinases, phosphatases, phospholipases, lipoxygenases, cyclooxygenases, oxidases, NO synthases, GTPases, proteases, isomerases, citrullinases, ubiquitin ligases, deubiquitinases, acetyltransferases, deacetylases, nucleotidyltransferases, ribonucleases), transcription factors, repressors, microRNAs (miRNAs), AU-rich element binding proteins (ARE-BPs; an ARE is a region with frequent A and U bases in a mRNA), and others. These myriad factors intersect in complex, intricate, and elegant networks. Signaling networks involve vast numbers of interactions with a substantially smaller number of hubs that are critical to determining the activity of the network (Ma'ayan 2009). We propose that two hubs critical to the signaling network of innate immune responses to microbes in the lung are nuclear factor kappa beta (NF-κB) and signal transducer and activator of transcription 3 (STAT3; Fig. 1). Diverse stimuli converge on these two transcription factors from myriad receptors through multiple signal transduction pathways, and these transcription factors then orchestrate responses by regulating and coordinating the expression of many genes that determine the outcome of infection. These hubs function locally, in cells within the infected lungs. In addition, these hubs are important for integrating innate immune responses throughout the organ systems, even for localized infections such as a non-bacteremic pneumonia. Here, we present evidence for the crucial roles of these two transcription factors during pneumonia, highlighting their roles in intrapulmonary and in extrapulmonary cells.

Roles of nuclear factor kappa beta (NF-κB) and signal transducer and activator of transcription 3 (STAT3) as signaling hubs mediating innate immunity during acute lower respiratory tract infection (iNOS inducible NO synthase, COX-2 cyclo-oxygenase-2, miRNAs microRNAs, ARE AU-rich element
Roles of NF-κB in lung innate immunity
NF-κB is a family of transcription factors including hetero- and homodimers of five different proteins (for a review, see Hayden and Ghosh 2008). At least some NF-κB proteins are expressed in all nucleated cells, being typically concentrated in the cytoplasm of resting cells by inhibitor kappa B (IκB) proteins. NF-κB becomes activated by a wide variety of stimuli relevant to infection and innate immunity, including pattern recognition receptors (PRRs) for microbial molecules, receptors for cytokines, and receptors for products released from damaged cells and tissues. Activation is mediated by the phosphorylation of IκB proteins, which leads to their degradation, after which NF-κB proteins become concentrated in the nucleus and bind κB sites in the DNA to regulate the transcription of nearby genes. To our knowledge, only two NF-κB proteins have been identified as translocating to the nucleus in response to microbial stimuli in the lungs, RelA (also known as p65) and p50 (Mizgerd et al. 2002); the other NF-κB proteins (c-Rel, RelB, and p52) have not been demonstrated to move into the nucleus during acute respiratory infection. Both IκB-α and IκB-β are degraded in mouse lungs after the instillation of lipopolysaccharide (LPS), and each of these IκB proteins associates with RelA in resting lungs (Mizgerd et al. 2002). These IκB proteins are probably regulatory steps that prevent spontaneous inflammation (Beg et al. 1995a; Cheng et al. 1998), with overcoming this inhibition being critical to innate immunity after microbial stimulation.
Targeted mutagenesis of the Rela gene causes embryonic lethality from which mice can be rescued by the additional interruption of tumor necrosis factor-α (TNF-α) or TNF receptor 1 (TNFR1; Alcamo et al. 2001). RelA-deficient mice on a TNFR1-deficient background are extremely susceptible to infection and die within weeks of birth unless they are kept under the most stringent barrier conditions and on an antibiotic regimen (Alcamo et al. 2001). In response to either LPS or Streptococcus pneumoniae in the lungs, RelA-deficient mice have extreme defects in the expression of multiple innate immunity genes, including chemokines and adhesion molecules mediating neutrophil recruitment (Alcamo et al. 2001; Quinton et al. 2007). Accordingly, these mice have decreased neutrophil recruitment in the lungs and impaired bacterial clearance from the lungs (Alcamo et al. 2001; Quinton et al. 2007). Thus, the induction of innate immunity genes in the lungs requires NF-κB RelA.
In contrast, targeted mutation of the Nfkb1 gene, which encodes NF-κB p50, neither results in infections nor compromises the health of mice in the absence of experimental challenges (Sha et al. 1995). In response to Escherichia coli or E. coli LPS in the lungs, p50 deficiency results in increased pulmonary expression of cytokines, including TNF-α, interleukin-1 (IL-1), IL-6, and chemokines (Mizgerd et al. 2003; Mizgerd et al. 2004a). The precise roles of p50 in limiting cytokine expression are complex and context-specific (Hayden and Ghosh 2008). The data available argue that p50 functions during acute bacterial pneumonia to repress (rather than promote) the expression of the multiple genes regulated by NF-κB. This can be disastrous for the health of the mice, resulting in increased lung injury and death despite no defect in intrapulmonary bacterial clearance during E. coli pneumonia (Mizgerd et al. 2003). Similarly, p50 deficiency results in increased cytokine expression, increased lung inflammation, and increased death after systemic administration of LPS (Gadjeva et al. 2004; Han et al. 2009). Thus, p50 is essential to braking the expression of innate immunity genes in the lungs in order to prevent or limit inflammatory injury during infection.
Whereas no humans have, to our knowledge, been identified with deficiencies of RelA or p50, human patient studies support the notion that diminished NF-κB activity increases susceptibility to infection and exaggerated NF-κB activity increases lung injury. For example, patients with deficiencies of MyD88 or interleukin-1 receptor-associated kinase 4 (IRAK-4) cannot activate NF-κB in response to IL-1 or diverse Toll-like receptor (TLR) ligands and are extremely susceptible to bacterial infections including pneumococus in particular (Ku et al. 2007; von Bernuth et al. 2008). These pathways of NF-κB activation might be especially important for host defense against pyogenic bacteria (Bousfiha et al. 2010), but they probably also contribute to host defense against viruses and fungi in the lungs. Supporting this, TLR4 polymorphisms that decrease NF-κB activation are associated with increased occurrences of severe lung infections with respiratory syncytial virus or Aspergillus (Awomoyi et al. 2007; Tal et al. 2004; Carvalho et al. 2008). Conversely, for patients with sepsis, a TLR1 polymorphism that increases NF-κB activation is associated with increased acute lung injury and increased death (Wurfel et al. 2008). Thus, NF-κB is a signaling hub critical to respiratory infection and lung innate immunity in many species including humans.
Cell-specific roles of NF-κB in lung innate immunity
Bone-marrow transplant studies suggest that innate immune responses to microbes in the lungs are dictated by NF-κB in both hematopoietic and non-hematopoietic cells. As with humans (von Bernuth et al. 2008), MyD88 deficiency predisposes mice to bacterial infections, including Pseudomonas aeruginosa pneumonia (Skerrett et al. 2004a). Remarkably, reciprocal bone-marrow transplants demonstrate that, whereas MyD88 deficiency in all cells results in 5-log more bacteria in the lungs, MyD88 deficiency in hematopoietic cells alone has no significant effect on bacterial burdens, and MyD88 deficiency in non-hematopietic cells alone has a significant but modest effect, increasing lung P. aeruginosa burdens by less than 1 order of magnitude (Hajjar et al. 2005). Although integrated host defenses are most markedly compromised by widespread MyD88 deficiency, phenotypes are apparent in the compartment-specific mutations. MyD88 deficiency in either the non-hematopietic cells alone or the hematopietic cells alone decreases neutrophil recruitment (Hajjar et al. 2005). Expression of the the ELR+CXC chemokines CXCL1 and CXCL2 is decreased by MyD88 deficiency in the non-hematopoietic cells, whereas expression of the early response cytokines TNF-α and IL-1β is diminished by MyD88 deficiency in hematopoietic cells (Hajjar et al. 2005). Thus, the widespread deficiency of MyD88 and downstream signaling such as NF-κB is disastrous, but deficiencies restricted to hematopoietic or non-hematopoietic cell types also show modest but significant phenotypes.
In which non-hematopoietic cells is the NF-κB signaling pathway important? Many studies implicate epithelial cells in the lung. Overexpression of a dominant negative (dn) IκB-α protein, in which an alanine residue replaces a serine whose phosphorylation by IκB kinase-β (IKK-β) is essential for signaling its proteasomal degradation and release of associated NF-κB proteins, is one strategy for diminishing the canonical pathway of NF-κB activation. Overexpressing dnIκB-α in alveolar epithelial cells (driven by the surfactant protein C [SPC] promoter) or in conducting airway epithelial cells (driven by the Clara Cell 10 [CC10] promoter) is sufficient to decrease the expression of cytokines elicited by LPS in the lungs, resulting in decreased neutrophil recruitment (Poynter et al. 2003; Skerrett et al. 2004b). Inhibition of NF-κB by SPC-driven dnIκB-α overexpression in alveolar epithelial cells compromises host defense during pneumococcal pneumonia, as measured by lung bacterial burdens (Quinton et al. 2007). Similarly, inhibition of NF-κB by CC10-driven deletion of IKK-β in airway epithelial cells compromises host defense during Group B Streptococcus (GBS) pneumonia, as measured by lung bacterial burdens (Fong et al. 2008). Thus, NF-κB activity is required in epithelial cells specifically for effective innate immunity in the lung. Conversely, the overexpression of a constitutively active IκB kinase in airway epithelial cells (driven by an inducible CC10 system) is sufficient to activate NF-κB in these cells, inducing pulmonary inflammation (including cytokine expression and neutrophil recruitment) and causing lung injury (including pulmonary edema, arterial hypoxemia, and death; Cheng et al. 2007). Thus, excessive NF-κB activity in airway epithelial cells alone results in acute lung injury. Together, these studies indicate epithelial cells as being relevant sites of the NF-κB signaling hub for lung innate immunity.
Several lines of evidence implicate NF-κB signaling in hematopietic cells. In mice, reciprocal bone-marrow chimera studies demonstrate that MyD88 deficiency in hematopoietic cells decreases cytokines and neutrophil recruitment during P. aeruginosa pneumonia (Hajjar et al. 2005), that TLR4 deficiency in hematopietic cells decreases cytokines and neutrophil recruitment after LPS aerosolization (Hollingsworth et al. 2005), and that deficiency of RIP2 (a NOD-like receptor that activates NF-κB) in hematopietic cells increases bacterial burdens during Chlamydiae pneumoniae pneumonia (Shimada et al. 2009). In human patients, stem cell transplantation resulting in hematopoietic cells with a haplotype conferring a hyporesponsive TLR4 (stimulating diminished NF-κB activation) increases the risk of invasive aspergillosis (Bochud et al. 2008). Conversely, if mice are reconstituted with hematopoietic cells deficient in some of the brakes on the NF-κB system (such as IκB-α or NF-κB p50), then LPS-induced pulmonary inflammation as measured by neutrophil recruitment is increased (Han et al. 2009). Thus, leukocyte NF-κB plays pivotal roles in lung innate immunity.
In some cases (Hollingsworth et al. 2005; Shimada et al. 2009), the effects of hematopoietic deficiencies in NF-κB signaling pathways can be ameliorated by intratracheal instillation of wild-type macrophages, suggesting that the macrophage might be one hematopoietic cell in which NF-κB is important to lung innate immunity. Consistent with this, and providing the most direct evidence to date that NF-κB in myeloid cells is a determinant of the outcome of lung infection, the mutation of RelA driven by the M lysozyme (LysM) locus decreases neutrophil recruitment and increases susceptibility of mice to P. aeruginosa pneumonia (Hess et al. 2010). The RelA products from macrophages that are essential to integrated host defenses against bacteria in the lungs have not been identified in these studies. In what seems to be stark contrast, the myeloid deletion of IKK-β via LysM targeting results in increased neutrophils and fewer bacteria during GBS pneumonia (Fong et al. 2008). Further studies are needed to clarify the roles of NF-κB signaling in myeloid cells, and to identify specific roles of NF-κB in distinct myeloid cell subsets in the lung (such as alveolar macrophages, dendritic cells, recruited neutrophils, and exudate monocyte/macrophages). Activation of NF-κB in myeloid cells outside of the lung may also be critical to host defense in the lung, as discussed below.
Of course, neither alveolar macrophages nor epithelial cells function alone. Signaling in each cell type may be influenced by the other. An important NF-κB pathway during pneumonia is the activation of epithelial cells by the macrophage-derived cytokines TNF-α, IL-1α, and IL-1β. Some microbes (such as pneumococcus) do not directly activate alveolar epithelial cells (Quinton et al. 2007). During pneumococcal pneumonia, alveolar epithelial cell NF-κB activation requires TNF-α, IL-1α, or IL-1β (Quinton et al. 2007; Jones et al. 2005). Blocking TNF-α or IL-1 signaling pathways individually has little or no effect during pneumococcal pneumonia, but their simultaneous blocking results in pronounced defects in NF-κB signaling, cytokine expression, and neutrophil recruitment, rendering mice highly susceptible to pneumococcal pneumonia (Jones et al. 2005). A clinical trial involving rheumatoid arthritis patients has demonstrated that combining a TNF receptor fusion protein (etanercept) with an IL-1 receptor antagonist (anakinra) significantly increases the risk of serious infections, particularly pneumonia, compared with single agent treatment (Genovese et al. 2004). Thus, in both mice and humans, the cytokines TNF and IL-1 have overlapping roles that are essential to host defense in the lungs, probably attributable to the activation of NF-κB in alveolar epithelial cells.
Roles of STAT3 in lung innate immunity
STAT3 is a transcription factor that becomes activated by tyrosine phosphorylation, which stimulates STAT3 homodimerization and the dimerization of STAT3 with other proteins such as STAT1 (Schindler et al. 2007). Dimers of tyrosine-phosphorylated STAT3 concentrate in the nucleus and bind DNA, regulating transcription. Tyrosine-phosphorylated STAT3 is observed in the lungs of mice with bacterial or viral pneumonia or acute inflammation elicited by endotoxemia, immune complexes, ozone, or hyperoxia (Severgnini et al. 2004; Gao et al. 2004; Hokuto et al. 2004; Matsuzaki et al. 2006; Quinton et al. 2008; Kida et al. 2008; Ikegami et al. 2008; Lang et al. 2008). In addition to functioning as a transcription factor in the nucleus, metabolic roles of STAT3 in the mitochondria have recently been demonstrated (Gough et al. 2009; Wegrzyn et al. 2009), but any relevance to lung innate immunity for mitochondrial STAT3 is purely speculative at present.
The most compelling evidence that STAT3 is important for acute lower respiratory infections comes from human studies. Hyper-IgE syndrome (HIES, also known as Job’s syndrome) is a rare genetic disease resulting from mutations in STAT3 rendering it transcriptionally inactive (Holland et al. 2007; Minegishi et al. 2007). Patients are heterozygous, and the dimerization of mutant allele products with themselves and with STAT3 from the functional allele results in ~25% normal STAT3 activity in patient cells (Minegishi et al. 2007). This defect in STAT3 signaling causes HIES patients to suffer from repeated bacterial pneumonias attributable to S. pneumoniae, Haemophilus influenzae, Staphylococcus aureus, and other common causes of community-acquired pneumonia, implicating a defect in lung host defense (Freeman and Holland 2009). In childhood, these patients develop pneumatoceles, suggesting a defect in lung injury repair, and these pneumatoceles then become sites of opportunistic infections of the lung, especially by Gram-negative bacteria and fungi (Freeman and Holland 2009). Pneumonia is the direct or indirect cause of death for HIES patients (Freeman et al. 2007).
HIES patients have a defect in Th17 cells (Milner et al. 2008; Ma et al. 2008), which may be one contributing factor in their susceptibility to lung infections. Th17 cells generate IL-17A, IL-17F, and IL-22, and signaling from these cytokines is important in lung host defense (Aujla et al. 2008; Ye et al. 2001). However, the sources of these cytokines in infected lungs have not been identified, and many cells other than Th17 cells can generate IL-17 and IL-22 (Mills 2008; Wolk et al. 2010). Whether Th17 cells specifically are important for lung host defense, or whether HIES patients have defects in IL-17 or IL-22 in their infected lungs is unclear. Mice deficient in STAT3 in all T cells or all CD4+ cells do not have a phenotype resembling HIES (Takeda et al. 1998; Harris et al. 2007), and bone-marrow transplantation in an HIES patient has failed to prevent immunodeficiency and infections (Gennery et al. 2000), results that, when taken together, suggest that STAT3 has roles beyond the development of Th17-like cells, which are essential to preventing lung infections and the resultant lung disease characteristic of HIES patients.
STAT3 becomes activated in the lungs of mice in response to microbial stimuli in the air spaces, including LPS, bacteria, or virus (Matsuzaki et al. 2006; Quinton et al. 2008; Ikegami et al. 2008), but STAT3 is not directly activated by signaling from PRRs for microbial molecules. IL-6 (which is induced by NF-κB during pneumonia; Quinton et al. 2007) is clearly one source of STAT3 activation during bacterial pneumonia, since the deficiency of IL-6 diminishes total levels of tyrosine-phosphorylated STAT3 in the lungs during pneumonia (Jones et al. 2006). IL-6 makes essential contributions to lung host defense, as mutation of IL-6 in mice leads to decreased neutrophil recruitment and bacterial clearance (Jones et al. 2006). These phenotypes do not correlate with the diminished expression of neutrophil chemokines or adhesion molecules. The molecular mechanisms responsible for decreased neutrophil recruitment and increased bacterial burdens in IL-6-deficient mice with pneumonia have yet to be identified.
All the IL-6 family cytokines can activate STAT3, and many IL-6 family cytokines are induced during bacterial pneumonia including oncostatin M (OsM), leukemia-inhibiting factor (LIF), and IL-11, in addition to IL-6 (Quinton et al. 2008). Using activation of an epithelial cell line as a reporter system for STAT3 activating stimuli in bronchoalveolar lavage fluid, Quinton et al. (2008) have demonstrated that the combination of IL-6 and LIF is responsible for virtually all STAT3 activation during the first 24 h of infection. Roles of LIF during lung infection are an important area for future research. By 48 h of infection with pneumonia, factors other than IL-6, LIF, and OsM are responsible for half of the STAT3-stimulating activity in the bronchoalveolar lavage fluid (Quinton et al. 2008), and the identity of these factors remains to be determined.
Several other cytokines that activate STAT3 are also essential determinants of lung innate immunity, although to our knowledge, no direct information is available about their roles in STAT3 activation during lung infection. IL-22 activates STAT3, and IL-22 blockade increases lung infection and lung injury during bacterial pneumonia (Aujla et al. 2008). The roles of IL-22 appear to involve epithelial cells particularly, with this cytokine enhancing barrier integrity and antimicrobial protein expression (Aujla et al. 2008). IL-23 activates STAT3, and IL-23 blockade increases lung infection during bacterial pneumonia (Happel et al. 2005). The role of IL-23 during pneumonia appears to be in driving the expression of IL-17 and IL-22 in the lungs, possibly from T cells (Aujla et al. 2008; Happel et al. 2005). IL-10 activates STAT3, and IL-10 receptor blockade increases lung injury elicited by influenza infection in the lungs (Sun et al. 2009a). The sites of IL-10 action may be the myeloid cells recruited to the infected lung, including monocyte/macrophages and neutrophils (Sun et al. 2009a). Increasing IL-10 expression can be protective against pneumonia-induced lung injury (Wang et al. 2005), but the anti-inflammatory effects of excess IL-10 compromise bacterial clearance (Sun et al. 2009b). STAT3 is essential to the anti-inflammatory actions of this cytokine in myeloid cells (Takeda et al. 1999), but the relevance to the lung of IL-10 signaling to macrophage STAT3 has not been specifically investigated. STAT3 can also be activated by other mediators implicated in lung host defense, such as leptin (Mancuso et al. 2002, 2006) or vascular endothelial growth factor (Yano et al. 2006), and others that may be speculated to influence lung host defense (albeit not yet tested, to our knowledge), such as IL-21, IL-27, or IL-35. Thus, STAT3 is a probable signaling hub for IL-6, LIF, IL-22, IL-23, IL-10, and many other possible mediators contributing to lung innate immunity to determine the outcome of acute lower respiratory infection.
STAT3 has been mutated in epithelial cells in the lung by using Cre transgenes driven by SPC or CC10. Such mice demonstrate increased susceptibility to lung injury induced by diverse stimuli, including E. coli pneumonia, LPS-induced pulmonary inflammation, adenoviral infection, hyperoxia, and naphthalene cytotoxicity (Hokuto et al. 2004; Matsuzaki et al. 2006; Quinton et al. 2008; Kida et al. 2008; Ikegami et al. 2008). Protection against lung injury is probably attributable to the multiple roles of STAT3 including the prevention of apoptosis, the promotion of repair processes involving cell migration and proliferation, and the regulation of surfactant homeostasis. STAT3 in the alveolar epithelium also contributes to neutrophil recruitment and anti-bacterial host defense (Quinton et al. 2008). Although modest in comparison, these findings are consistent with such phenotypes resulting from IL-6 deficiency or HIES and suggest that STAT3 in the epithelium is one mechanism by which IL-6 contributes to host defense, and that the alveolar epithelial cell is one of the cells in which STAT3 functions to prevent lung infection.
Emerging extrapulmonary roles of NF-κB and STAT3 signaling in lung innate immunity
Effective immune responses within the confines of the intrapulmonary space are critical for both promoting lung host defenses and limiting the possibility of systemic disease. Integral components of the local milieu are, however, systemically derived blood constituents such as hematopoietic cells and extravasated plasma proteins. Much of our existing knowledge regarding innate immune responses within infected airspaces is born from research conducted to identify the functions and regulation of locally synthesized antimicrobial factors and/or factors enabling the movement of key extrapulmonary mediators into the lungs. Although the importance of these systemic elements is appreciated within the context of lung infections, our understanding of the way that extrapulmonary tissues respond to lung infections and of the extent of these responses is remarkably limited. The hepatic acute phase response (APR) and hematopoiesis represent two classical systemic reactions to infection and injury. As with local innate immunity, we propose that NF-κB and STAT3 serve as particularly important signaling hubs for these systemic innate immune responses during lung infection.
Roles of NF-κB and STAT3 in the hepatic APR
The APR was first recognized nearly 80 years ago in patients with pneumococcal pneumonia (Tillett and Francis 1930) and is now considered a hallmark of infection and injury. It is defined by significant changes (>25%) in circulating concentrations of acute phase proteins (APPs), of which there are at least 40 (Gabay and Kushner 1999). Select members of this family are routinely used as biomarkers of disease severity in patients, including those with pneumonia (Almirall et al. 2004; Smith et al. 1995; Yip et al. 2005). These proteins are typically liver-derived, based largely on studies revealing a sensitive capacity of hepatocytes to synthesize them (Andus et al. 1988; Ruminy et al. 2001) and seemingly disproportionate expression levels in the liver as compared with other tissues (Meek and Benditt 1986). The net influence of APPs on outcome during infections such as those in the lung is speculative, but known functions ascribed to individual APPs imply that their presence is generally beneficial to host defense and tissue protection. These functions are diverse, including but not limited to bacterial opsonization, bacteriostatic and bactericidal effects, cytokine induction, increased chemotaxis, anti-oxidant activity, metal transport, and protease inhibition (Gabay and Kushner 1999).
The quantity and diversity of APPs make the dissection of the regulatory mechanisms and functions of the APR, as an integrated biological response, a complex undertaking. Several transcription factors involved in the APR have been identified (Ruminy et al. 2001). Amongst these, NF-κB RelA and STAT3, the latter of which was originally known as the “acute phase response factor” (Akira et al. 1994; Zhong et al. 1994), might represent particularly important regulatory nodes for APP transcription in response to bacterial stimuli in the lungs (Quinton et al. 2009). Both transcription factors can directly mediate APP expression and are exquisitely responsive to cytokines that are present and essential during pneumonia, namely TNFα and IL-1 (activating RelA) and IL-6 (activating STAT3; Andus et al. 1988; Thorn et al. 2004). Requirements for RelA and STAT3 have been reported in vitro for APP gene expression (Quinton et al. 2009; Betts et al. 1993; Patel et al. 2007). RelA and STAT3 have also been shown to interact physically in a manner that is required for the transcriptional activity of at least some APPs (Hagihara et al. 2005; Uskokovic et al. 2007).
In vivo, the determination of the direct physiological roles of RelA and STAT3 during the APR or otherwise has historically been challenging, in large part because of the embryonic lethality of mice lacking functional genes for either factor (Beg et al. 1995b; Takeda et al. 1997). Conditional mutation, however, has revealed STAT3 as a requirement for maximal APP expression in mouse liver during endotoxemia and polymicrobial sepsis (Alonzi et al. 2001; Sakamori et al. 2007). Site-specific targeting of NF-κB signaling has also been accomplished but has not yet been used to interrogate APP expression specifically. Interestingly, mutation of NF-κB RelA, IKKβ, or NEMO (NF-κB essential modulator), all of which are critical components of the canonical NF-κB signaling pathway (Hayden and Ghosh 2008), promotes liver injury in some contexts of severe inflammation (Geisler et al. 2007; Luedde et al. 2005), possibly influencing the extrapulmonary pathophysiology manifested during pneumonia or other conditions.
The early response cytokines TNFα and IL-1 together with IL-6 are well-recognized activators of APP synthesis in hepatocytes (Andus et al. 1988; Thorn et al. 2004; Kopf et al. 1994; Zahedi and Whitehead 1993). Moreover, as discussed above, these three cytokines are essential for host defense during pneumonia, albeit for reasons that are incompletely understood (Jones et al. 2005, 2006; Mizgerd et al. 2004b; van der Poll et al. 1997). It is plausible that hepatocytes are important targets of TNFα, IL-1, and IL-6 during an acute pulmonary inflammation. In response to bacteria or bacterial products administered into the lungs of mice, APPs are expressed in liver in association with the increased activation of NF-κB and STAT3 (Quinton et al. 2009; Mizgerd et al. 2001). In mice lacking a functional gene for IL-6, the induction of some APPs and STAT3 activation are nearly eliminated during bacterial pneumonia compared with the responses observed in their wild-type counterparts (Quinton et al. 2009). IL-6-dependent APP expression has also been demonstrated in response to intratracheal LPS (Vernooy et al. 2005; Gamble et al. 2008). Interestingly, the reliance upon IL-6 for STAT3 activation during a type of pneumonia elicited by Gram-positive or Gram-negative bacteria is considerably more pronounced in the liver compared with the lung (Jones et al. 2006; Quinton et al. 2009), reinforcing the possibility that hepatocytes are an important facilitator of IL-6-mediated immune responses. Mice devoid of all signaling receptors for TNFα and IL-1 also lack maximal liver APP expression (Quinton et al. 2009). Furthermore, signaling from TNFα and IL-1 receptors is required for NF-κB RelA activation in liver during pneumonia (Quinton et al. 2009; Mizgerd et al. 2001), suggesting that, like IL-6, immune properties conferred by early response cytokines during pneumonia may extend to hepatocytes.
The net physiological consequence of the APR in the context of lung innate immunity or otherwise is unknown. Myriad APPs are involved, all which exhibit different and sometimes opposing effects on immune function and/or tissue protection. Studies have, however, implied important roles for individual APPs following an intrapulmonary challenge. Serum amyloid P (SAP), a prominent short pentraxin APP in mice similar in structure and function to human C-reactive protein (CRP), promotes the deposition of complement component C3 on the surface of pneumococcus and is required for bacterial clearance and survival in pneumonic mice (Yuste et al. 2007). In these studies, defects in SAP-deficient mice could be rescued by administration of human SAP (Yuste et al. 2007). Similarly, human CRP administration is protective in mice infected with S. pneumoniae (Mold et al. 1981). LPS-binding protein (LBP) facilitates the binding of LPS with CD14, thereby enhancing inflammatory responses during infections. LBP levels are elevated in mice in response to bacterial stimuli, and its presence is required for maximal innate immunity and host defense (Gamble et al. 2008; Branger et al. 2004; Brass et al. 2004; Fan et al. 2002; Knapp et al. 2003). As the field progresses, major challenges for investigators include: (1) determining the influence of all APP changes in response to a given physiological stress, rather than to single APPs, which are rarely if ever regulated as a lone entity; and (2) discriminating between the functions of baseline APP concentrations and those produced outside of homeostatic conditions. Most APPs are present in abundant concentrations in healthy subjects, and APP deletion may reveal distinct functions for APPs compared with eliminating acute phase changes in APP concentrations. Identifying and manipulating critical points of APP regulation will be necessary to gain true insight regarding the function and significance of the APR during lung innate immune responses. To date, research in this field suggests that NF-κB, STAT3, and the cytokines promoting their activity represent the foundation of a lung-liver axis and an important circuit through which the hepatocytes respond to infectious or other stimuli in the lung.
Roles of NF-κB and STAT3 in granulopoiesis
As potential pathogens subvert the resident defense mechanisms of the respiratory tract, recruitment of neutrophils becomes a critical innate immune component for preventing overwhelming infection. In animal models, the clearance of bacterial, viral, and fungal pathogens from the lung is markedly diminished in settings of reduced neutrophilic inflammation (Greenberger et al. 1996; Rehm et al. 1980; Tsai et al. 2000), and neutropenia or neutrophil dysfunction predisposes patients to opportunistic lung infections (Pennington 1986; Winkelstein et al. 2000). However, neutrophils are short-lived in the circulation (4–8 h), requiring a constant release of newly differentiated cells from the bone marrow to maintain normal baseline quantities and even greater demand in response to infections (Bicknell et al. 1994). So how does the supply meet the demand? During a pulmonary innate immune response, the bone marrow must be able to respond, directly or indirectly, to stimuli elaborated within the airspaces. Further, cells within the bone marrow must be appropriately tuned to respond to pathogens at the site of infection. Evidence is now available supporting the idea that signaling from NF-κB and STAT3 is central amongst the processes required for eliciting the extrapulmonary neutrophil response.
Granulocyte colony-stimulating factor (G-CSF) is a hematopoietic growth factor that strongly influences the differentiation, mobilization, and function of neutrophils (Demetri and Griffin 1991). The most well-recognized biological role of G-CSF is to control the maturation and release of granulocytes from the bone marrow under both normal and emergency conditions (Panopoulos and Watowich 2008; Lieschke et al. 1994; Liu et al. 1996). Indeed, the administration of recombinant G-CSF has long been used to re-establish blood neutrophil levels in neutropenic patients (Welte et al. 1996). Although signaling downstream of the G-CSF receptor involves numerous transcription factors, STAT3 activity has been shown to mediate its hematopoietic effects. Mutation of the STAT3-binding domain of the G-CSF receptor renders mice neutropenic, a phenotype that is ameliorated by overexpression of constitutively active STAT3 (McLemore et al. 2001). In addition, mice lacking functional STAT3 in myeloid progenitor cells have a reduced granulopoietic response following administration of G-CSF or Listeria monocytogenes (Panopoulos et al. 2006). STAT3-deficient neutrophils are also less chemotactic toward CXCR2 ligands, which are critical mediators of alveolar neutrophil recruitment during lung infections (Tsai et al. 2000), indicating that G-CSF is involved in both the quantity and quality of circulating neutrophils (Panopoulos et al. 2006).
G-CSF levels are highly elevated in the lungs and blood of both mice and humans in response to bacteria or bacterial products in the airspaces (O'Grady et al. 2001; Quinton et al. 2002). Indeed, the robust increase in circulating G-CSF content during an acute pulmonary inflammation stands somewhat in contrast to that of other cytokines, which are more compartmentalized within challenged airspaces (Boutten et al. 1996; Nelson et al. 1989). Following the administration of either recombinant G-CSF or E. coli into mouse lungs, circulating and bone-marrow neutrophil progenitors are increased in association with elevated bone-marrow STAT3 activity, suggesting that lung-derived G-CSF is sufficient to initiate STAT3-mediated granulopoiesis in the marrow (Shahbazian et al. 2004). During P. aeruginosa pneumonia, mice lacking the G-CSF receptor exhibit significant reductions in neutrophil numbers in the bone marrow, blood, and consequently air spaces of the lung (Gregory et al. 2007). This hematopoietic defect results in impaired bacterial clearance, worsened lung histopathology, and increased numbers of apoptotic neutrophils (which may reflect roles for STAT3 in cell survival), indicating that G-CSF signaling to STAT3 is critical for the innate immune response to lung infections (Gregory et al. 2007). G-CSF is a crucial means through which the lungs communicate with bone marrow and circulating neutrophils, allowing them to respond to localized infections in the lung.
NF-κB promotes the expression of numerous factors important for pulmonary innate immunity, including TNFα, IL-1, IL-6, and G-CSF (Quinton et al. 2007; Dunn et al. 1994; Pahl 1999), all of which, as described above, target extrapulmonary tissues in response to stimuli in the lung. In this capacity, NF-κB activity in the lung is a key initiator of extrapulmonary responses during an acute pulmonary inflammation. Recent evidence also identifies an additional extrapulmonary role for NF-κB tuning neutrophils in the bone marrow to be more effective at killing lung pathogens including S. pneumoniae and S. aureus (Clarke et al. 2010). The microbial flora of mice releases bacterial products that circulate through the blood and activate NF-κB in bone-marrow neutrophils. This NF-κB activation primes neutrophils and increases their ability to kill bacteria. In such a way, NF-κB signaling in the bone marrow might be essential to innate immune host defense in the lungs. The degree to which the NF-κB-mediated microbiome-to-bone-marrow circuit directly impacts pulmonary host defense and whether and when signals from the infected lung influence NF-κB activity and neutrophil priming in the marrow are compelling questions that remain to be addressed.
Roles of NF-κB and STAT3 in directing post-transcriptional regulation of lung innate immunity?
Whereas NF-κB and STAT3 are hubs of transcriptional regulation, innate immunity is also regulated at post-transcriptional levels (Anderson 2010; O'Connell et al. 2010). Roles of the major mediators of post-transcriptional regulation, miRNAs and ARE-BPs, in tuning innate immunity are complex and only beginning to be elucidated. Clearly, miRNAs and ARE-BPs regulate the expression of many extracellular and intracellular innate immunity mediators (Jing et al. 2005; Jones et al. 2009; Taylor et al. 1996), often to limit but also sometimes to enhance protein expression from targeted transcripts. The types of miRNA and ARE-BP change in response to receptor recognition of microbial products, and some of these changes alter innate immunity gene expression. Excitingly, many of these miRNAs and ARE-BPs are themselves under transcriptional regulation by NF-κB or STAT3. As but a few examples from the rapidly advancing field of miRNA regulation of innate immunity, TLR4 signaling can, in distinct settings, mediate NF-κB-dependent induction of mir-146 (which targets TRAF6 ad IRAK-1 to tune TLR signaling; Taganov et al. 2006), of mir-9 (which targets Nfkb1 to diminish NF-κB p50 and p105; Bazzoni et al. 2009), of mir-147 (which limits cytokine expression by as yet unknown means; Liu et al. 2009), of mir-21 (which targets PDCD4 to enhance pro-inflammatory cytokines; Sheedy et al. 2010), and of mir-155 (which targets SHIP1 to enhance pro-inflammatory kinase activity; McCoy et al. 2010). Similarly, the STAT3-mediated transcription of mir-21 can be essential to its anti-apoptotic function (Loffler et al. 2007), and the STAT3-mediated repression of mir-155 can be essential to the protective anti-inflammatory effects of IL-10 (McCoy et al. 2010). Although most of this review has focused on innate immune responses to pathogens in the lung, data about if, when, or how the regulation of miRNAs and/or ARE-BPs by NF-κB or STAT3 contributes to effective pulmonary host defense without excessive lung injury are virtually lacking. We suggest this is the crucial next frontier that needs to be crossed to further our understanding of innate immunity to pathogens in the lung.
Concluding remarks
NF-κB and STAT3 integrate inputs from diverse stimuli including microbial ligands and host-derived cytokines, and they mediate transcription levels for myriad genes including those for cytokines, chemokines, colony-stimulating factors, adhesion molecules, acute phase proteins, antimicrobial proteins, and anti-apoptotic proteins. As such, these signaling hubs coordinate and orchestrate innate immune responses to microbes in the lungs (Fig. 1). A current area of interest is the activity of these signaling hubs outside of the lung, as recent data suggest that extrapulmonary sites of transcriptional regulation contribute to innate immunity functions during acute lower respiratory infection. Finally, whereas NF-κB and STAT3 are the best recognized such hubs at present, other hubs are likely to emerge. Important future goals will to identify post-transcriptional hubs governing lung innate immunity and to determine how much these post-transcriptional hubs are themselves influenced by NF-κB and STAT3 activities during lung infection.
References
Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T (1994) Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77:63–71
Alcamo EA, Mizgerd JP, Horwitz BH, Bronson R, Beg AA, Scott M, Doerschuk CM, Hynes RO, Baltimore D (2001) Targeted mutation of tumor necrosis factor 1 rescues the RelA-deficient mouse and reveals a critical role for NF-kB in leukocyte recruitment. J Immunol 167:1592–1600
Almirall J, Bolibar I, Toran P, Pera G, Boquet X, Balanzo X, Sauca G (2004) Contribution of C-reactive protein to the diagnosis and assessment of severity of community-acquired pneumonia. Chest 125:1335–1342
Alonzi T, Maritano D, Gorgoni B, Rizzuto G, Libert C, Poli V (2001) Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol Cell Biol 21:1621–1632
Anderson P (2010) Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol 10:24–35
Andus T, Geiger T, Hirano T, Kishimoto T, Heinrich PC (1988) Action of recombinant human interleukin 6, interleukin 1 beta and tumor necrosis factor alpha on the mRNA induction of acute-phase proteins. Eur J Immunol 18:739–746
Armstrong GL, Conn LA, Pinner RW (1999) Trends in infectious disease mortality in the United States during the 20th century. JAMA 281:61–66
Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK (2008) IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 14:275–281
Awomoyi AA, Rallabhandi P, Pollin TI, Lorenz E, Sztein MB, Boukhvalova MS, Hemming VG, Blanco JC, Vogel SN (2007) Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J Immunol 179:3171–3177
Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M (2009) Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci USA 106:5282–5287
Beg AA, Sha WC, Bronson RT, Baltimore D (1995a) Constitutive NF-kB activation, enhanced granulopoiesis, and neonatal lethality in IkBa-deficient mice. Genes Dev 9:2736–2746
Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D (1995b) Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 376:167–170
Bernuth H von, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y, Vasconcelos J, Sirvent N, Guedes M, Vitor AB, Herrero-Mata MJ, Arostegui JI, Rodrigo C, Alsina L, Ruiz-Ortiz E, Juan M, Fortuny C, Yague J, Anton J, Pascal M, Chang HH, Janniere L, Rose Y, Garty BZ, Chapel H, Issekutz A, Marodi L, Rodriguez-Gallego C, Banchereau J, Abel L, Li X, Chaussabel D, Puel A, Casanova JL (2008) Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321:691–696
Betts JC, Cheshire JK, Akira S, Kishimoto T, Woo P (1993) The role of NF-kappa B and NF-IL6 transactivating factors in the synergistic activation of human serum amyloid A gene expression by interleukin-1 and interleukin-6. J Biol Chem 268:25624–25631
Bicknell S, Eeden S van, Hayashi S, Hards J, English D, Hogg JC (1994) A non-radioisotopic method for tracing neutrophils in vivo using 5′-bromo-2′-deoxyuridine. Am J Respir Cell Mol Biol 10:16–23
Bochud PY, Chien JW, Marr KA, Leisenring WM, Upton A, Janer M, Rodrigues SD, Li S, Hansen JA, Zhao LP, Aderem A, Boeckh M (2008) Toll-like receptor 4 polymorphisms and aspergillosis in stem-cell transplantation. N Engl J Med 359:1766–1777
Bousfiha A, Picard C, Boisson-Dupuis S, Zhang SY, Bustamante J, Puel A, Jouanguy E, Ailal F, El-Baghdadi J, Abel L, Casanova JL (2010) Primary immunodeficiencies of protective immunity to primary infections. Clin Immunol 135:204–209
Boutten A, Dehoux MS, Seta N, Ostinelli J, Venembre P, Crestani B, Dombret MC, Durand G, Aubier M (1996) Compartmentalized IL-8 and elastase release within the human lung in unilateral pneumonia. Am J Respir Crit Care Med 153:336–342
Branger J, Florquin S, Knapp S, Leemans JC, Pater JM, Speelman P, Golenbock DT, Poll T van der (2004) LPS-binding protein-deficient mice have an impaired defense against Gram-negative but not Gram-positive pneumonia. Int Immunol 16:1605–1611
Brass DM, Savov JD, Whitehead GS, Maxwell AB, Schwartz DA (2004) LPS binding protein is important in the airway response to inhaled endotoxin. J Allergy Clin Immunol 114:586–592
Carvalho A, Pasqualotto AC, Pitzurra L, Romani L, Denning DW, Rodrigues F (2008) Polymorphisms in toll-like receptor genes and susceptibility to pulmonary aspergillosis. J Infect Dis 197:618–621
Cheng JD, Ryseck RP, Attar RM, Dambach D, Bravo R (1998) Functional redundancy of the nuclear factor kB inhibitors IkB-a and IkB-b. J Exp Med 188:1055–1062
Cheng DS, Han W, Chen SM, Sherrill TP, Chont M, Park GY, Sheller JR, Polosukhin VV, Christman JW, Yull FE, Blackwell TS (2007) Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J Immunol 178:6504–6513
Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN (2010) Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16:228–231
Demetri GD, Griffin JD (1991) Granulocyte colony-stimulating factor and its receptor. Blood 78:2791–2808
Dunn SM, Coles LS, Lang RK, Gerondakis S, Vadas MA, Shannon MF (1994) Requirement for nuclear factor (NF)-kappa B p65 and NF-interleukin-6 binding elements in the tumor necrosis factor response region of the granulocyte colony-stimulating factor promoter. Blood 83:2469–2479
Fan MH, Klein RD, Steinstraesser L, Merry AC, Nemzek JA, Remick DG, Wang SC, Su GL (2002) An essential role for lipopolysaccharide-binding protein in pulmonary innate immune responses. Shock 18:248–254
Fong CH, Bebien M, Didierlaurent A, Nebauer R, Hussell T, Broide D, Karin M, Lawrence T (2008) An antiinflammatory role for IKKbeta through the inhibition of "classical" macrophage activation. J Exp Med 205:1269–1276
Freeman AF, Holland SM (2009) Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes. Pediatr Res 65:32R–37R
Freeman AF, Kleiner DE, Nadiminti H, Davis J, Quezado M, Anderson V, Puck JM, Holland SM (2007) Causes of death in hyper-IgE syndrome. J Allergy Clin Immunol 119:1234–1240
Gabay C, Kushner I (1999) Acute-phase proteins and other systemic responses to inflammation. N Engl J Med 340:448–454
Gadjeva M, Tomczak MF, Zhang M, Wang YY, Dull K, Rogers AB, Erdman SE, Fox JG, Carroll M, Horwitz BH (2004) A role for NF-kappa B subunits p50 and p65 in the inhibition of lipopolysaccharide-induced shock. J Immunol 173:5786–5793
Gamble L, Bagby GJ, Quinton LJ, Happel KI, Mizgerd JP, Zhang P, Nelson S (2008) The systemic and pulmonary lipopolysaccharide binding protein response to intratracheal lipopolysaccharide. Shock 31:212–217
Gao H, Guo RF, Speyer CL, Reuben J, Neff TA, Hoesel LM, Riedemann NC, McClintock SD, Sarma JV, Van Rooijen N, Zetoune FS, Ward PA (2004) Stat3 activation in acute lung injury. J Immunol 172:7703–7712
Geisler F, Algul H, Paxian S, Schmid RM (2007) Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology 132:2489–2503
Gennery AR, Flood TJ, Abinun M, Cant AJ (2000) Bone marrow transplantation does not correct the hyper IgE syndrome. Bone Marrow Transplant 25:1303–1305
Genovese MC, Cohen S, Moreland L, Lium D, Robbins S, Newmark R, Bekker P (2004) Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum 50:1412–1419
Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE (2009) Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324:1713–1716
Greenberger MJ, Strieter RM, Kunkel SL, Danforth JM, Laichalk LL, McGillicuddy DC, Standiford TJ (1996) Neutralization of macrophage inflammatory protein-2 attenuates neutrophil recruitment and bacterial clearance in murine Klebsiella pneumonia. J Infect Dis 173:159–165
Gregory AD, Hogue LA, Ferkol TW, Link DC (2007) Regulation of systemic and local neutrophil responses by G-CSF during pulmonary Pseudomonas aeruginosa infection. Blood 109:3235–3243
Hagihara K, Nishikawa T, Sugamata Y, Song J, Isobe T, Taga T, Yoshizaki K (2005) Essential role of STAT3 in cytokine-driven NF-kappaB-mediated serum amyloid A gene expression. Genes Cells 10:1051–1063
Hajjar AM, Harowicz H, Liggitt HD, Fink PJ, Wilson CB, Skerrett SJ (2005) An essential role for non-bone marrow-derived cells in control of Pseudomonas aeruginosa pneumonia. Am J Respir Cell Mol Biol 33:470–475
Han W, Joo M, Everhart MB, Christman JW, Yull FE, Blackwell TS (2009) Myeloid cells control termination of lung inflammation through the NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol 296:L320–L327
Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, Odden AR, Shellito JE, Bagby GJ, Nelson S, Kolls JK (2005) Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med 202:761–769
Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardoll DM, Drake CG (2007) Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol 179:4313–4317
Hayden MS, Ghosh S (2008) Shared principles in NF-kappaB signaling. Cell 132:344–362
Hess C, Herr C, Beisswenger C, Zakharkina T, Schmid RM, Bals R (2010) Myeloid RelA regulates pulmonary host defense networks. Eur Respir J 35:343–352
Hokuto I, Ikegami M, Yoshida M, Takeda K, Akira S, Perl AK, Hull WM, Wert SE, Whitsett JA (2004) Stat-3 is required for pulmonary homeostasis during hyperoxia. J Clin Invest 113:28–37
Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B (2007) STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357:1608–1619
Hollingsworth JW, Chen BJ, Brass DM, Berman K, Gunn MD, Cook DN, Schwartz DA (2005) The critical role of hematopoietic cells in lipopolysaccharide-induced airway inflammation. Am J Respir Crit Care Med 171:806–813
Ikegami M, Falcone A, Whitsett JA (2008) STAT-3 regulates surfactant phospholipid homeostasis in normal lung and during endotoxin-mediated lung injury. J Appl Physiol 104:1753–1760
Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J (2005) Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 120:623–634
Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP (2005) Lung NF-{kappa}B activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J Immunol 175:7530–7535
Jones MR, Quinton LJ, Simms BT, Lupa MM, Kogan MS, Mizgerd JP (2006) Roles of interleukin-6 in activation of STAT proteins and recruitment of neutrophils during Escherichia coli pneumonia. J Infect Dis 193:360–369
Jones MR, Quinton LJ, Blahna MT, Neilson JR, Fu S, Ivanov AR, Wolf DA, Mizgerd JP (2009) Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat Cell Biol 11:1157–1163
Kida H, Mucenski ML, Thitoff AR, Le Cras TD, Park KS, Ikegami M, Muller W, Whitsett JA (2008) GP130-STAT3 regulates epithelial cell migration and is required for repair of the bronchiolar epithelium. Am J Pathol 172:1542–1554
Knapp S, Vos AF de, Florquin S, Golenbock DT, Poll T van der (2003) Lipopolysaccharide binding protein is an essential component of the innate immune response to Escherichia coli peritonitis in mice. Infect Immun 71:6747–6753
Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kohler G (1994) Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368:339–342
Ku CL, Bernuth H von, Picard C, Zhang SY, Chang HH, Yang K, Chrabieh M, Issekutz AC, Cunningham CK, Gallin J, Holland SM, Roifman C, Ehl S, Smart J, Tang M, Barrat FJ, Levy O, McDonald D, Day-Good NK, Miller R, Takada H, Hara T, Al-Hajjar S, Al-Ghonaium A, Speert D, Sanlaville D, Li X, Geissmann F, Vivier E, Marodi L, Garty BZ, Chapel H, Rodriguez-Gallego C, Bossuyt X, Abel L, Puel A, Casanova JL (2007) Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med 204:2407–2422
Lang JE, Williams ES, Mizgerd JP, Shore SA (2008) Effect of obesity on pulmonary inflammation induced by acute ozone exposure: role of interleukin-6. Am J Physiol Lung Cell Mol Physiol 294:L1013–L1020
Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR (1994) Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 84:1737–1746
Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC (1996) Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity 5:491–501
Liu G, Friggeri A, Yang Y, Park YJ, Tsuruta Y, Abraham E (2009) miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci USA 106:15819–15824
Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, Cvijic H, Ullmann AK, Stadler PF, Horn F (2007) Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 110:1330–1333
Luedde T, Assmus U, Wustefeld T, Meyer zu Vilsendorf A, Roskams T, Schmidt-Supprian M, Rajewsky K, Brenner DA, Manns MP, Pasparakis M, Trautwein C (2005) Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. J Clin Invest 115:849–859
Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC (2008) Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med 205:1551–1557
Ma'ayan A (2009) Insights into the organization of biochemical regulatory networks using graph theory analyses. J Biol Chem 284:5451–5455
Mancuso P, Gottschalk A, Phare SM, Peters-Golden M, Lukacs NW, Huffnagle GB (2002) Leptin-deficient mice exhibit impaired host defense in Gram-negative pneumonia. J Immunol 168:4018–4024
Mancuso P, Huffnagle GB, Olszewski MA, Phipps J, Peters-Golden M (2006) Leptin corrects host defense defects after acute starvation in murine pneumococcal pneumonia. Am J Respir Crit Care Med 173:212–218
Matsuzaki Y, Xu Y, Ikegami M, Besnard V, Park KS, Hull WM, Wert SE, Whitsett JA (2006) Stat3 is required for cytoprotection of the respiratory epithelium during adenoviral infection. J Immunol 177:527–537
McCoy CE, Sheedy FJ, Qualls JE, Doyle SL, Quinn SR, Murray PJ, O'Neill LA (2010) IL-10 inhibits miR-155 induction by toll-like receptors. J Biol Chem 285:20492–20498
McLemore ML, Grewal S, Liu F, Archambault A, Poursine-Laurent J, Haug J, Link DC (2001) STAT-3 activation is required for normal G-CSF-dependent proliferation and granulocytic differentiation. Immunity 14:193–204
Meek RL, Benditt EP (1986) Amyloid A gene family expression in different mouse tissues. J Exp Med 164:2006–2017
Mills KH (2008) Induction, function and regulation of IL-17-producing T cells. Eur J Immunol 38:2636–2649
Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O'Shea J, Holland SM, Paul WE, Douek DC (2008) Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452:773–776
Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H (2007) Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448:1058–1062
Mizgerd JP (2006) Lung infection—a public health priority. PLoS Med 3:e76
Mizgerd JP (2008) Acute lower respiratory tract infection. N Engl J Med 358:716–727
Mizgerd JP, Spieker MR, Doerschuk CM (2001) Early response cytokines and innate immunity: essential roles for TNF receptor 1 and type I IL-1 receptor during Escherichia coli pneumonia in mice. J Immunol 166:4042–4048
Mizgerd JP, Scott ML, Spieker MR, Doerschuk CM (2002) Functions of IkB proteins in inflammatory responses to E. coli LPS in mouse lungs. Am J Respir Cell Mol Biol 27:575–582
Mizgerd JP, Lupa MM, Kogan MS, Warren HB, Kobzik L, Topulos GP (2003) Nuclear factor-kB p50 limits inflammation and prevents lung injury during Escherichia coli pneumonia. Am J Respir Crit Care Med 168:810–817
Mizgerd JP, Lupa MM, Spieker MS (2004a) NF-kappaB p50 facilitates neutrophil accumulation during LPS-induced pulmonary inflammation. BMC Immunol 5:10
Mizgerd JP, Lupa MM, Hjoberg J, Vallone JC, Warren HB, Butler JP, Silverman ES (2004b) Roles for early response cytokines during Escherichia coli pneumonia revealed by mice with combined deficiencies of all signaling receptors for TNF and IL-1. Am J Physiol Lung Cell Mol Physiol 286:L1302–L1310
Mold C, Nakayama S, Holzer TJ, Gewurz H, Du Clos TW (1981) C-reactive protein is protective against Streptococcus pneumoniae infection in mice. J Exp Med 154:1703–1708
Nelson S, Bagby GJ, Bainton BG, Wilson LA, Thompson JJ, Summer WR (1989) Compartmentalization of intraalveolar and systemic lipopolysaccharide-induced tumor necrosis factor and the pulmonary inflammatory response. J Infect Dis 159:189–194
O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D (2010) Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 10:111–122
O'Grady NP, Preas HL, Pugin J, Fiuza C, Tropea M, Reda D, Banks SM, Suffredini AF (2001) Local inflammatory responses following bronchial endotoxin instillation in humans. Am J Respir Crit Care Med 163:1591–1598
Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18:6853–6866
Panopoulos AD, Watowich SS (2008) Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and “emergency” hematopoiesis. Cytokine 42:277–288
Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, Watowich SS (2006) STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood 108:3682–3690
Patel DN, King CA, Bailey SR, Holt JW, Venkatachalam K, Agrawal A, Valente AJ, Chandrasekar B (2007) Interleukin-17 stimulates C-reactive protein expression in hepatocytes and smooth muscle cells via p38 MAPK and ERK1/2-dependent NF-kappaB and C/EBPbeta activation. J Biol Chem 282:27229–27238
Pennington JE (1986) Gram-negative bacterial pneumonia in the immunocompromised host. Semin Respir Infect 1:145–150
Poll T van der, Keogh CV, Guirao X, Buurman WA, Kopf M, Lowry SF (1997) Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis 176:439–444
Poynter ME, Irvin CG, Janssen-Heininger YM (2003) A prominent role for airway epithelial NF-kappa B activation in lipopolysaccharide-induced airway inflammation. J Immunol 170:6257–6265
Quinton LJ, Nelson S, Boe DM, Zhang P, Zhong Q, Kolls JK, Bagby GJ (2002) The granulocyte colony-stimulating factor response after intrapulmonary and systemic bacterial challenges. J Infect Dis 185:1476–1482
Quinton LJ, Jones MR, Simms BT, Kogan MS, Robson BE, Skerrett SJ, Mizgerd JP (2007) Functions and regulation of NF-kappaB RelA during pneumococcal pneumonia. J Immunol 178:1896–1903
Quinton LJ, Jones MR, Robson BE, Simms BT, Whitsett JA, Mizgerd JP (2008) Alveolar epithelial STAT3, IL-6 family cytokines, and host defense during Escherichia coli pneumonia. Am J Respir Cell Mol Biol 38:699–706
Quinton LJ, Jones MR, Robson BE, Mizgerd JP (2009) Mechanisms of the hepatic acute-phase response during bacterial pneumonia. Infect Immun 77:2417–2426
Rehm SR, Gross GN, Pierce AK (1980) Early bacterial clearance from murine lungs. Species-dependent phagocyte response. J Clin Invest 66:194–199
Ruminy P, Gangneux C, Claeyssens S, Scotte M, Daveau M, Salier JP (2001) Gene transcription in hepatocytes during the acute phase of a systemic inflammation: from transcription factors to target genes. Inflamm Res 50:383–390
Sakamori R, Takehara T, Ohnishi C, Tatsumi T, Ohkawa K, Takeda K, Akira S, Hayashi N (2007) Signal transducer and activator of transcription 3 signaling within hepatocytes attenuates systemic inflammatory response and lethality in septic mice. Hepatology 46:1564–1573
Schindler C, Levy DE, Decker T (2007) JAK-STAT signaling: from interferons to cytokines. J Biol Chem 282:20059–20063
Severgnini M, Takahashi S, Rozo LM, Homer RJ, Kuhn C, Jhung JW, Perides G, Steer M, Hassoun PM, Fanburg BL, Cochran BH, Simon AR (2004) Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol 286:L1282–L1292
Sha WC, Liou H, Tuomanen EI, Baltimore D (1995) Targeted disruption of the p50 subunit of NF-kB leads to multifocal defects in immune responses. Cell 80:321–330
Shahbazian LM, Quinton LJ, Bagby GJ, Nelson S, Wang G, Zhang P (2004) Escherichia coli pneumonia enhances granulopoiesis and the mobilization of myeloid progenitor cells into the systemic circulation. Crit Care Med 32:1740–1746
Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O'Leary JJ, Ruan Q, Johnson DS, Chen Y, O'Neill LA (2010) Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol 11:141–147
Shimada K, Chen S, Dempsey PW, Sorrentino R, Alsabeh R, Slepenkin AV, Peterson E, Doherty TM, Underhill D, Crother TR, Arditi M (2009) The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog 5:e1000379
Skerrett SJ, Liggitt HD, Hajjar AM, Wilson CB (2004a) Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol 172:3377–3381
Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB (2004b) Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol 287:L143–L152
Smith RP, Lipworth BJ, Cree IA, Spiers EM, Winter JH (1995) C-reactive protein. A clinical marker in community-acquired pneumonia. Chest 108:1288–1291
Sun J, Madan R, Karp CL, Braciale TJ (2009a) Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med 15:277–284
Sun L, Guo RF, Newstead MW, Standiford TJ, Macariola DR, Shanley TP (2009b) Effect of IL-10 on neutrophil recruitment and survival after Pseudomonas aeruginosa challenge. Am J Respir Cell Mol Biol 41:76–84
Taganov KD, Boldin MP, Chang KJ, Baltimore D (2006) NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA 103:12481–12486
Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S (1997) Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA 94:3801–3804
Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S (1998) Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol 161:4652–4660
Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S (1999) Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 10:39–49
Tal G, Mandelberg A, Dalal I, Cesar K, Somekh E, Tal A, Oron A, Itskovich S, Ballin A, Houri S, Beigelman A, Lider O, Rechavi G, Amariglio N (2004) Association between common Toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J Infect Dis 189:2057–2063
Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ (1996) A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4:445–454
Thorn CF, Lu ZY, Whitehead AS (2004) Regulation of the human acute phase serum amyloid A genes by tumour necrosis factor-alpha, interleukin-6 and glucocorticoids in hepatic and epithelial cell lines. Scand J Immunol 59:152–158
Tillett WS, Francis T (1930) Serological reactions in pneumonia with non-protein somatic fraction of pneumococcus. J Exp Med 52:561–571
Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ (2000) CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun 68:4289–4296
Uskokovic A, Dinic S, Mihailovic M, Grigorov I, Ivanovic-Matic S, Bogojevic D, Grdovic N, Arambasic J, Vidakovic M, Martinovic V, Petrovic M, Poznanovic G (2007) STAT3/NFkappaB interplay in the regulation of alpha2-macroglobulin gene expression during rat liver development and the acute phase response. IUBMB Life 59:170–178
Vernooy JH, Reynaert N, Wolfs TG, Cloots RH, Haegens A, Vries B de, Dentener MA, Buurman WA, Wouters EM (2005) Rapid pulmonary expression of acute-phase reactants after local lipopolysaccharide exposure in mice is followed by an interleukin-6 mediated systemic acute-phase response. Exp Lung Res 31:855–871
Wang E, Bergeron Y, Bergeron MG (2005) Ceftriaxone pharmacokinetics in interleukin-10-treated murine pneumococcal pneumonia. J Antimicrob Chemother 55:721–726
Ware LB, Matthay MA (2000) The acute respiratory distress syndrome. N Engl J Med 342:1334–1349
Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC (2009) Function of mitochondrial Stat3 in cellular respiration. Science 323:793–797
Welte K, Gabrilove J, Bronchud MH, Platzer E, Morstyn G (1996) Filgrastim (r-metHuG-CSF): the first 10 years. Blood 88:1907–1929
Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H (2000) Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 79:155–169
Wolk K, Witte E, Witte K, Warszawska K, Sabat R (2010) Biology of interleukin-22. Semin Immunopathol 32:17–31
Wurfel MM, Gordon AC, Holden TD, Radella F, Strout J, Kajikawa O, Ruzinski JT, Rona G, Black RA, Stratton S, Jarvik GP, Hajjar AM, Nickerson DA, Rieder M, Sevransky J, Maloney JP, Moss M, Martin G, Shanholtz C, Garcia JG, Gao L, Brower R, Barnes KC, Walley KR, Russell JA, Martin TR (2008) Toll-like receptor 1 polymorphisms affect innate immune responses and outcomes in sepsis. Am J Respir Crit Care Med 178:710–720
Yano K, Liaw PC, Mullington JM, Shih SC, Okada H, Bodyak N, Kang PM, Toltl L, Belikoff B, Buras J, Simms BT, Mizgerd JP, Carmeliet P, Karumanchi SA, Aird WC (2006) Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J Exp Med 203:1447–1458
Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK (2001) Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194:519–527
Yip TT, Chan JW, Cho WC, Yip TT, Wang Z, Kwan TL, Law SC, Tsang DN, Chan JK, Lee KC, Cheng WW, Ma VW, Yip C, Lim CK, Ngan RK, Au JS, Chan A, Lim WW (2005) Protein chip array profiling analysis in patients with severe acute respiratory syndrome identified serum amyloid a protein as a biomarker potentially useful in monitoring the extent of pneumonia. Clin Chem 51:47–55
Yuste J, Botto M, Bottoms SE, Brown JS (2007) Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae. PLoS Pathog 3:1208–1219
Zahedi K, Whitehead AS (1993) Regulation of mouse serum amyloid P gene expression by cytokines in vitro. Biochim Biophys Acta 1176:162–168
Zhong Z, Wen Z, Darnell JE Jr (1994) Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264:95–98
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Quinton, L.J., Mizgerd, J.P. NF-κB and STAT3 signaling hubs for lung innate immunity. Cell Tissue Res 343, 153–165 (2011). https://doi.org/10.1007/s00441-010-1044-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-010-1044-y