
Abstract
Neurod1 is a crucial basic helix-loop-helix gene for most cerebellar granule cells and mediates the differentiation of these cells downstream of Atoh1-mediated proliferation of the precursors. In Neurod1 null mice, granule cells die throughout the posterior two thirds of the cerebellar cortex during development. However, Neurod1 is also necessary for pancreatic β-cell development, and therefore Neurod1 null mice are diabetic, which potentially influences cerebellar defects. Here, we report a new Neurod1 conditional knock-out mouse model created by using a Tg(Atoh1-cre) line to eliminate Neurod1 in the cerebellar granule cell precursors. Our data confirm and extend previous work on systemic Neurod1 null mice and show that, in the central lobules, granule cells can be eradicated in the absence of Neurod1. Granule cells in the anterior lobules are partially viable and depend on as yet unknown genes, but the Purkinje cells show defects not previously recognized. Interestingly, delayed and incomplete Tg(Atoh1-cre) upregulation occurs in the most posterior lobules; this leads to near normal expression of Neurod1 with a concomitant normal differentiation of granule cells, Purkinje cells, and unipolar brush cells in lobules IX and X. Our analysis suggests that Neurod1 negatively regulates Atoh1 to ensure a rapid transition from proliferative precursors to differentiating neurons. Our data have implications for research on medulloblastoma, one of the most frequent brain tumors of children, as the results suggest that targeted overexpression of Neurod1 under Atoh1 promoter control may initiate the differentiation of these tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The production of neurons requires the complex integration of extra- and intracellular factors to control the balance between proliferation, differentiation, migration, and survival. Since the cerebellum was first used by Ramón y Cajal to establish the neuron theory (Sotelo 2008), this regular cytology has provided insights into neuroanatomical and physiological problems. In recent decades, the cerebellum has evolved into a system for studying central nervous system (CNS) development at the molecular level, by combining morphogenetic movements, neuronal cytological maturation, and the establishment of neuronal layers (Hatten and Heintz 1995; Sgaier et al. 2005). This increased interest has in part been triggered by the derailment of proliferation regulation in medulloblastoma cases (Leung et al. 2004; Barisone et al. 2008; Schuller et al. 2008). Despite the complexity of the cerebellum at the level of the molecular coding of development, the cerebellar cortex is histologically uniform and divided into three distinct cellular layers in the adult: the molecular layer (ML), the Purkinje cell layer (PCL), and the granule cell layer (GCL; Mugnaini et al. 1997). The most superficial ML contains stellate and basket cell interneurons, dendrites of Purkinje cells (PCs), axons of granule cells (GCs; parallel fiber), and Bergmann glia processes (Voogd and Glickstein 1998). The next layer (PCL) comprises the somata of PCs and also the somata of the Bergmann glia (Voogd and Glickstein 1998). The innermost GCL lying above the white matter consists of the most numerous neuronal cell type of the brain, GCs, in the majority of the layer and the somata of Golgi cell and unipolar brush cells (UBCs) predominantly in the posterior lobules (lobules IX and X; Mugnaini et al. 1997).
The formation of the cerebellum spans embryonic and postnatal development, initiating at embryonic day 9 (E9) in the mouse (Hatten and Heintz 1995; Larramendi 1969; Fujita et al. 1966; Fujita 1969). Two primary regions are thought to give rise to the neurons that make up the cerebellum. The first region is the ventricular zone in the roof of the fourth ventricle, this region produces γ-aminobutyric acid (GABA)ergic neurons, including PCs, Golgi, basket, stellate cells, and small deep cerebellar nuclei neurons (Hatten and Heintz 1995; Hoshino et al. 2005). Precursors of the cerebellar GCs are generated in the second germinal zone, which is called the rhombic lip (Englund et al. 2006; Machold and Fishell 2005; Wang et al. 2005). They proliferate and migrate anteromedially from the lateral caudal portion to the outer pial surface of the developing cerebellum between E13 and E16 to form the external granular layer (EGL; Alder et al. 1996; Altman and Bayer 1978; Hatten et al. 1982). The cells of the outer layer of the EGL continue to proliferate after birth, when the postmitotic cells in the inner EGL begin to differentiate and migrate radially into the cerebellar cortex along the processes of the Bergmann glia cells to form the internal granular layer (IGL; Fujita et al. 1966; Hatten et al. 1982). Cerebellar GCs form up to 60% of the total CNS neurons in mammals and derailment of proliferation leads to the most common brain tumor in children, forming up to 30% of pediatric tumor cases (Barisone et al. 2008). Understanding the regulation and differentiation in the cerebellum is thus of significant clinical and basic scientific importance. We focus here on the molecular basis of the differentiation pathway of cerebellar GCs on the premise that the initiation of differentiation might combat uncontrolled proliferation as found in medulloblastomas.
Proteins of the basic helix-loop-helix (bHLH) class play a central role in the determination of neuronal lineages in the peripheral nervous system (PNS) and CNS (Bertrand et al. 2002; Kageyama et al. 2007). The bHLH protein Neurod1 is widely expressed in both CNS and PNS in vertebrates (Lee et al. 1995). Ectopic expression of Neurod1 in Xenopus neural ectoderm leads to neuronal differentiation, suggesting that Neurod1 regulates the formation of neurons from neural precursors (Lee et al. 1995). In the mouse CNS, a high level of Neurod1 expression is found in differentiating neurons and in mature neurons such as the GCs of the cerebellum, inferior colliculus, and hippocampus, and the neurons of the limbic system (Lee 1997; Gurung and Fritzsch 2004). In the PNS, Neurod1 expression is found in developing and mature sensory neurons (Lee et al. 1995; Kim et al. 2001). For example, during inner ear development, Neurod1 expression is first detected in the sensory neuroblast precursors as early as E8.75 (Ma et al. 1998). Consistently, Neurod1 null mutants have severe reduction of vestibular neurons and near complete loss of cochlear neurons in the ear (Kim et al. 2001).
Neurod1 expression in the cerebellum correlates with the differentiation of GCs (Dahmane and Ruiz-i-Altaba 1999; Goldowitz and Hamre 1998). Neurod1 is highly expressed in both EGL and IGL of the cerebellum between postnatal day 5 (P5) and P13, and the IGL expression persists at a stable level until adulthood (Lee et al. 2000; Miyata et al. 1999). Despite the near uniform histology throughout its lobules, the cerebellum has divergent expression pattern of certain genes in various lobules of the cerebellum. For example, the anterior lobules of the cerebellum are able to undergo some near normal development without Neurod1 (Miyata et al. 1999; Cho and Tsai 2006). In contrast, an absence of Neurod1 leads to a near total loss of GCs and disarrangement of PCs in the posterior lobules (Miyata et al. 1999; Cho and Tsai 2006). Atoh1, another bHLH gene, is expressed in cerebellar GC precursors (GCPs) at the rhombic lip and in the outer EGL in the developing cerebellum (Akazawa et al. 1995; Ben-Arie et al. 1996, 1997; Helms et al. 2000). Genomic disruption has established that Atoh1 is essential for the proper development of cerebellar GCs, as Atoh1 null mice lack the EGL (Ben-Arie et al. 1997; Helms et al. 2000) and other Atoh1-dependent brain areas (Wang et al. 2005; Fritzsch et al. 2006a). However, overexpression of Atoh1 disrupts neural differentiation in normal cerebellar development (Gazit et al. 2004; Helms et al. 2001; Isaka et al. 1999), implying an interaction (not as yet fully characterized) with Neurod1 in the regulation of proliferation and differentiation.
In the present study, we have used a Tg(Atoh1-cre) (Matei et al. 2005) to eliminate a loxP-flanked Neurod1 (Goebbels et al. 2005) in the developing cerebellum to sidestep the diabetic condition in Neurod1 null mice, thus avoiding the additional systemic effects of low or limited insulin production known to cause various neuropathies. Previous work has demonstrated that delayed elimination of floxed Neurod1 in mature cerebellar GCs does not cause any defects despite the continued expression of Neurod1 in those GCs (Goebbels et al. 2005). In contrast to this absence of any recognizable defects in near adult Neurod1 conditional null mice, we have found a gradient of Neurod1 loss and concomitant defects in cerebellar development in our conditional model that eliminates Neurod1 prior to cerebellar GC differentiation in most lobules. However, the posterior lobules (1/2VIII + IX + X) do not lose all of the Neurod1 expression and show near normal morphology. In agreement with the more recent insights into the complexity of transcription initiation (Buratowski 2008), we suspect that insufficient and delayed upregulation of Tg(Atoh1-cre) in the posterior lobules is incompatible with the excision of the floxed Neurod1. The central lobules (VI + VII + 1/2VIII) show complete absence of Neurod1 and massive loss of all GCs. In situ hybridization analysis indicates that Atoh1 acts in these lobules in a negative feedback loop with Neurod1. Atoh1 expands its expression in continuously proliferating precursor populations in the absence of Neurod1. These GCs eventually all die without ever migrating in a coordinated fashion into the IGL. We also find disorganization of PC distribution and orientation in these cerebellar lobules of Neurod1 conditional mutant mice. As expected based on previous work (Miyata et al. 1999; Cho and Tsai 2006), loss of Neurod1 in the anterior lobules is partially rescued by an as yet unknown transcription factor that compensates the need for Neurod1 in differentiating GCs. We confirm this previous finding and our more detailed analysis shows aberrations in the PC dendritic trees previously unrecognized in systemic Neurod1 null mice in these lobules.
Materials and methods
Mice and genotyping
Generation of mice variously genotyped as Neurod1 null (Miyata et al. 1999), floxed Neurod1 (Goebbels et al. 2005), and Tg(Atoh1-cre) with a ROSA26 reporter (Matei et al. 2005) was as described previously. Conditional Neurod1 null mice were generated by breeding homozygotic floxed Neurod1 mice (Neurod1 f/f) with Neurod1 f/+ ,Tg(Atoh1-cre) mice. The resulting Neurod1 f/f ,Tg(Atoh1-cre) mice were used as Neurod1 conditional mutant, and the Neurod1 f/+ ,Tg(Atoh1-cre) heterozygous siblings were used as controls. Tail biopsies were used for genomic DNA preparation, and polymerase chain reaction analysis was performed for genotyping. Animal procedures were performed in accordance with IACUC guidelines for use of laboratory animals in biological research.
X-gal staining
Heads of mice perfused with 4% paraformaldehyde were hemisected, and cerebella were dissected and then briefly washed with phosphate buffer. The samples were stained in a solution containing 0.1 M phosphate buffer, 1% deoxycholic acid, 2% NP40, 1 mM magnesium chloride, 0.5 M potassium ferricyanide, 0.5 M potassium ferrocyanide, and 2 mg/ml X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactoside) for 24 h at room temperature (Matei et al. 2006).
In situ hybridization
In situ hybridization was performed with the RNA probe labeled with digoxigenin. The plasmids containing the cDNAs were used to generate the RNA probe by in vitro transcription. The dissected cerebella were fixed in 0.4% paraformaldehyde and digested briefly with 10 µg/ml Proteinase K (Ambion, Austin, Tex., USA) for 15–20 min. The samples were then hybridized overnight at 60°C to the riboprobe in hybridization solution containing 50% formamide, 50% 2× saline sodium citrate, and 6% dextran sulfate. After the unbound probe had been washed off, the samples were incubated overnight with an anti-digoxigenin antibody (Roche Diagnostics, Mannheim, Germany) conjugated with alkaline phosphatase. After a series of washes, the samples were reacted with nitroblue phosphate/5-bromo, 4-chloro, 3-indolil phosphate (BM purple substrate, Roche Diagnostics, Germany), which is enzymatically converted to a purple-colored product. Reacted parts were mounted flat in glycerol and viewed in a Nikon Eclipse 800 microscope by using differential interference contrast microscopy. Images were captured with Image-Pro software.
Immunocytochemistry
For immunocytochemical staining, 100-µm sagittal sections of cerebellum were defatted in 70% ethanol overnight and blocked with 5% normal goat serum in PBS containing 0.1% Triton X-100 for 2 h. The primary antibodies for active caspase 3 (Cell Signaling Technology), calbindin (Sigma), and calretinin (Chemicon) were then used at dilutions of 1:100, 1:500, and 1:400, respectively and incubated on the sections for 48 h at 4˚C. After several washes with PBS, corresponding secondary antibodies (1:500; conjugated to Alexa dyes) were added and incubated on the sections overnight at 4°C. The sections were washed with PBS, counter-stained with Hoechst stain, and mounted in glycerol. Images were taken via a Leica TCS SP5 confocal microscope.
Lipophilic dye tracing
Lipophilic-dye-soaked filter strips (Fritzsch et al. 2005) were inserted into the cerebellar folia parasagittally to label parallel fibers. We also labeled PCs dendrites by shooting lipophilic dye bullets coated with tungsten particles into the 200-µm parasagittal cerebellar sections (Benediktsson et al. 2005).
Plastic embedding and Stevenel’s blue staining
Thick parasagittal sections of cerebellum were fixed in 2.5% glutaraldehyde overnight followed by several washes with 0.1 M phosphate buffer and then fixed with 1% osmium tetroxide for 1 h. Samples were then dehydrated in graded ethanol and propylene oxide, embedded with Epon 812 in beam capsules, and baked at 60°C for 48 h. Sections (2 µm) were cut by using an Ultratome and stained with Stevenel’s blue (del Cerro et al. 1980), composed of 2% potassium permanganate and 1.3% methylene blue, at 60°C.
Body weight and blood glucose measurement
Mice were weighed at the specified age, and tail vein blood was then used to determine the blood glucose concentration by the FreeStyle flash blood glucose monitoring system (Therasense).
Area measurement
We took three different sagittal sections of cerebellum of various ages to measure the area of total cerebellum and the area of lobule X and then calculated the percentage between them. We also measured the thickness of EGL in micrometers in three different areas and calculated the mean. We counted GC numbers in a 40-μm region along the thickness of the EGL and calculated the mean of three counts in three different areas. We used Image-Pro software to perform the calibrated measurements.
Results
Conditional Neurod1 knock-out mice
Previous studies have shown that Neurod1 can convert non-neuronal ectodermal cells into fully differentiated neurons. In addition, Neurod1 is expressed abundantly in mature neurons in adult brain structures, including the hippocampus and cerebellum, which suggests a secondary role of Neurod1 in fully differentiated neurons (Lee et al. 1995).
In this study, we analyzed the function of Neurod1 in postnatal murine cerebellum by using Neurod1 conditional knock-out mice [Neurod1 f/f ,Tg(Atoh1-cre),ROSA26]. We employed an Atoh1 promoter fragment to drive Cre recombinase. Cre-mediated excision of the floxed Neurod1 gene results in deletion of Neurod1 only in the region where Tg(Atoh1-cre) is expressed, which can be monitored by β-galactosidase staining resulting from the Cre-mediated activation of the ROSA26-lacZ reporter (Matei et al. 2005). Neurod1 heterozygous littermates [Neurod1 f/+ ,Tg(Atoh1-cre ROSA26] were used as controls. In contrast to the systemic Neurod1 null mice, which died within a few days after birth because of severe hyperglycemia, our Neurod1 conditional knock-out mice survived to adulthood. We measured their body weight and blood glucose and found no abnormality when compared with control Neurod1 heterozygous mice (Table 1). Therefore, we could successfully circumvent the effect of Neurod1 in pancreatic β-cell development in our conditional mouse model.
Effect of delayed Tg (Atoh1-cre) upregulation in cerebellum
Previous work has shown that Atoh1 is necessary for cerebellum formation (Wang et al. 2005; Bermingham et al. 2001). We first examined Tg(Atoh1-cre) expression in the cerebellum by analyzing the β-galactosidase distribution in both Neurod1 conditional knock-out and control heterozygous mice aged P2, P11, P15, and P21 and in adults over one year of age. At P2, Tg(Atoh1-cre) showed expression from lobules I through VIII. This expression had progressed to lobule IX by P11, to one third of lobule of X by P15, and to half of lobule X by the adult stage (Fig. 1a-e). The onset of Tg(Atoh1-cre) upregulation was delayed in the posterior lobules (IX + X), and this insufficient Tg(Atoh1-cre) upregulation apparently led to the failure of excision of floxed Neurod1 in these lobules, as demonstrated by in situ hybridization for Neurod1 (Fig. 1f’-j’). Indeed, Neurod1 expression remained unchanged at later stages, even after Tg(Atoh1-cre) started to be expressed in the posterior lobules, suggesting that Tg(Atoh1-cre) was effectively recombining at the embryonic expression levels, with the later increase shown by ROSA26-lacZ being ineffective.

Inadequate Tg(Atoh1-cre) upregulation revealed by ROSA26 (a-e, f-j) failed to excise Neurod1 mRNA (a’-e’, f’-j’) in lobule IX and X in Neurod1 mutant cerebellum revealed by in situ hybridization of Neurod1. a-e’ Neurod1 f/+, Tg(Atoh1-cre)[control]. f-j’ Neurod1 f/f, Tg(Atoh1-cre)[mutant]. a, f In P2 control and mutant cerebella, Tg(Atoh1-cre) was prominently expressed from lobule I to VIII, weakly expressed in lobule IX in heterozygous, but completely absent in lobule X. b, g Tg(Atoh1-cre) expression progressed further to lobule IX by P11 and was homogeneously expressed in heterozygous cerebellum but misplaced and condensed in EGL of central lobules in mutant cerebellum. c, h Tg(Atoh1-cre) was expressed uniformly in up to one third of lobule X by P15 in heterozygous cerebellum and dispersed in central lobules in mutant cerebellum. d, i, e, j Tg(Atoh1-cre) expression progressed to half of lobule X in P21 and in adult heterozygous cerebellum (d, e) and was lost in central lobules in mutants (i, j). a’-e’ Neurod1 was expressed uniformly in all the lobules in heterozygous cerebellum as shown by in situ hybridization of Neurod1. f’-j’ Expression of Neurod1 was absent in lobules I to half of VIII in mutant from P2 to adult when Tg(Atoh1-cre) upregulation was prominent. Despite the late upregulation in lobule IX and X, Tg(Atoh1-cre) failed to excise Neurod1 in lobules X, IX, and 1/2VIII in mutant cerebellum. The recombination of Neurod1 by using Tg(Atoh1-cre) must happen in embryonic stages and results in no apparent additional recombination after further upregulation of Tg(Atoh1-cre) as revealed by the expansion of ROSA26-lacZ. h’, i’ Boxed regions are shown at higher magnification in h’’, i’’, respectively. Bars 250 µm (a-j, a’-j’), 10 µm (h’’, i’’)
We have previously shown that, in developing cochlear nucleus and spiral neurons, Tg(Atoh1-cre) is expressed only days after Neurod1 is upregulated (Kim et al. 2001; Fritzsch et al. 2006a; Matei et al. 2005; Bulfone et al. 2000). Consistent with this, the floxed Neurod1 could not be excised by delayed Tg(Atoh1-cre) upregulation, and we detected normal expression of Neurod1 in these cochlear nuclei and neurons in mutant ears (Fig. 2).

Expression of Tg(Atoh1-cre) and Neurod1 in the cochlear nucleus and in inner ear sensory neurons with inadequate upregulation of Tg(Atoh1-cre) consistent with normal development of cochlear nucleus and inner ear in Neurod1 conditional null mice. a-a’ Atoh1 was strongly expressed in both dorsal and ventral cochlear nucleus in adults as shown by lacZ expression. b Neurod1 was also expressed in both cochlear nuclei more prominently in dorsal cochlear nucleus as revealed by lacZ. c However, the cochlear nucleus developed normally in the absence of Neurod1, as shown in Neurod1 conditional null mice. d-d’’ Tg(Atoh1-cre) expression in spiral ganglia revealed by ROSA26 was insufficient and delayed as shown in P7 Neurod1 conditional null mice. e-e’’ Inner ear sensory neurons were normal as shown by ROSA26 in mutant mice. Note that ROSA26 shows lacZ for all of past and present Atoh1 upregulation. In contrast, Atoh1-lacZ and Neurod1-lacZ show only actual expression of Atoh1 and Neurod1, respectively (DCN dorsal cochlear nucleus, AVCN antero-ventral cochlear nucleus, PVCN postero-ventral cochlear nucleus, Spgl spiral ganglion, OC organ of Corti, HC horizontal crista, AC anterior crista, Vgl vestibular ganglion). Bars 100 µm (a, a’, d-e’’), 250 µm (b, c)
Severe loss of GCs in central lobules
All GCs in the central lobules (1/2VI–1/2VIII) of the cerebellum were lost in adult Neurod1 conditional mutant mice (Figs. 1, 3e, f). However, this loss was not attributable to the failure of GC to proliferate. At P11, the EGL was near normal in thickness, but GCs dispersed and migrated radically in these lobules from the EGL to the IGL. All GCs degenerated with progression of time (Fig. 3a-f), and the majority of GCs had disappeared by P21 (Fig. 3d). We further evaluated the degeneration of GCs through analysis of pyknotic nuclei by Hoechst stain, by Stevenel’s stain in thin sections, and by activated caspase 3 immunocytochemistry. In the central lobules of mutant cerebellum, the degeneration of GCs was evident by the presence of many pyknotic nuclei in the EGL (Fig. 4b, d-d’’, Table 2, see also Changes of EGL over time). In addition, these pyknotic nuclear profiles co-localized with activated caspase 3 (Fig. 4d-d’’), clearly demonstrating that GCs underwent apoptosis in the absence of Neurod1. Although the anterior lobules were organized more orderly, many pyknotic changes were observed with activated caspase-3-positive nuclei in GCs (Fig. 4a, Table 2, see also Changes of EGL over time), consistent with the reduction of GCs reported in systemic Neurod1 null mice (Miyata et al. 1999).

Absence of Neurod1 led to massive loss of granule cells (GCs) in the central lobules of Neurod1 mutant cerebellum. a, b GCs shown by ROSA26 were abnormally shifted from the external granular layer (EGL) to the internal granular layer (IGL) in central lobules (half of VI-VIII) in P11 Neurod1 mutant cerebellum, whereas GCs were almost normally organized in EGL and IGL in other lobules (boxed area in a shown at higher magnification in b). c By P15, GCs were dispersed and scattered from EGL to IGL in central lobules (asterisks). d At P21, the majority of GCs were lost in lobule VII and half of lobule VIII (asterisks). e, f Almost all the GCs were lost in central lobules in adult (asterisks; GCL granule cell layer; boxed area in e shown at higher magnification in f). Bars 100 µm

Granule cells (GCs) degenerate via apoptotic cell death in the absence of Neurod1, as demonstrated by activated caspase 3 staining in P11 mice. a GC losses were partial in the anterior lobules of Neurod1 mutant cerebellum with few activated caspase-3-positive cells, as shown by caspase 3 antibody staining. b, d-d’’ Severe degeneration of GCs occurred in the central lobules of Neurod1 mutant cerebellum analyzed by appearance of pyknotic nuclei shown with Hoechst stain. Such apoptotic cells showed co-localization (arrows) with the activated caspase 3 (d’’). c Occasional caspase-3-positive cells were present in the Neurod1 heterozygous cerebellum. Bars 100 µm (a-c), 10 µm (d-d’’)
The GC depletion in central lobules was also seen in Neurod1 f/Z, Tg(Atoh1-cre) mice, which carry a Neurod1 null allele that contains the lacZ reporter gene in place of the Neurod1 coding region at the Neurod1 locus. The X-gal staining reflected the endogenous Neurod1 expression pattern and showed a near total loss of GCs in the central lobules in the adult mutant cerebellum (Fig. 5b-b’’). In Neurod1 heterozygous mice, the cerebellum developed uniformly in all lobules (Fig. 5a, a’), indicating the lack of dosage effect of Neurod1 on GC differentiation.

Loss of granule cells in the central lobules of adult Neurod1 f/z, Tg(Atoh1-cre) mice observed by Neurod1-lacZ expression. a, a’ Neurod1-lacZ was expressed uniformly in all the lobules in heterozygous [Neurod1 +/Z, Tg(Atoh1-cre)] cerebellum. b-b’’ Granule cell loss demonstrated by the absence of Neurod1-lacZ expression in the central lobules in Neurod1 f/Z mice, which carry Neurod1 null allele containing the lacZ reporter gene in place of the Neurod1 coding region (GCL granule cell layer). Note that the lacZ expression reflects endogenous Neurod1 expression. Almost all the granule cells were lost in lobule VII (asterisk in b’’). Bars 10 µm (a-b’), 50 µm (b’’)
Presence of Neurod1 rescued lobules IX and X in mutant cerebellum
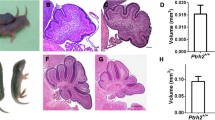
In contrast to the severe loss of GCs in the central lobules, lobules IX and X in mutant cerebellum had near normal cytoarchitecture, and lobule X was the closest to wildtype size among all the lobules. Since the total area of the cerebellum was reduced to about half in the mutant, the relative area of lobule X was nearly doubled in mutant compared with the heterozygous control (Fig. 6). The GCs in the EGL and IGL were normally arranged in lobules IX and X and in up to half of the lobule VIII in mutant mice; this was consistent with the residual expression of Neurod1, as shown by in situ hybridization in these lobules (Fig. 7a). Massive disorganization of GCs was observed from the point at which the Neurod1 gene was deleted in the mutant (Fig. 7a, c). Moreover, no distinct demarcation was seen between the EGL, ML, PCL, and IGL in central lobules in the mutant (Fig. 7b), although in lobules IX and X, the organization of these layers was almost normal compared with that in control cerebellum (Fig. 7d). This demonstrates that the presence of Neurod1 can save the development of one lobule in the same cerebellum in which the others are affected severely by the absence of Neurod1. This internal control allows us to correlate directly the histological differences in the various lobules with the conditional deletion of Neurod1 and the requirement of GCs for Neurod1 for their differentiation.

Normal growth of lobule-X in Neurod1 mutant cerebellum in comparison with other lobules. a Area of near mid-sagittal sections was measured by using Image-Pro in P7 and P21 in Neurod1 heterozygous and mutant cerebellum. The area in the mutant was comparable with in heterozygotes at P7 but drastically reduced by P21. b Area measurement of lobule X showed higher growth in mutant than heterozygous cerebellum at P7, but at P21, the growth reduced slightly in mutant compared with heterozygous cerebellum. c Percentage of growth of lobule X compared with that of total cerebellum was greatly increased in the mutant and was almost double in mutant compared with heterozygous P21 cerebellum. The presence of Neurod1 in lobule IX and X rescued normal growth, whereas absence of Neurod1 caused a massive size reduction in the other lobules of the mutant cerebellum. Bar 250 µm

Presence of Neurod1 rescued the posterior lobules in Neurod1 conditional mutant cerebellum, as shown in P15 mice. a Retention of Neurod1 in the posterior lobules (1/2VIII-X) of Neurod1 conditional null mice, as shown by in situ hybridization of Neurod1. b EGL was thicker in central lobules compared with that of lobules IX and X, as shown by Hoechst stain (white arrows). The boxed area in b is shown at higher magnification in c (EGL external granular layer, ML molecular layer, IGL internal granular layer). c In mutants, migration of GCs was dispersed and scattered from EGL to IGL in central lobules starting from the point (red arrow in c) in lobule VIII from which the Neurod1 gene was excised (red arrow in a). The layers of cerebellum were severely disorganized in central lobules in the mutant (b), whereas in posterior lobules of same cerebellum (b), they were arranged in a pattern similar to heterozygous cerebellum (d). Bars 50 µm
Neurod1 mutation influences survival of GCs but not UBCs
Consistent with the degree of activated caspase-3-positive GC nuclei in neonate (Fig. 4), we have seen reduced survival to massive loss of GCs in anterior to central lobules, respectively, in adult mutant cerebellum (Fig. 8d) analyzed by Hoechst stain. The density of GCs only in the posterior lobules of the mutant (Fig. 8d’) were comparable with heterozygous mice (Fig. 8a, a’), whereas the loss of GCs in other lobules was only clearly noticeable.

Absence of Neurod1 caused reduction of GCs in the anterior lobules but normal organization of UBCs in Neurod1 conditional null cerebellum, as shown by Hoechst and calretinin staining, respectively, in adult mice. a, a’ Normal organization and equal distribution of GCs along all the lobules in heterozygous cerebellum, as shown by Hoechst stain. d, d’ Density of GCs in lobules 1/2VIII-X was comparable with heterozygous cerebellum, whereas there was partial loss of GCs in the anterior lobules (I-1/2VI) and severe loss in central lobules (1/2VI-1/2VIII). b-c’ Abundant UBCs were found only in lobules X and half of IX in heterozygous cerebellum shown by calretinin staining (red). e-f’ UBCs were normally distributed in lobules X and half of IX in mutant cerebellum with no defect attributable to retention of Neurod1 in these lobules with rescued normal morphology. Note that the mutant cerebellum section lies near the hemispheres resulting in a smaller size of all lobules (boxed areas in a-f shown at higher magnification in a’–f’). Bars 100 µm
We also analyzed the morphology of UBCs in Neurod1 mutant cerebellum by immunocytochemistry with calretinin, a specific marker for UBCs. The distribution of UBCs was mainly restricted to lobules X and half of IX (Fig. 8b-c’), consistent with a previous report (Dino et al. 1999). The morphology and distribution of UBCs has not changed in the mutant cerebellum because of the retention of Neurod1 in the posterior lobules, which rescued UBCs (Fig. 8e-f’) together with GCs and PCs. The presence of a normal posterior lobule provided intrinsic controls for sagittal slice results, and thus, this mouse model allowed us directly to assess the postnatal functions of the GCs in the cerebellar cortex in the same preparation.
Changes of EGL over time
We next measured the thickness of the EGL and semi-quantified the number of GCs in the EGL of various ages and in various lobules (Table 2). In the heterozygous cerebellum, the thickness of the EGL was reduced linearly with age as development proceeded, and as GCs migrated to the IGL (Fig. 9a-a’’’). However, in Neurod1 conditional mutant mice, both the thickness of the EGL and the number of GCs in lobule VII were increased in P11 compared with P7 (Fig. 9c-c’), whereas in the posterior half of lobule VIII or in lobules IX or X (Fig. 9b-b’’’), they were comparable with the heterozygous cerebellum (Table 2). At all ages, the EGL, ML, PCL, and IGL were distinctly developed and could be distinguished clearly in the heterozygous cerebellum (Fig. 9a-a’’’). In the conditional Neurod1 mutant cerebellum, all layers of the cerebellum were severely disrupted in the central lobules. In lobule VII of the mutant cerebellum, there was no ML, as all the GCs were scattered and dispersed in and out of the EGL (Fig. 9c-c’’’). Pyknotic nuclei were most apparent in lobules VI-VIII (Fig. 9c-c’’) but also found in lobules I-V (Fig.9d-d’). In the posterior half of lobule VIII, in which Neurod1 gene was retained, the layers were less disorganized than in the rest of the central lobules.

Changes in the different layers in the different lobules over time, as shown by Stevenel’s staining. a-a’’’ All the layers of the cerebellum were distinct and well organized in control mice, as shown in lobule IX in which the thickness (blue arrow) of the external granular layer (EGL), and numbers of GC were reduced linearly with progression of age (ML molecular layer, PCL Purkinje cell layer, IGL internal granular layer). b-b’’’ In lobule X of mutant cerebellum, all the layers were organized normally at all ages. c-c’’’ In central lobules shown in lobule VII of Neurod1 mutant cerebellum, the EGL was expanded (blue arrow), and the number of GCs were abnormally increased in P11 (c’) from P7 (c), although by P15, GCs were dispersed and scattered and many pyknotic nuclei (red arrows) and mitotic nuclei (green arrowheads) were observed in lobule VII, with no ML and no definite PCL (P). d-d’’’ In the anterior lobules shown in lobule V of Neurod1 mutant mice, the layers were reasonably well-organized, but pyknotic nuclei and mitotic nuclei were observed in EGL in P7 and P11 mice (d, d’). Bars 10 µm
GCs depletion is directly proportional to organization of PCs and their dendritic arborization
PCs are an integral component of the cytoarchitecture of the cerebellar cortex, with their orderly alignment in the monolayer and their planar dendritic arbor. In the mouse, PCs cease to divide at about E12 and form a monolayer at about postnatal day 10 (Miale and Sidman 1961). Dendritic trees of PCs develop dramatically during the second and third postnatal weeks (Larramendi 1969; Weiss and Pysh 1978; Mugnaini 1969), and this development appears to depend on the proper formation of parallel fibers, the axons of GCs (Bradley and Berry 1978).
To assess the morphology of PCs in the Neurod1 conditional null mice, we performed immunocytochemistry with an antibody against calbindin in the adult cerebellum (Fig. 10). We found severely disorganized PCL in the anterior and central lobules (lobules I–VIII) of the cerebellum of Neurod1 mutant mice (Fig. 10c-d’’, e’, e’’). In contrast, the PCs formed a monolayer with a normal orientation of their dendrites in lobules IX and X (Fig. 10b-b’’), as in the heterozygous cerebellum (Fig. 10a-a’’, e). In central lobules (lobules VI-1/2VIII), PCs were dispersed randomly and showed no definite polarity of their dendrites (Fig. 10c-c’’, e’). Interestingly, although the organization of the GCL and the ML of the anterior lobules was nearly normal with only some GC loss, PCs formed a disorganized monolayer, with either weeping-willow-like dendrites or bidirectional dendrites projecting toward both the pia surface and the GCL (Fig. 10d-d’’, e’’).

Aberration of Purkinje cell (PC) dendrites was directly proportional to granule cell loss in Neurod1 conditional mutant cerebellum, as shown by calbindin staining (a-d’’) and dye tracing (e-e’’) in adult mice (ML molecular layer, PCL PC layer, GCL granule cell layer). a-a’’, e PCs were oriented in a monolayer with nicely organized dendrites in ML throughout all the lobules in control cerebellum. b-b’’ In Neurod1 mutant cerebellum, normal organization of PCs were observed only in lobules X and IX, where PCs were arranged in a single layer, and their dendrites projected toward the ML. c-c’’, e’ In the central lobules (1/2VI-1/2VIII) of mutant cerebellum, PCs were randomly organized with no specific direction of their dendrites. d-d’’, e’’ In the anterior lobules (I-1/2VI) of mutant cerebellum, PCs almost formed a single layer, but their dendrites were either inversely polarized (arrow) toward GCL or Bifurcated and bidirectional (e’’) to both the pial surface (dotted blue line) and GCL. Bars 100 µm (a-b’, c-c’, d-d’), 50 µm (c’’, d’’), 10 µm (e-e’’)
Some disorganization of PCs found in lobules VI-VIII has previously been reported in the posterior region of the cerebellum in systemic Neurod1 null mutants (Miyata et al. 1999; Cho and Tsai 2006) and has been suggested to be secondary to GC loss. By lipophilic dye tracing, we have, for the first time, demonstrated the abnormal orientation of PC dendrites in the cerebellum of Neurod1 mutant mice (Fig. 10e’, e’’) and shown that, in the anterior lobules, many PCs have bifurcated dendrites (Fig. 10e’’).
Loss of GCs affects distribution of mossy fibers
Cerebellar GCs are innervated by mossy fibers and thus relate mossy fiber input to PCs. Agranular cerebella have previously been generated by other means (Berry and Bradley 1976; Sotelo 1975; Arsenio Nunes et al. 1988), but the effect of the absence of GCs on the mossy fiber input into the cerebellum has not been systematically investigated with modern tracing techniques. The differential effects of our conditional deletion of Neurod1 in various lobules allow us directly to compare the innervations of mossy fibers in the neighboring lobules by using lipophilic dye tracing.
Parasagittal insertions of lipophilic dye-soaked filter strips resulted in the labeling of parallel fibers along a given folium (Fig. 11) for a variable distance parallel to the insertion site. However, in mutant cerebellum, parallel fibers could not be traced in lobule VI through the anterior part of lobule VIII, thus confirming the complete absence of parallel fibers because of the complete absence of GCs in these lobules. Interestingly, instead of having parallel fibers extending medio-lateral, these lobules showed mossy fibers extending laterally beyond the point at which they could be traced in lobules IX and X. This implies that, in the absence of GCs, the parasagittal organization of mossy fibers is in part disrupted. The extent to which they are disrupted requires additional analysis through direct tracing of these inputs from their sources with lipophilic dyes.

Distribution of mossy fibers affected by GC loss in central lobules of mutant cerebellum, as shown in adult mice by lipophilic dye tracing. a Distribution of PCs and GCs in lobule VIII of mutant cerebellum shown by calbindin staining and ROSA26, respectively (ML molecular layer, IGL internal granular layer, PCL Purkinje cell layer). b-d Mossy fibers (MF) in the central lobules directly pass toward the pial surface (dotted blue line) instead of synapsing with the GCs because of the nonexistence of the latter (CF climbing fiber, PF parallel fiber). Moreover, parallel fibers could not be traced in the central lobule consistent with GC loss (asterisk). Bars 50 µm (a, c), 20 µm (d), 10 µm (b)
The organization of mossy fibers was not only disrupted in a medio-lateral plane but also within a given folium. Whereas only climbing fibers could be traced into the ML in wildtype cerebella and folia IX + X in the conditional Neurod1 mutants, we found many fibers running, some even partially myelinated, throughout the folium and underneath the pia surface (Fig. 11b). These data imply that the contact between mossy fibers and GCs is needed to stop extension of those fibers into the ML and to restrict them to this layer. As previously indicated in agranular cerebella (Sotelo 1975; Arsenio Nunes et al. 1988), mossy fibers might be able to contact PC dendrites directly in the absence of GCs. We are currently investigating this possibility with appropriate double- and triple-color tracing with lipophilic dyes (Jensen-Smith et al. 2007).
In summary, GCs may not only be relevant for the normal formation of cerebellar layers, the alignment of PCs, and the proper development of PC dendrites, but may also play a role in the organization of the cerebellar mossy and climbing fiber input. In the absence of GCs, mossy fibers might behave like climbing fibers, an idea that we are currently testing in a longitudinal study of mossy and climbing fiber development in postnatal conditional Neurod1 null mice.
Atoh1 expression in Neurod1-depleted area
Neural bHLH transcription factors have specific roles in cerebellar development, and their expression needs to be precisely controlled both spatially and temporally as neuronal precursors mature. Atoh1 is a bHLH transcriptional factor homologous to the proneural factor atonal in Drosophila. Atoh1 is one of the earliest markers of cerebellar GC progenitors, and its expression is turned off as the GCs in EGL exit the cell cycle and begin to differentiate and migrate inward to become mature GCs in the IGL (Alder et al. 1999).
In our Neurod1 heterozygous mice, Atoh1 was expressed in the outer EGL with a gradient that was higher in the anterior lobules than in the posterior lobules at P7 and P11 (Fig. 12a–b’). At P15, Atoh1 expression was abolished in almost all lobules, except for some retention in lobule VII (Fig. 12c, c’), the last lobule to differentiate (Joyner and Zervas 2006). No Atoh1 expression was detectable by in situ hybridization at P21 or older (data not shown). However, in our Neurod1 mutant mice, Atoh1 expression remained turned on for longer and was expanded compared with the control cerebellum. At P7, Atoh1 was expressed in a similar pattern in mutant cerebellum as in heterozygous cerebellum (Fig. 12a, d). However, in the central lobules in P11 mutants, Atoh1 was abnormally expanded inwardly toward the ill-defined inner EGL and IGL (Fig. 12e, e’). Atoh1 expression was also discernable in the P15 mutant, with a gradient that was higher in the anterior and central lobules and almost absent in lobules IX and X, the lobules in which Neurod1 expression was preserved because of delayed upregulation of Tg(Atoh1-cre) (Fig. 12f, f’).

Atoh1 expression started normally but expanded and stayed turned on for longer in Neurod1 mutant cerebellum, as shown by in situ hybridization of Atoh1. a–b’ Atoh1 was expressed in the outer EGL with a gradient that was higher in the anterior lobules than in the posterior lobules at P7 and P11 heterozygous mice. c, c’ By P15, Atoh1 expression was abolished in almost all lobules, except for some retention in lobule VII in heterozygous mice. d, d’ In P7 Neurod1 mutant cerebellum, Atoh1 was expressed in a similar pattern as that in P7 heterozygous cerebellum. e, e’ In the P11 mutant, Atoh1 was abnormally expanded inwardly toward the inner EGL and IGL in the central lobules. f, f’ Atoh1 expression was still maintained in P15 mutant cerebellum, with the gradient being higher in the anterior and central lobules and almost diminished in lobules IX and X. The boxed areas in a-f are shown at higher magnification in a’-f’. Bars 250 µm (a-f), 50 µm (a’-f’)
Barhl1 expression in Neurod1 mutant cerebellum
The transcription factor Barhl1 is one of the few known downstream genes of Atoh1 and is expressed in the developing inner ear hair cells, cerebellar GCs, precerebellar neurons, and collicular neurons (Bulfone et al. 2000; Chellappa et al. 2008; Higashijima et al. 1992; Kojima et al. 1991; Li et al. 2002). Barhl1 plays a crucial role in the migration and survival of cerebellar GCs and certain precerebellar neurons by governing the expression of Neurotrophin3 (Ntf3; Li et al. 2004). Li et al. (2004) have shown that the absence of Barhl1 causes increased GC death and concomitant loss of mossy-fiber-forming precerebellar neurons. Therefore, we have analyzed the expression of Barhl1 in the Neurod1 mutant cerebellum.
In the Neurod1 heterozygous cerebellum, Barhl1 was homogeneously expressed in all lobules in the EGL, whereas in the IGL, a gradient was present showing more prominent expression in lobules I-VI and lower expression in lobules IX and X (Fig. 13a-c). In Neurod1 conditional mutant cerebellum, Barhl1 was expressed prominently in the EGL, but the expression in IGL was almost abolished from P11 onward (Fig. 13d, d’). In P15 mutants, Barhl1 was expressed in EGL and expanded inwardly from EGL (Fig. 13e, e’). At P21, when no EGL was left in control littermates, Barhl1 persisted in EGL of the central lobules of the mutant cerebellum (Fig. 13f, f’). The Barhl1 expression pattern matched that of Atoh1 in Neurod1 mutant cerebellum, but with a delay of 1–4 days (Figs. 12, 13).

Spatial and temporal expression of Barhl1 changes in absence of Neurod1 in cerebellum, as shown by in situ hybridization of Barhl1. a, a’ Barhl1 was expressed in EGL in all lobules and in IGL, was highly expressed in anterior lobules I-1/2VI, absent in lobules VII and VIII, and low in lobules IX and X in P11 heterozygous cerebellum. b-b’’ By P15, Barhl1 expression was downregulated in IGL and EGL in heterozygous cerebellum. c By P21, Barhl1 expression was diminished in heterozygous mice. d, d’ In P11 mutant cerebellum, IGL was almost devoid of Barhl1 expression, except for some in lobule X; EGL showed expanded Barhl1 expression. e, e’ Barhl1 expression persisted and expanded in EGL predominantly in central lobules in mutant P15 cerebellum, instead of being expressed in IGL. f, f’ Barhl1 abnormally continued to be expressed in EGL of central lobules in P21 mutant cerebellum. The boxed areas in a-f are shown at higher magnification in a’-f’. Bars 250 µm (a-f), 50 µm (a’-f’, b’’)
In summary, the expression of both Atoh1 and Barhl1 supports the notion of an expansion of the EGL in lobules VI-VIII and a delayed downregulation of proliferative precursors in these lobules. Because Atoh1 and Barhl1 expression remained turned on for a prolonged period in the EGL in order to continue proliferation of the GCPs in Neurod1 mutant mice, the EGL became thicker in the central lobules compared with lobules IX and X in the same cerebellum (Figs. 7b, 9). The GCs in lobule VIII seemed to lack coordinated migration from EGL to IGL in mutant mice (Fig. 7c).
Discussion
In agreement with our earlier work and that of others (Miyata et al. 1999; Cho and Tsai 2006), we demonstrate that Neurod1 is indispensible for the differentiation and maintenance of GCs of cerebellar lobules VI-VIII. In contrast to the systemic Neurod1 null mutant, we have now generated a mouse model in which some lobules develop almost normally, some lobules are consistently agranular, and some lobules show certain GC and PC defects not previously recognized. Our data also suggest that Neurod1 antagonizes Atoh1 and thus may aid in moving proliferative GCPs into differentiation.
Lack of Neurod1 excision in posterior lobules is related to delayed and incomplete Tg(Atoh1-cre) expression
In the cerebellum of our conditional Neurod1 null mouse, we find Neurod1 mRNA to be present in lobules IX and X, and this lack of Neurod1 excision is correlated with the inadequate and delayed upregulation of Tg(Atoh1-cre) in these lobules. Our data from the other lobules show that Tg(Atoh1-cre) can successfully eliminate Neurod1, thus indicating the success of the genetic manipulation involved in constructing the floxed Neurod1. We also show that, in the cochlear nuclei and spiral sensory neurons of the mutant mice, the expression of Tg(Atoh1-cre) is even more delayed compared with Neurod1 gene upregulation (Kim et al. 2001; Ohyama and Groves 2004), resulting in the continued presence of the Neurod1 transcript. The molecular mechanism underlying these differences needs further investigation.
Previous studies have shown a gradient of ROSA26-eYFP expression driven by Tg(Atoh1-cre), with YFP expression being high from lobule I through lobule VIII, weak in lobule IX, and almost absent in lobule X (Schuller et al. 2008). This gradient of YFP expression is consistent with the gradient of Tg(Atoh1-cre)-mediated ROSA26-LacZ expression shown in our study and thus reflects a consistent expression profile on a different background.
Our observation of cerebellar defects in Neurod1 conditional null mice is in apparent contrast to that in the normal cerebellum shown in a previous study by using the GABAA receptor α6 subunit promoter-driving Cre to delete the Neurod1 gene (Goebbels et al. 2005). However, the GABAA receptor α6 subunit is expressed only in the post-migratory mature GCs around P14 (Aller et al. 2003). Therefore, deletion of Neurod1 after the GCs become mature apparently does not interfere with GC viability and produces a near normal cerebellar phenotype. Our data show that posterior lobules develop earlier than the anterior lobules, and that the degree of Neurod1 dependency is more profound in posterior lobules than in anterior lobules. Combined, our present and previous data suggest that, within each lobule, the susceptibility of GCs to loss of Neurod1 decreases with the progression of development, leaving the differentiated GCs unaffected by Neurod1 elimination, but introduces apoptosis if eliminated in postmitotic cells prior to the onset of differentiation. We are currently testing this hypothesis by analyzing the effects of Neurod1 deletion in a Pax2-cre line that shows Cre expression throughout the cerebellum and the ear prior to the upregulation of Neurod1 (Kim et al. 2001; Ohyama and Groves 2004; Soukup et al. 2009).
Differential defects in cerebellar lobules in Neurod1 conditional knock-out mice
Previous work on systemic Neurod1 null mice have shown that the absence of Neurod1 leads to a lack of foliation and the complete loss of GCs in the posterior half of the cerebellum, whereas a substantial number of GCs survive and differentiate in the anterior lobules (Miyata et al. 1999; Cho and Tsai 2006). Several other studies on mutations involving GC depletion, such as leaner and meander tail mutations, have also found that the degree of defect in the cerebellum differs considerably between the anterior and posterior lobules with the compartment boundary lying in lobule VI (Herrup and Wilczynski 1982). A gradient of defects in the cerebellar lobules is also observed in medulloblastoma formation. Olig2, a bHLH protein, preferentially contributes to GC formation in the posterior lobules of the cerebellum. With conditional overexpression of activated Smoothened under the control of Olig2, the mice develop medulloblastoma restricted to the posterior lobules only (Schuller et al. 2008). Tg(Atoh1-cre)-driven overexpression of Smoothened or the conditional deletion of the sonic hedgehog receptor Patched is known to produce medulloblastoma followed by severe hyperplasia in the central lobules (Schuller et al. 2008; Yang et al. 2008). Another gene showing a clear delineation of expression is Tlx3, which can also result in restricted medulloblastoma (Schuller et al. 2008). Overall, these more recent molecular data indicate a sophisticated molecular subdivision of the cerebellar cortex that may underlie specific targeting of mossy fibers such as the vestibular fibers to the nodulus and uvula (Maklad and Fritzsch 2003).
In our current study, we not only confirm the previous notion of an anterior-posterior differential requirement of Neurod1 for GC maintenance, but also show that the combination of a delayed posterior upregulation of Tg(Atoh1-cre), combined with the anterior-posterior requirement of Neurod1, results in a mosaic of GC developmental loss leading to three distinct areas in the mature cerebellum.
-
1)
Lobules I-V in which some GCs differentiate, and some fail to survive in the absence of Neurod1; these lobules nevertheless show a distinct disorganization of PC dendrites into a bipolar pattern that has not previously been reported.
-
2)
Lobules VI-VIII with a near complete loss of all GCs, severe disorganization of PC distribution and dendrites, and diminished foliation.
-
3)
Lobules IX and X with near normal organization of cerebellar architecture and GC development because of the sparing of Neurod1 recombination.
This newly developed model created by using genetic engineering allows us, for the first time, to investigate GC-dependent aspects of cerebellar cytoarchitecture development that cannot be fully assessed with varied or complete destruction of GCs through other means (Berry and Bradley 1976; Sotelo 1975; Arsenio Nunes et al. 1988). Our model is reproducible and is free of systemic defects that could influence overall viability and thus indirectly affect cerebellar development. Moreover, this is the first model of an agranular cerebellum that provides, in addition to the defective lobules, normal control development in the same animal, albeit in different lobules. Below, we provide an initial assessment of this new model system on hitherto unexplored aspects of the influence of GC axons on PC dendrites and the role of the coordinated transition of bHLH genes for GC proliferation and maturation.
GC survival and differentiation regulate cerebellar layering and PC orientation and distribution
Our data demonstrate, for the first time, that the degree of layer formation is directly tied to the degree of GC differentiation, whereas the normal patterning of PCs depends both on the proper formation of GC axons, the parallel fibers, and on some unrecognized GC properties dependent on Neurod1. In support of this idea, the normal formation of layers is found in lobules I-V and IX-X, but normal PC dendrites exist only in lobules IX and X. Thus, the mere presence of GCs and their parallel fiber axons is necessary for the development of the typical cerebellar layers, but dendritic branching requires additional features that are not provided by the GCs in the Neurod1-depleted anterior lobules. This has not previously been noticed, as we and others (Miyata et al. 1999; Cho and Tsai 2006) did not analyze PC development in simple Neurod1 null mice in sufficient detail.
The absence of any layers and the random distribution of PCs and their dendrites in the poorly defined lobules VI-VIII are consistent with previous work on the function of GCs in agranular cerebella generated by irradiation, viral infection, or GC-specific mutations (Berry and Bradley 1976; Sotelo 1975; Arsenio Nunes et al. 1988). In contrast to those early attempts to identify the developmental significance of GC formation on PC dendritic development, we provide here a model that not only confirms the generality of those findings, but does so with unprecedented reproducibility because it is based on a genetically engineered mouse. We are currently exploring the missing steps in PCs development and their innervations through mossy and climbing fibers, which may also be severely disrupted in their ability to segregate properly to GCs and PCs, respectively (Arsenio Nunes et al. 1988). Should our developmental data confirm previous suggestions (Arsenio Nunes et al. 1988) and show that the segregation of mossy and climbing fibers depends on the presence of GCs and a differential affinity of mossy fibers and climbing fibers to GCs, we should be able to establish the evolution and development of GCs as a central hub for the cerebellum and its development.
In this study, the absence of Neurod1 severely affects the central lobules, resulting in both GC depletion and PC disorganization. The abnormal arborization of PCs is dependent on GCs, as the degeneration of GCs leads to the abolition of parallel fibers, which, in turn, cannot provide input to the PCs. The monolayer alignment of PCs with the polarity and planar organization of their dendrites perpendicular to parallel fibers represents the functional efficiency implied in central neural tissues, as it allows the maximal divergence and convergence of connections from parallel fibers to PCs.
Some interesting relationships emerge in comparison with existing data. The Foxp2 missense mutation causes specific language deficiency originally identified in the KE family in which half of the family members have a speech disorder. Fujita et al. (2008) have demonstrated severe ultrasonic vocalization and motor impairment in Foxp2 knockin mice. They have also found immature development of PCs with poor dendrite formation and overall growth reduction of the cerebellum. In our Neurod1 conditional null mice, the PC dendrites are disorganized. Further investigations to analyze speech- and language-related impairment by means of our conditional Neurod1 mouse model are warranted.
Role of bHLH genes in regulation of proliferation and differentiation of neurons
Previous work has shown that many developing systems require the coordinated transition of various bHLH genes with specific aspects of proliferation of neuronal progenitors and their differentiation (Bertrand et al. 2002; Helms et al. 2000). For example, in the ear, the first bHLH gene to be expressed is Neurog1. It regulates the proliferation of neuronal precursors, which then differentiate into neurons and hair cells through the upregulation of Neurod1 or Atoh1, respectively (Bulfone et al. 2000; Fritzsch et al. 2006b, 2007; Raft et al. 2007).
In the cerebellum, Atoh1 functions as a neuronal precursor factor to promote the proliferation of GCPs in the outer EGL (Fig. 14). This is supported by the loss of GC formation in Atoh1 null mice (Wang et al. 2005; Bermingham et al. 2001) and the additional precursor proliferation in Atoh1-overexpressing mice (Helms et al. 2001). Specifically, overexpression of Atoh1 causes misregulation of Neurod1 and other markers of differentiating cerebellar GCs, thus leading to reduced viability of GCs, suggesting that balanced levels of Atoh1 and some other transcription factors are required for GC development. Our data agree with this scenario, show that normal levels of Neurod1 are indeed needed for most GC differentiation, and also provide a novel mechanism of regulating Atoh1 expression to ensure a coordinated transition from an Atoh1-positive proliferating neuroblast to a differentiating granule neuron (Fig. 14).

Differential control of granule cell progenitor (GCP) proliferation and differentiation in cerebellar development and tumorigenesis. Overview of the signaling pathways for proliferation and differentiation of the GCP from neural stem cells. Atoh1 expressed in GCP promotes GCP proliferation, whereas Neurod1 facilitates GC differentiation, both by forming heterodimers with ubiquitously expressed E proteins and activating downstream E-box-containing genes. Our data suggest that Neurod1 regulates Atoh1 via a negative feedback loop. After upregulation of Neurod1, Atoh1 shuts down, thereby promoting transition from proliferation to differentiation. In the absence of Neurod1, continued expression of Atoh1 causes enhanced proliferation of GCP, aberrant migration, and failure of differentiation, eventually leading to extensive cell death. Sonic hedgehog (Shh) plays a key role in GCP proliferation by positively regulating cyclin D1 and cyclin D2 and by inducing expression of Mycn, Mxd3 and Bmi1. Overexpression of Shh and its receptor Smoothened (Smo), Mycn, or Mxd3 gives rise to the uncontrolled proliferation of GCP and the formation of medulloblastoma (Schuller et al. 2008; Yang et al. 2008). Id proteins also affect the balance between proliferation and differentiation by binding to and inhibiting bHLH proteins and also by binding to retinoblastoma (Rb) proteins, key regulators of cell cycle progression (Zebedee and Hara 2001)
We show here that Atoh1 is rapidly downregulated when Neurod1 is upregulated to allow GCPs in the innermost layer of the EGL to differentiate and migrate into their proper topological position in the IGL. In Neurod1 conditional null mice, the absence of Neurod1 leads to prolonged expression of Atoh1 in these precursors, suggesting a negative feedback loop within a given cell as previously suggested for other systems such as the olfactory system (Kawauchi et al. 2004) or the ear (Fritzsch et al. 2006b). Consistent with this idea, we have observed the expansion of Atoh1 in the inner EGL and continued proliferation of GCPs (as demonstrated by mitotic figures near the ML; Fig. 9c, c’). Because of this delay in turning off Atoh1 and the absence of Neurod1 in the central lobules, the migration of GCs is aberrant resulting in their massive degeneration. The precise way that Atoh1 and Neurod1 interact with other bHLH genes to regulate both proliferation and differentiation (Barisone et al. 2008) and the specific binding differences that exist in Atoh1 compared with Neurod1 in this context remain unclear. However, close examination of the medulloblastoma data base show elevated levels of Atoh1 and decreased levels of Neurod1, consistent with a differential role of those two bHLH genes in mediating either proliferation or differentiation. Recent data on viral-induced overexpression support this notion for Neurod1 in the hippocampus (Roybon et al. 2009).
Barhl1 is dependent on Atoh1 expression and lies downstream of Atoh1 expressed in cerebellar GCs (Chellappa et al. 2008). In our mutant cerebellum, Barhl1 shows an expansion of its expression similar to that of Atoh1 expression, as Atoh1 is a key upstream regulator of Barhl1. Barhl1 expression is maintained longer in the absence of Neurod1 in the central lobules. In the anterior lobules of the Neurod1 mutant cerebellum, Barhl1 may, in part, be responsible for rescuing GCs to some degree. Barlh1 cannot however save the central lobules. The feedback upregulation of Atoh1 and Barhl1 cannot guide the GCs into appropriate migration or differentiation, thereby indicating the critical need of Neurod1 in these lobules.
In summary, our data on the conditional deletion of Neurod1 by using the Tg(Atoh1-cre) line have generated a novel viable mouse model for the study of agranular cerebellar cortex development. Since only some lobules are massively defective, this model allows intrinsic controls of specific defects within adjacent normally developed lobules. Our data also suggest that Neurod1 antagonizes Atoh1 and thus might aid in moving proliferative precursors into differentiation. We are currently testing whether the expression of Neurod1 under Atoh1 promoter control can force proliferative GCPs into differentiation as a possible way to combat the fairly frequently occurring childhood tumor medulloblastoma (Fig. 14).
References
Akazawa C, Ishibashi M, Shimizu C, Nakanishi S, Kageyama R (1995) A mammalian helix-loop-helix factor structurally related to the product of Drosophila proneural gene atonal is a positive transcriptional regulator expressed in the developing nervous system. J Biol Chem 270:8730–8738
Alder J, Cho NK, Hatten ME (1996) Embryonic precursor cells from the rhombic lip are specified to a cerebellar granule neuron identity. Neuron 17:389–399
Alder J, Lee KJ, Jessell TM, Hatten ME (1999) Generation of cerebellar granule neurons in vivo by transplantation of BMP-treated neural progenitor cells. Nat Neurosci 2:535–540
Aller MI, Jones A, Merlo D, Paterlini M, Meyer AH, Amtmann U, Brickley S, Jolin HE, McKenzie AN, Monyer H, Farrant M, Wisden W (2003) Cerebellar granule cell Cre recombinase expression. Genesis 36:97–103
Altman J, Bayer SA (1978) Prenatal development of the cerebellar system in the rat. I. Cytogenesis and histogenesis of the deep nuclei and the cortex of the cerebellum. J Comp Neurol 179:23–48
Arsenio Nunes ML, Sotelo C, Wehrle R (1988) Organization of spinocerebellar projection map in three types of agranular cerebellum: Purkinje cells vs. granule cells as organizer element. J Comp Neurol 273:120–136
Barisone GA, Yun JS, Diaz E (2008) From cerebellar proliferation to tumorigenesis: new insights into the role of Mad3. Cell Cycle 7:423–427
Ben-Arie N, McCall AE, Berkman S, Eichele G, Bellen HJ, Zoghbi HY (1996) Evolutionary conservation of sequence and expression of the bHLH protein Atonal suggests a conserved role in neurogenesis. Hum Mol Genet 5:1207–1216
Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, Matzuk MM, Zoghbi HY (1997) Math1 is essential for genesis of cerebellar granule neurons. Nature 390:169–172
Benediktsson AM, Schachtele SJ, Green SH, Dailey ME (2005) Ballistic labeling and dynamic imaging of astrocytes in organotypic hippocampal slice cultures. J Neurosci Methods 141:41–53
Bermingham NA, Hassan BA, Wang VY, Fernandez M, Banfi S, Bellen HJ, Fritzsch B, Zoghbi HY (2001) Proprioceptor pathway development is dependent on Math1. Neuron 30:411–422
Berry M, Bradley P (1976) The growth of the dendritic trees of Purkinje cells in irradiated agranular cerebellar cortex. Brain Res 116:361–387
Bertrand N, Castro DS, Guillemot F (2002) Proneural genes and the specification of neural cell types. Nat Rev Neurosci 3:517–530
Bradley P, Berry M (1978) The Purkinje cell dendritic tree in mutant mouse cerebellum. A quantitative Golgi study of Weaver and Staggerer mice. Brain Res 142:135–141
Bulfone A, Menguzzato E, Broccoli V, Marchitiello A, Gattuso C, Mariani M, Consalez GG, Martinez S, Ballabio A, Banfi S (2000) Barhl1, a gene belonging to a new subfamily of mammalian homeobox genes, is expressed in migrating neurons of the CNS. Hum Mol Genet 9:1443–1452
Buratowski S (2008) Transcription. Gene expression—where to start? Science 322:1804–1805
Chellappa R, Li S, Pauley S, Jahan I, Jin K, Xiang M (2008) Barhl1 regulatory sequences required for cell-specific gene expression and autoregulation in the inner ear and central nervous system. Mol Cell Biol 28:1905–1914
Cho JH, Tsai MJ (2006) Preferential posterior cerebellum defect in BETA2/NeuroD1 knockout mice is the result of differential expression of BETA2/NeuroD1 along anterior-posterior axis. Dev Biol 290:125–138
Dahmane N, Ruiz-i-Altaba A (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126:3089–3100
del Cerro M, Cogen J, Cerro C del (1980) Stevenel's blue, an excellent stain for optical microscopical study of plastic embedded tissues. Microsc Acta 83:117–121
Dino MR, Willard FH, Mugnaini E (1999) Distribution of unipolar brush cells and other calretinin immunoreactive components in the mammalian cerebellar cortex. J Neurocytol 28:99–123
Englund C, Kowalczyk T, Daza RA, Dagan A, Lau C, Rose MF, Hevner RF (2006) Unipolar brush cells of the cerebellum are produced in the rhombic lip and migrate through developing white matter. J Neurosci 26:9184–9195
Fritzsch B, Muirhead KA, Feng F, Gray BD, Ohlsson-Wilhelm BM (2005) Diffusion and imaging properties of three new lipophilic tracers, NeuroVue Maroon, NeuroVue Red and NeuroVue Green and their use for double and triple labeling of neuronal profile. Brain Res Bull 66:249–258
Fritzsch B, Pauley S, Feng F, Matei V, Nichols DH (2006a) The evolution of the vertebrate auditory system: transformations of vestibular mechanosensory cells for sound processing is combined with newly generated central processing neurons. Int J Comp Psychol 19:1–24
Fritzsch B, Pauley S, Beisel KW (2006b) Cells, molecules and morphogenesis: the making of the vertebrate ear. Brain Res 1091:151–171
Fritzsch B, Beisel KW, Pauley S, Soukup G (2007) Molecular evolution of the vertebrate mechanosensory cell and ear. Int J Dev Biol 51:663–678
Fujita S (1969) Autoradiographic studies on histogenesis of the cerebellar cortex. In: Llinas R (ed) Neurobiology of cerebellar evolution and development. American Medical Association, Chicago, pp 743–747
Fujita S, Shimada M, Nakamura T (1966) H3-thymidine autoradiographic studies on the cell proliferation and differentiation in the external and the internal granular layers of the mouse cerebellum. J Comp Neurol 128:191–208
Fujita E, Tanabe Y, Shiota A, Ueda M, Suwa K, Momoi MY, Momoi T (2008) Ultrasonic vocalization impairment of Foxp2 (R552H) knockin mice related to speech-language disorder and abnormality of Purkinje cells. Proc Natl Acad Sci USA 105:3117–3122
Gazit R, Krizhanovsky V, Ben-Arie N (2004) Math1 controls cerebellar granule cell differentiation by regulating multiple components of the Notch signaling pathway. Development 131:903–913
Goebbels S, Bode U, Pieper A, Funfschilling U, Schwab MH, Nave KA (2005) Cre/loxP-mediated inactivation of the bHLH transcription factor gene NeuroD/BETA2. Genesis 42:247–252
Goldowitz D, Hamre K (1998) The cells and molecules that make a cerebellum. Trends Neurosci 21:375–382
Gurung B, Fritzsch B (2004) Time course of embryonic midbrain and thalamic auditory connection development in mice as revealed by carbocyanine dye tracing. J Comp Neurol 479:309–327
Hatten ME, Heintz N (1995) Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci 18:385–408
Hatten ME, Furie MB, Rifkin DB (1982) Binding of developing mouse cerebellar cells to fibronectin: a possible mechanism for the formation of the external granular layer. J Neurosci 2:1195–1206
Helms AW, Abney AL, Ben-Arie N, Zoghbi HY, Johnson JE (2000) Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development 127:1185–1196
Helms AW, Gowan K, Abney A, Savage T, Johnson JE (2001) Overexpression of MATH1 disrupts the coordination of neural differentiation in cerebellum development. Mol Cell Neurosci 17:671–682
Herrup K, Wilczynski SL (1982) Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience 7:2185–2196
Higashijima S, Michiue T, Emori Y, Saigo K (1992) Subtype determination of Drosophila embryonic external sensory organs by redundant homeo box genes BarH1 and BarH2. Genes Dev 6:1005–1018
Hoshino M, Nakamura S, Mori K, Kawauchi T, Terao M, Nishimura YV, Fukuda A, Fuse T, Matsuo N, Sone M, Watanabe M, Bito H, Terashima T, Wright CV, Kawaguchi Y, Nakao K, Nabeshima Y (2005) Ptf1a, a bHLH transcriptional gene, defines GABAergic neuronal fates in cerebellum. Neuron 47:201–213
Isaka F, Ishibashi M, Taki W, Hashimoto N, Nakanishi S, Kageyama R (1999) Ectopic expression of the bHLH gene Math1 disturbs neural development. Eur J Neurosci 11:2582–2588
Jensen-Smith H, Gray B, Muirhead K, Ohlsson-Wilhelm B, Fritzsch B (2007) Long-distance three-color neuronal tracing in fixed tissue using NeuroVue dyes. Immunol Invest 36:763–789
Joyner AL, Zervas M (2006) Genetic inducible fate mapping in mouse: establishing genetic lineages and defining genetic neuroanatomy in the nervous system. Dev Dyn 235:2376–2385
Kageyama R, Ohtsuka T, Kobayashi T (2007) The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development 134:1243–1251
Kawauchi S, Beites CL, Crocker CE, Wu HH, Bonnin A, Murray R, Calof AL (2004) Molecular signals regulating proliferation of stem and progenitor cells in mouse olfactory epithelium. Dev Neurosci 26:166–180
Kim WY, Fritzsch B, Serls A, Bakel LA, Huang EJ, Reichardt LF, Barth DS, Lee JE (2001) NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development 128:417–426
Kojima T, Ishimaru S, Higashijima S, Takayama E, Akimaru H, Sone M, Emori Y, Saigo K (1991) Identification of a different-type homeobox gene, BarH1, possibly causing Bar (B) and Om (1D) mutations in Drosophila. Proc Natl Acad Sci USA 88:4343–4347
Larramendi L (1969) Analysis of synaptogenesis in the cerebellum of the mouse. In: Llinas R (ed) Neurobiology of cerebellar evolution and development. American Medical Association, Chicago, pp 803–843
Lee JE (1997) Basic helix-loop-helix genes in neural development. Curr Opin Neurobiol 7:13–20
Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H (1995) Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science 268:836–844
Lee JK, Cho JH, Hwang WS, Lee YD, Reu DS, Suh-Kim H (2000) Expression of neuroD/BETA2 in mitotic and postmitotic neuronal cells during the development of nervous system. Dev Dyn 217:361–367
Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P, Van Lohuizen M, Marino S (2004) Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 428:337–341
Li S, Price SM, Cahill H, Ryugo DK, Shen MM, Xiang M (2002) Hearing loss caused by progressive degeneration of cochlear hair cells in mice deficient for the Barhl1 homeobox gene. Development 129:3523–3532
Li S, Qiu F, Xu A, Price SM, Xiang M (2004) Barhl1 regulates migration and survival of cerebellar granule cells by controlling expression of the neurotrophin-3 gene. J Neurosci 24:3104–3114
Ma Q, Chen Z, Barco Barrantes I del, Pompa JL de la, Anderson DJ (1998) Neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron 20:469–482
Machold R, Fishell G (2005) Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron 48:17–24
Maklad A, Fritzsch B (2003) Partial segregation of posterior crista and saccular fibers to the nodulus and uvula of the cerebellum in mice, and its development. Brain Res Dev Brain Res 140:223–236
Matei V, Pauley S, Kaing S, Rowitch D, Beisel KW, Morris K, Feng F, Jones K, Lee J, Fritzsch B (2005) Smaller inner ear sensory epithelia in Neurog1 null mice are related to earlier hair cell cycle exit. Dev Dyn 234:633–650
Matei VA, Feng F, Pauley S, Beisel KW, Nichols MG, Fritzsch B (2006) Near-infrared laser illumination transforms the fluorescence absorbing X-Gal reaction product BCI into a transparent, yet brightly fluorescent substance. Brain Res Bull 70:33–43
Miale IL, Sidman RL (1961) An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp Neurol 4:277–296
Miyata T, Maeda T, Lee JE (1999) NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev 13:1647–1652
Mugnaini E (1969) Ultrastructural studies on the cerebellar histogenesis. II. Maturation of nerve cell populations and establishment of synaptic connections in the cerebellar cortex of the chick. In: Llinas R (ed) Neurobiology of cerebellar evolution and development. American Medical Association, Chicago, pp 749–782
Mugnaini E, Dino MR, Jaarsma D (1997) The unipolar brush cells of the mammalian cerebellum and cochlear nucleus: cytology and microcircuitry. Prog Brain Res 114:131–150
Ohyama T, Groves AK (2004) Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis 38:195–199
Raft S, Koundakjian EJ, Quinones H, Jayasena CS, Goodrich LV, Johnson JE, Segil N, Groves AK (2007) Cross-regulation of Ngn1 and Math1 coordinates the production of neurons and sensory hair cells during inner ear development. Development 134:4405–4415
Roybon L, Hjalt T, Stott S, Guillemot F, Li JY, Brundin P (2009) Neurogenin2 directs granule neuroblast production and amplification while NeuroD1 specifies neuronal fate during hippocampal neurogenesis. PLoS ONE 4:e4779
Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL (2008) Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14:123–134
Sgaier SK, Millet S, Villanueva MP, Berenshteyn F, Song C, Joyner AL (2005) Morphogenetic and cellular movements that shape the mouse cerebellum; insights from genetic fate mapping. Neuron 45:27–40
Sotelo C (1975) Anatomical, physiological and biochemical studies of the cerebellum from mutant mice. II. Morphological study of cerebellar cortical neurons and circuits in the weaver mouse. Brain Res 94:19–44
Sotelo C (2008) Viewing the cerebellum through the eyes of Ramon y Cajal. Cerebellum 7:517–522
Soukup GA, Fritzsch B, Pierce ML, Weston MD, Jahan I, McManus MT, Harfe BD (2009) Residual microRNA expression dictates the extent of inner ear development in conditional Dicer knockout mice. Dev Biol 328:328–341
Voogd J, Glickstein M (1998) The anatomy of the cerebellum. Trends Neurosci 21:370–375
Wang VY, Rose MF, Zoghbi HY (2005) Math1 expression redefines the rhombic lip derivatives and reveals novel lineages within the brainstem and cerebellum. Neuron 48:31–43
Weiss GM, Pysh JJ (1978) Evidence for loss of Purkinje cell dendrites during late development: a morphometric Golgi analysis in the mouse. Brain Res 154:219–230
Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH, Wainwright BJ, Wechsler-Reya RJ (2008) Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 14:135–145
Zebedee Z, Hara E (2001) Id proteins in cell cycle control and cellular senescence. Oncogene 20:8317–8325
Acknowledgements
We express our thanks to Drs. K.-A. Nave and S. Goebbels, Max-Planck-Institute of Experimental Medicine, for providing the floxed Neurod1 mice used in this study. We also thank the Bioimaging Facility in the Department of Biology at University of Iowa for assisting with the Leica TCS SP5 confocal microscope. We are grateful to J. Kersigo for extensive help in breeding and genotyping and in the various steps during sample preparation and to K. Thompson for proofreading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ning Pan and Israt Jahan contributed equally to this paper.
This work was supported by an NIH grant (R01 DC 005590) to B.F.
Rights and permissions
About this article
Cite this article
Pan, N., Jahan, I., Lee, J.E. et al. Defects in the cerebella of conditional Neurod1 null mice correlate with effective Tg(Atoh1-cre) recombination and granule cell requirements for Neurod1 for differentiation. Cell Tissue Res 337, 407–428 (2009). https://doi.org/10.1007/s00441-009-0826-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-009-0826-6