
Abstract
Autosomal recessive polycystic kidney disease (ARPKD) belongs to a group of congenital hepatorenal fibrocystic syndromes characterized by dual renal and hepatic involvement of variable severity. Despite the wide clinical spectrum of ARPKD (MIM 263200), genetic linkage studies indicate that mutations at a single locus, PKHD1 (polycystic kidney and hepatic disease 1), located on human chromosome region 6p21.1–p12, are responsible for all phenotypes of ARPKD. Identification of cystic disease genes and their encoded proteins has provided investigators with critical tools to begin to unravel the molecular and cellular mechanisms of PKD. PKD cystic epithelia share common phenotypic abnormalities despite the different genetic mutations that underlie the disease. Recent studies have shown that many cyst-causing proteins are expressed in multimeric complexes at distinct subcellular locations within epithelia. This co-expression of cystoproteins suggests that cyst formation, regardless of the underlying disease gene, results from perturbations in convergent and/or integrated signal transduction pathways. To date, no specific therapies are in clinical use for ameliorating cyst growth in ARPKD. However, studies noted in this review suggest that therapeutic targeting of the cAMP and epidermal growth factor receptor (EGFR)-axis abnormalities in cystic epithelia may translate into effective therapies for ARPKD and, by analogy, autosomal dominant polycystic kidney disease (ADPKD). A particularly promising approach appears to be the targeting of downstream intermediates of both the cAMP and EGFR axis. This review focuses on ARPKD and presents a concise summary of the current understanding of the molecular genetics and cellular pathophysiology of this disease. It also highlights phenotypic and mechanistic similarities between ARPKD and ADPKD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Renal cystic diseases (RCD) include a group of monogenic kidney abnormalities that cause significant morbidity and mortality (Dell et al. 2004). Histopathologically, renal cysts are fluid-filled epithelia-lined dilated saccular lesions that generally arise from tubular segments. Although isolated renal cysts may arise as degenerative lesions in adults, the most common cause of nondysplastic nonsyndromal multiple renal cysts are autosomal dominant (ADPKD; MIM 601313 and MIM 173910; PKD1 and PKD2) or autosomal recessive (ARPKD; MIM 263200) polycystic kidney disease.
ADPKD is the most common renal monogenic disease with an incidence of 1/400–1000. ARPKD comprises 5%–8% of all individuals requiring renal replacement therapy (dialysis and/or kidney transplantation). Approximately 80%–85% of ADPKD patients have mutations in the PKD1 gene, located on chromosome 16p13, which encodes polycystin-1 (PC-1). The remaining 10%–15% of ADPKD cases have mutations in the PKD2 gene, which is located on chromosome 4q21, and which encodes polycystin-2 (PC-2). ADPKD is usually asymptomatic until the middle decades. However, 2% to 5% of ADPKD patients present with a severe neonatal course and significant morbidity and mortality.
ARPKD is a significant cause of renal and liver-related morbidity and mortality in childhood. Estimates of disease prevalence vary widely, but an overall frequency of 1 in 20,000 live births and a carrier level of up to 1:70 have been recently proposed (Zerres et al. 1998).
The majority of ARPKD patients present clinically as newborn or young children. Despite dramatic improvements in neonatal and intensive care over the past decade, neonatal mortality is 25%–35%. The clinical spectrum of surviving patients is considerably more variable. Principal manifestations of the disease involve the fusiform dilation of renal collecting tubules or ducts (CT) and dysgenesis of the hepatic portal triad attributable to ductal plate abnormalities.
The ARPKD disease gene, PKHD1 (polycystic kidney and hepatic disease 1), is a large gene located on chromosome 6p21.1–p12; it spans 470 kb of genomic DNA and produces a cDNA of 16 kb. A minimum of 86 exons is assembled into a variety of alternatively spliced transcripts. The longest continuous open reading frame (ORF) is predicted to yield a novel 4,074-amino-acid multidomain integral membrane protein of unknown function called fibrocystin (Ward et al. 2002) or polyductin (Onuchic et al. 2002).
Historically, inherited renal diseases were characterized by the description of clinical symptoms, age of presentation, and the history of disease progression. Advances in molecular genetics have led to the identification of a number of cyst-causing genes including the three PKD genes (PKD1, PKD2, and PKHD1). This has allowed previous descriptive cataloging of many renal diseases to be classified and confirmed at the genetic or molecular level. Identification of RCD genes and their encoded cysto-proteins has provided investigators with the basic tools to begin to unravel the molecular and cellular mechanisms of PKD. Recent studies have led to the discovery that many of these proteins are expressed in multimeric complexes at distinct subcellular locations within renal epithelia (Wilson 2004b). This co-expression of cystoproteins in complexes suggests that cyst formation in many different diseases may result from similar perturbations in convergent and/or integrated signal transduction pathways (Wilson 2004b). This may provide a molecular basis for the perplexing clinical observation that diseases caused by mutations in many different genes may produce similar, and at times, identical clinical phenotypes.
This review will focus on ARPKD and present a concise summary of the current understanding of the molecular genetics and cellular pathophysiology of this disease. It will also highlight phenotypic and mechanistic similarities between ARPKD and ADPKD.
Clinical spectrum and pathology
ARPKD (MIM 263200) was first recognized as a distinct morphologic form of cystic disease in 1902 (Osathanondh and Potter 1964a). In a landmark study, Blyth and Ockenden (1971) classified ARPKD into four distinct phenotypes (perinatal, neonatal, infantile, and juvenile) on the basis of clinical manifestations and the age at presentation. The classifications focused on the wide spectrum of renal CT and biliary ductal plate abnormalities. These ranged from nephromegaly and the oligohydramnois sequence with minimal biliary abnormalities (“perinatal”) to severe portal hypertension with minimal renal cystic disease (“juvenile”). Prior to advances in molecular genetics and the identification of the ARPKD disease gene, the four groups were thought to be distinct disease entities caused by different mutant genes.
ARPKD belongs to a group of congenital hepatorenal fibrocystic syndromes characterized by dual renal and hepatic involvement of variable severity. Renal manifestations are characterized by both ectasia and cystic dilation of renal CT. The fusiform dilated CT are lined by undifferentiated epithelium and surrounded by abnormal deposition of extracellular matrix. Unlike ADPKD cysts, renal cystic lesions in ARPKD retain both their afferent and efferent connections (Bernstein and Slovis 1992; Dell et al. 2004; Osathanondh and Potter 1964c). The kidney appears spongy, and there is no clear separation of the cortex and medulla. From 10% to 90% of CT are affected, resulting in wide variability of renal dysfunction. Depending on the number of CT involved, the kidneys may be massively enlarged. Histological analysis of the kidney reveals multiple subcapsular cystic lesions corresponding to radially oriented, ectatic CTs (Osathanondh and Potter 1964a, 1964b).
Liver disease is an invariable feature of ARPKD. The manifestations generally vary according to the patient’s age at presentation (Blyth and Ockenden 1971; Dell et al. 2004). The liver disease consists of biliary dysgenesis and periportal fibrosis (congenital hepatic fibrosis; CHF). Dilation of both intrahepatic (Caroli’s syndrome) and extra-hepatic bile ducts (extra-HBD) and CHF lead to recurrent ascending cholangitis and portal hypertension with splenomegaly and esophageal varices (Blyth and Ockenden 1971; Davis et al. 2003; Dell and Avner 2003; Desmet 1992; Guay-Woodford and Desmond 2003; Harris and Rossetti 2004; Jorgensen 1977).
Despite advances in neonatal care, the short-term and long-term morbidity and mortality of ARPKD remain substantial. Notwithstanding the variable clinical spectrum of ARPKD, the majority of patients are identified either in utero or at birth (Dell et al. 2004; Guay-Woodford and Desmond 2003). The most severely affected fetuses have enlarged echogenic kidneys and oligohydramnios attributable to poor fetal renal output. The only signs potentially detectable in utero are enlargement and increased echogenicity of both kidneys. However such findings are often not present during second trimester fetal sonography (Zerres et al. 1998). Pulmonary hypoplasia, resulting from oligohydramnios, occurs in the majority of affected infants and is a major cause of morbidity and mortality in the newborn period. Improved respiratory treatment leads to increased neonatal survival, but death still occurs in the neonatal period in approximately 25%–30% of affected individuals, primarily because of respiratory insufficiency (Kaariainen et al. 1988; Kaplan et al. 1989; Roy et al. 1997). The prognosis is more optimistic for ARPKD patients surviving the neonatal period (Roy et al. 1997).
Approximately 50% of affected individuals progress to end-stage renal disease (ESRD) within the first decade of life (Cole et al. 1987; Roy et al. 1997). Modern neonatal respiratory support and renal replacement therapies have improved the 10-year survival rate of patients who survive the first year to 82% (Dell and Avner 2003). For infants who survive the perinatal period, a wide range of associated morbidities can evolve, including systemic hypertension, renal failure, portal hypertension, and fibrosis of both the liver and kidneys (Davis et al. 2003; Dell et al. 2004; Guay-Woodford 1996). The 15-year survival rate is estimated to be 67%–79% (Dell and Avner 2003).
A minority of individuals present as older children, usually with hepatosplenomegaly as the presenting feature. Later childhood presentation is usually associated with less renal enlargement and more variability in cyst size (Blyth and Ockenden 1971). As described above, these patients are more likely to develop complications of CHF and Caroli’s disease and may require porto-systemic shunting. In rare cases, sequential or simultaneous liver-kidney transplants can be considered a viable therapeutic option (Davis et al. 2003). The clinical spectrum of ARPKD has been expanded by a recent study involving molecular characterization. Almost 1/3 of the patients presenting with classical hepatic phenotype and documented mutations in the PKHD1 gene are over 20 years old at the time of diagnosis (Adeva et al. 2006).
Hypertension may occur in up to 80% of children with ARPKD, is frequently severe, and is generally correlated with decreased renal function. The mechanisms of hypertension are unknown, although increased intravascular volume secondary to dysregulation of renal sodium transport and activation of the renin-angiotension axis have been implicated (Kaplan et al. 1989). Therapies with angiotension converting enzyme inhibitors or ATII receptor inhibitors are generally effective (Guay-Woodford and Desmond 2003; Jafar et al. 2005; Kaplan et al. 1989). Additional clinical complications include nephrogenic diabetes insipidus, failure to thrive, and hyponatremia (Dell et al. 2004; Guay-Woodford and Desmond 2003; Zerres et al. 1996).
Genetics
Localization, identification, and expression of PKHD1
Despite the variable clinical spectrum of ARPKD (MIM 263200), genetic linkage studies indicate that mutations at a single locus, PKHD1, mapped to human chromosome region 6p21.1–p12 are responsible for all phenotypes of ARPKD (Guay-Woodford et al. 1995; Zerres et al. 1998). PKHD1 has recently been cloned by several independent groups (Onuchic et al. 2002; Ward et al. 2002; Xiong et al. 2002).
PKHD1 is amongst the largest disease genes identified to date in the human genome, with a complicated transcription profile that probably generates multiple variants of the fibrocystin protein. The PKHD1 gene spans approximately 470 kb of genomic DNA, and an estimated minimum of 86 exons (12,222 bp) are assembled into a variety of alternatively spliced transcripts ranging from 9–16 kb (Losekoot et al. 2005; Onuchic et al. 2002). Both PKHD1 and its mouse ortholog (Pkhd1) encode a complex and extensive array of splice variants, with most abundant transcriptional expression in fetal and adult kidney and weaker expression in other tissues including liver and pancreas (Nagasawa et al. 2002; Onuchic et al. 2002; Ward et al. 2002). Northern and reverse transcription/polymerase chain reaction (RT-PCR) analyses of the murine transcript have also detected low level expression in the lung, testis, vascular smooth muscle, sympathetic ganglia, and trachea, most likely the result of alternative splice variants (Nagasawa et al. 2002). The degree of alternative splicing and differential tissue-specific expression of the various splice forms requires further analysis.
The longest PKHD1 transcript includes 67 exons with an ORF composed of 66 exons extending from exons 2–67 and encodes a unique 4074-amino-acid protein. The longest ORF of the mouse ortholog (Pkhd1) encodes a protein of 4059 amino acids; the mouse and human protein sequences are 73% identical overall and 55% identical in the carboxyl-terminal tail (Nagasawa et al. 2002). Although both the human and mouse proteins have large ORFs, the expressed proteins appear to represent a number of splice variants with tissue-specific patterns of expression (Ward et al. 2003). In addition, a truncated form of PKHD1, missing the final seven exons and a novel terminal exon, has been reported (Xiong et al. 2002).
Characterization of the PKHD1 mutational spectrum
An anticipated benefit from the identification of a human disease gene is the ability to understand the molecular basis of clinical variations in disease presentation, progression, and outcome. Genotype-phenotype correlations should enhance the understanding of the molecular pathogenesis of the disease and provide improved patient care by predicting disease progression. However, the combination of the large size of PKHD1, allelic heterogeneity, the high level of missense mutations, and the complicated pattern of splicing poses significant challenges to DNA-based diagnostic testing.
The mutation detection rate obtained in separate studies has varied substantially, from 42% to 65%, based primarily on the cohort of patients screened within the respective studies (Bergmann et al. 2003, 2004a,b; Onuchic et al. 2002; Ward et al. 2002). These studies have demonstrated that mutations are scattered throughout the gene without evidence of clustering at specific sites, and neutral polymorphisms are common. Most mutations are unique to single families (“private mutations”), and most affected patients represent compound heterozygotes. Approximately 45% of the changes are predicted to truncate the protein. All missense mutations are non-conservative, with the affected amino acid residues found to be conserved in the murine fibrocystin ortholog. One recurrent mutation, thr36 to met (T36M; 606702.0001), is thought to represent a mutation hotspot and has been found in a variety of populations (Bergmann et al. 2003). Two founder mutations, arg496 to thr (R496×; 606702.0007) and val3471 to gly (V3471G; 606702.0008), comprise approximately 60% of PKHD1 mutations in a Finnish population (Bergmann et al. 2003).
However, more recent studies with new algorithms have yielded detection rates of 85% for the entire clinical spectrum of ARPKD patients (Bergmann et al. 2005a,c; Losekoot et al. 2005; Sharp et al. 2005). Strategies based on denaturing high performance liquid chromatography (DHPLC) and PCR have been successfully used for mutation screening. In general, sequence analysis, combined with haplotype analysis in multiply affected families, has proven to provide the most reliable and comprehensive results. To date, however, only micromutations in the 66 exons encoding the longest ORF have been described and account for approximately 85% of mutations (Bergmann et al. 2005b). As of January 22, 2006, 305 different PKHD1 micromutations (point mutations and small deletions/duplications/insertions) on 670 mutated alleles had been listed in the locus-specific database http://www.humgen.rwthaachen.de).
Bergmann et. al. recently described the use of DHPLC for alternatively spliced exons and quantitative real time polymerase chain reaction to detect genomic imbalances (Bergmann et al. 2005b). In 58 patients, three different heterozygous PKHD1 deletions and several single nucleotide changes in alternatively spliced exons were identified, suggesting that gross deletions in PKHD1 account for a detectable proportion of ARPKD cases.
Prenatal diagnosis and genotype-phenotype correlations
Because of the significant morbidity and mortality of ARPKD, many parents of ARPKD children seek prenatal diagnosis to guide future family planning. Prior to the identification of the PKHD1 gene, prenatal diagnosis was only feasible by indirect genotyping (haplotype-based analysis). However, interpretation of haplotype-based analysis is difficult in cases without an unambiguous clinicopathologic diagnosis. If parental renal, hepatic, or pancreatic ultrasound reveals cyst formation, severe, early onset ADPKD must be considered. Thus, in families with diagnostic uncertainties, characterization of PKHD1 mutations by direct sequencing is the only option for accurate genetic counseling and prenatal diagnosis. Prenatal diagnoses of ARPKD based on PKHD1 mutation analysis has been successfully accomplished (Zerres et al. 2004), and clinical gene-based testing will probably become more widely available as the number of diagnostic centers increases. In addition, in vitro fertilization following pre-implantation genetic diagnosis will also become available.
Although the improved algorithms provide high detection rates, the ability definitively to assess the likelihood of micromutations being pathogenic is still lacking. In order to predict whether a variant is likely to be disease-causing, several factors are taken into account: evolutionary conservation of the amino acids, class of amino acid, no other mutations in cis, co-segregation with the disease, and/or previously published pathogenic variant.
Obviously, the correct classification of missense variants as pathogenic mutations or harmless polymorphisms is crucial for a correct diagnosis. In addition, the roles of the various splice forms in determining disease severity have yet to be determined. A potential biological function of the alterations identified in alternatively spliced exons must await the confirmation and definition of transcripts containing these alternative spliced exons and their predicted reading frames. Continued cataloging of mutations, especially pathogenic missense variants, and the resulting disease phenotypes will be required to define genotype-phenotype correlations fully.
However, the bulk of mutational data currently available permit broad categorization of missense mutations as severe, moderate, or mild changes. Therefore, genotype-phenotype correlations can be drawn for the type of mutation rather than for the site of individual mutations. All patients carrying two truncating mutations display a severe phenotype with peri- or neonatal demise (Bergmann et al. 2003), whereas patients surviving the neonatal period have, on average, at least one missense mutation (Furu et al. 2003). This indicates that some missense changes may not entirely inactivate the product, but rather generate a hypomorphic allele. Additionally, some missense mutations may only disrupt specific splice forms of PKHD1 without affecting other functional variants (Rossetti et al. 2003).
Regulation of PKHD1 transcription
Insights into the tissue-specific transcriptional regulation of PKHD1 are becoming available with the recent discovery of an evolutionarily conserved transcription factor (TCF)-binding site in the proximal promoter of the mouse Pkhd1 gene (Hiesberger et al. 2004). TCF-2, or hepatocyte nuclear factor-1β (HNF-1β), is a Pit-1/Oct-1/Unc-86 (POU)/homeodomain-containing transcription factor that regulates tissue-specific gene expression in the kidney, liver, pancreas, and other epithelial organs (Igarashi et al. 2005). Mutations of HNF-1β have been shown to produce maturity-onset diabetes of the young type 5 (MODY5) and are associated with congenital cystic abnormalities of the kidney. Igarashi et. al (2005) have recently demonstrated that kidney-specific deletion of HNF-1β by using Cre/loxP recombination results in renal cyst formation paralleled by a 70% decrease in Pkhd1 expression. These studies demonstrate that HNF-1β is required for the development of the mammalian kidney, and that HNF-1β directly regulates the Pkhd1 promoter.
The ARPKD protein, fibrocystin
Fibrocystin is a novel integral membrane protein of 4074 amino acids (4059 amino acids in the mouse) with a signal peptide, an extensive (3860 amino acid) highly glycosylated, N-terminal extracellular region, a single transmembrane (TM) domain, and a short (192 amino acid) cytoplasmic tail containing four putative cAMP/cGMP phosphorylation sites (Nagasawa et al. 2002; Onuchic et al. 2002; Ward et al. 2002; Fig. 1). However, Northern expression analysis suggests the likelihood that splice forms of PKHD1 generate alternative proteins, including secreted products (Nagasawa et al. 2002; Onuchic et al. 2002).

Predicted structure of fibrocystin, a large integral membrane protein, with the largest known open reading frame, translating a 4074-amino-acid novel peptide (4059 amino acids in mouse). Fibrocystin has homologies to several known proteins, including TIG/IPT (immunoglobulin-like fold shared by plexins and transcription factors) or TIG-like domains in the extracellular region. This domain has 80–100 amino acids and is found in several receptor molecules, including Met, various plexins, and Ron, although the large number of domains seems unique to the fibrocystins. The structure of fibrocystin as an integral membrane protein with a large extracellular portion containing multiple glycosylation sites, and a short intracellular C-terminal domain containing potential protein kinase A and PCK phosphorylation sites suggest that this protein acts as a transducer of extracellular information into the cell by eliciting signal transduction cascades resulting in the modulation of gene transcription (aa amino acids)F
Alternatively spliced transcripts of PKHD1 are predicted to fall into two broad categories. The first is predicted to contain the single TM element but to vary with respect to the insertion of other peptide domains. The second category would include transcripts that lack the transmembrane-spanning domain and may therefore be secreted. This complex pattern of splicing has been found to be highly conserved in the murine ortholog (Nagasawa et al. 2002). Multiple variants of fibrocystin generated from alternatively spliced transcripts of PKHD1 may provide a mechanism for regulating the temporal and spatial functions of fibrocystin isoforms in a tissue-specific manner (Onuchic et al. 2002; Ward et al. 2002).
Fibrocystin has homologies to several known proteins; this provides some clues to the function of the protein. The large amino-terminal extracellular portion contains domains with TIG, TIG-like, TMEM2, and DKFZ homologs (Wang et al. 2004). The predicted extracellular domain contains 10–12 TIG/IPT (IgG-like, plexin, transcription factor) or TIG-like domains that are shared by RON, the hepatocyte growth factor receptor, MET, and the plexin superfamily involved in the regulation of cellular adhesion, repulsion, and proliferation. In addition, the extracellular region of fibrocystin contains TMEM2 and DFKZ homologies, plus multiple PbH1 (parallel beta-helix 1) repeats that may bind to carbohydrate moieties such as glycoproteins on the cell surface or in the basement membrane.
The function of this protein remains to be determined, but the structure and homologies suggest a role as a receptor protein involved in the regulation of cellular adhesion, repulsion, and proliferation and/or the regulation and maintenance of renal CT and HBD (Bergmann et al. 2003; Onuchic et al. 2002; Ward et al. 2002). Delineation of the normal role of fibrocystin in the development, differentiation, and maintenance of renal CT and HBD will provide important insights into the way that mutations in a single gene cause cyst formation with a wide variation in cellular and clinical phenotype.
When fibrocystin was initially identified, there were no known related proteins until the identification of fibrocystin-L (Hogan et al. 2003). This protein is encoded by the PKHDL1 gene at chromosome region 8q23. Fibrocystin-L (4243 amino acids; 466 kDa) has homology (overall identity of 25.0% and similarity of 41.5%) within the extracellular region to fibrocystin. Despite the similarity of this protein to fibrocystin, mutation analysis indicates that it is not associated with ARPKD or any renal or biliary phenotype. Indeed, expression studies suggest a possible role in cellular immunity with up-regulation in activated T-cells (Hogan et al. 2003).
Fibrocystin expression
During embryogenesis, PKHD1 is expressed in epithelial derivatives, including ureteric bud branches, intra- and extra-HBD, and pancreatic ducts (Menezes et al. 2004; Ward et al. 2003). This temporal and spatial expression pattern of PKHD1 parallels the CT and biliary epithelial pathology of ARPKD.
Immunohistohemical analysis demonstrates that fibrocystin is expressed along the apical and upper lateral cell surface and within the cytoplasm in cortical and medullary CT epithelia (Menezes et al. 2004; Nagano et al. 2005; Wang et al. 2004), biliary and pancreatic ductal epithelia, and thick ascending limbs of Henle (Menezes et al. 2004; Zhang et al. 2004). Immunofluorescence studies have revealed that, like other cystoproteins including the ADPKD proteins PC-1 and PC-2, fibrocystin co-localizes in centrosomes, basal bodies or primary apical cilia in renal epithelial cells (Menezes et al. 2004; Wang et al. 2004; Ward et al. 2003; Zhang et al. 2004) and in the cilia of cholangiocytes of intra-HBD (Masyuk et al. 2003).
The precise pattern of staining appears to vary with the stage of cellular differentiation (Menezes et al. 2004; Nagano et al. 2005; Zhang et al. 2004). This variation may be attributable solely to differences in antibody specificity and technical aspects of the fixation and/or staining procedures employed. Alternatively, differences in antibody specificity combined with the temporal and spatial expression of alternatively spliced isoforms of fibrocystin may represent a regulatory mechanism inherent to the biological function of fibrocystin in renal and biliary epithelia.
The general genotype-phenotype correlations outlined above suggest that severe manifestations of ARPKD are generally attributable to the presence of two truncating mutations in PKHD1 resulting in the absence of fibrocystin expression. This appears to be supported by study reports that ARPKD tissue samples lack fibrocystin expression (Ward et al. 2002; Zhang et al. 2004). Evidence from our laboratory does not support this generalization. Immunohistochemical staining of freshly processed renal samples from severely affected ARPKD patients with well characterized monoclonal antibodies generously provided by Dr. Christopher Ward (Ward et al. 2003) and affinity-purified polyclonal antibodies generated in our laboratory has demonstrated equivalent robust fibrocystin expression in CT cystic lesions (Fig. 2). These results suggest that not all severely affected patients lack fibrocystin expression and that collection, processing, and fixation are critical variables that may hamper the detection of fibrocystin. Additional immunohistological and Western analysis, especially from severely affected patients, in conjunction with mutational analysis, may provide important insights into the complexity of temporal and spatial expression of fibrocystin splicing variants.

Immunohistochemical localization of fibrocystin. Sections of human fetal kidneys from severe ARPKD demonstrate robust membrane localization on the apical and basolateral membranes of lectin-identified collecting tubular cysts with two different antibodies to fibrocystin. Cell and tissue lysates subjected to Western analysis demonstrate that both antibodies recognize equivalent MW proteins of >440 kDa and 200 kDa (data not shown). Thus, fibrocystin expression is not limited to cilia, basal bodies, or centrosomes and can be expressed even in severe ARPKD
Cilia and multimeric protein complexes in PKD
Observations initially made in animal models of RCD showing that protein products of cyst-causing genes, such as polaris (Tg737), cystin (cpk), and inversin (Nphp2), were in part localized to the primary cilia of renal epithelia led to the “primary cilia” hypothesis (Guay-Woodford 2003). Simply stated, the hypothesis is that structural or functional abnormalities in the primary apical cilia of tubular epithelia play a role in renal cyst development (Pazour and Rosenbaum 2002). This suggests that the cilia serve as an organizing center for the early steps of signal transduction pathways that are responsible for monitoring the integrity of the nephron and bile ducts and participate in regulating epithelial proliferation and differentiation.
Structurally, cilia consist of a microtubule-based axoneme covered by a specialized plasma membrane. The ciliary axoneme is built from one of two basal bodies that form the core of the centrosome and extend from the cell surface into the extracellular space (Praetorius and Spring 2005). Cilia are defined as motile, primary, or nodal (for a comprehensive review, see Praetorius and Spring 2005).
Primary cilia are non-motile hair-like structures that emerge as single projections from one of the two basal bodies or centrioles (Pazour 2004). Researchers have recognized for decades that individual cilia appear, at least temporarily, on most cells in the body, but most cell biologists have dismissed them as evolutionary remnants. This assertion has recently been challenged by the observations that the three proteins associated with human PKD, including the ADPKD proteins, PC-1 and PC-2, and the ARPKD protein fibrocystin, are expressed in cilia and in other apical, lateral, and basal locations.
Additional cystoproteins responsible for non-PKD forms of human RCD, such as nephronophthisis (nephrocystin-1, nephrocystin-2, and nephrocystin-4) and Bardet-Biedl syndrome (BBS 1–8), are also partially expressed in the basal bodies and/or the primary cilia (Hildebrandt and Otto 2005). These findings indicate a striking association between proteins that are involved in RCD (cystoproteins) and cilia (Igarashi and Somlo 2002).
Additional compelling data linking cilia structure and/or function in renal cyst formation cyst development have been reported in studies of mice with mutations in Kif3a. Lack of Kif3a in mice produces embryonic lethality; however, conditional disruption of the floxed allele of Kif3a in the kidney gives viable offspring with cystic lesions in the distal nephron and CTs, specifically where cre recombinase is expressed (Lin et al. 2003). This study has demonstrated that tissue-specific inactivation of Kif3A in renal tubular epithelial cells results in viable offspring that develop rapidly progressive CT cystic lesions leading to ESRD and death by postnatal day 21. The epithelial cells lining the cystic lesions exhibit the distinctive abnormal cellular phenotype associated with PKD, including increased proliferation, apical mislocalization of the epidermal growth factor receptor, and increased expression of β-catenin and c-Myc, and inhibition of p21CIP1 (Lin et al. 2003).
The pathogenic link between cystoprotein expression in cilia, basal bodies, and centrosomes, on the one hand, and the renal cystic phenotype, on the other hand, remains unknown. However, recent studies have demonstrated that physical manipulation of the primary cilium, including bending or removal, elicits changes in Ca2+ flux (Praetorius et al. 2003, 2004; Praetorius and Spring 2001, 2003a,b) demonstrating that cilia can function as mechanosensors or chemosensors to sense fluid movement or ionic composition in the kidney. The physiological consequence of alterations of calcium concentrations in the renal CT epithelium remains to be clearly defined, particularly in ARPKD. Of note, the only binding partner for fibrocystin identified to date is calcium-modulating cyclophilin ligand, a protein involved in Ca2+ signaling (Nagano et al. 2005).
Cyst development and growth is a complex multi-mechanistic process. Immunohistological and immunofluorescent analyses of fibrocystin expression demonstrate that fibrocystin is only partially localized to the basal body and primary ciliary of renal epithelia and that, like ADPKD proteins PC1 and PC2, fibrocystin is localized at additional sites. The expression of PKD proteins in cilia, basal bodies, and intercellular junctions and at focal adhesions suggests that there may be common signaling pathways for cyst formation through the abnormal integration of signal transduction pathways, as shown in Fig. 3 (Nauli et al. 2003; Wilson 2004a).

PKD proteins in multimeric complexes. Summary of recent advances suggesting that cystoproteins exist in multimeric protein complexes and function at distinct sites within epithelial cells that include: basal cell-matrix focal adhesion sites to regulate epithelial cell adhesion and migration; apical-lateral cell-cell adherens sites to regulate differentiated cell shape; within Golgi; apical-central ciliary sites to sense flow and regulate tubule diameter. Expression and signaling from multiple sites establishes signaling platforms and feedback loops that regulate cell growth, proliferation, differentiation, and dedifferentiation. Elucidation of the dynamic regulated signaling from these multiple sites are a current focus of investigation (AJ adherens junction, TJ tight junction)
Cellular pathophysiology of ARPKD, with comments on ADPKD
Despite the identification of the genes responsible for ADPKD and ARPKD, the precise function of these genes and their protein products remains incompletely characterized, because of the novel attributes of the genes (including the complexity of their structure), the large size of PKD1 and PKHD1, and the multiple transcripts produced by these genes. Despite these difficulties, the development of antibodies to PKD proteins coupled with the study of non-orthologous animal models of PKD have provided important insights regarding cystogenic pathways and cellular pathophysiology common to many cystic diseases (Guay-Woodford 2003; Nauli and Zhou 2004; Pazour et al. 2000, 2002; Phillips et al. 2004; Praetorius et al. 2003; Sun et al. 2004; Witzgall 2005; Yoder et al. 2002).
PKD cystic renal epithelia share common phenotypic abnormalities despite the different genetic mutations that underlie the disease. Numerous animal models and in vitro cell culture systems utilizing human and animal CT cells have established that the development of PKD is characterized by a switch from a well-differentiated non-proliferative reabsorptive epithelia to a partially dedifferentiated, secretory epithelia characterized by polarization defects and high rates of proliferation and apoptosis (Dell et al. 2004; Harris and Rossetti 2004; Murcia et al. 1999; Wilson 2004a,b). This is a critical functional change, since mathematical modeling of epithelia demonstrates that proliferation and secretion are necessary and sufficient to account for cyst growth in PKD (Welling and Grantham 1991). The fluid secreted from these abnormal cells is rich in cytokines, lipid factors, and epithelial growth factors, which further stimulate epithelial proliferation resulting in significant ductal ectasia (Grantham et al. 1995; MacRae Dell et al. 2004). Cultured epithelial cells from patients or animal models of PKD have consistently demonstrated an increased intrinsic capacity for proliferation and survival (Gabow 1993; Grantham 1996, 1987; Wilson 2004a).
cAMP-mediated proliferation
Mounting evidence suggests that the adenylyl cyclase-adenosine 3′,5′-cyclic monophosphate (cAMP) pathway promotes both fluid secretion and cell proliferation in both ADPKD and ARPKD renal epithelia. Mutated PKD proteins are thought to disrupt intracellular Ca2+ homeostasis or Ca2+ signaling leading to cellular dedifferentiation and hyperproliferation through an abnormal cAMP-mediated proliferative pathway.
In normal human and mouse renal epithelial cells, cAMP has been shown to inhibit the Ras/Raf-1/MEK/ERK pathway at the level of Raf-1 and to decrease cell proliferation. In contrast, cAMP has been shown to stimulate B-Raf and activate the MEK/ERK pathway in ARPKD and ADPKD cells leading to increased cell proliferation (Grantham 1997a,b; Sullivan and Grantham 1996; Wallace et al. 2002; Yamaguchi et al. 2000, 2003, 2004, 2006). Therefore, cAMP can be either mitogenic or anti-mitogenic.
In renal epithelia, the switch in cAMP from an anti-mitogen to a mitogenic stimulus has been shown to be directly correlated with intracellular calcium levels [Ca2+]i (Yamaguchi et al. 2004). The PI 3-K/Akt pathway has been shown to regulate cAMP-dependent activation of B-Raf/MEK/ERK and cell proliferation in CT principal cells (M-1; Yamaguchi et al. 2003). Phosphorylation of B-Raf by Akt has been shown to regulate B-Raf activity negatively (Guan et al. 2000; Zhang and Guan 2000). Yamaguchi et. al (2004) have demonstrated that reducing intracellular [Ca2+]i in immortalized mouse M-1 CT cells or primary human kidney epithelial (NHK) cells for 3–5 h converts both cell types from a normal cAMP growth-inhibited phenotype to an abnormal cAMP growth-stimulated phenotype, characteristic of PKD renal epithelia. Reduction of [Ca2+]i decreases Akt activity, allows cAMP-dependent B-Raf activation, and stimulates cell proliferation. Direct inhibition of either PI 3-K or Akt causes cAMP-dependent ERK activation and cell proliferation, thus inducing a phenotypic switch that imitates Ca2+ restriction (Yamaguchi et al. 2004). Pharmacological elevation of Ca2+ increases P-Akt levels, whereas Ca2+ channel blockers decrease P-Akt (Yamaguchi et al. 2006). This observation that the reduction of intracellular [Ca2+] in M-1 or NHK cells with calcium channel blockers replicates the abnormal proliferative response of PKD cells to cAMP establishes a link between the PKD proliferative phenotype, reduction in [Ca2+]i, and cAMP activity. Evidence for the role of [Ca2+]i-mediated proliferation has been further strengthen by studies showning that the elevation of [Ca2+]i levels in ADPKD and ARPKD cultured cells increases Akt activity and blocks cAMP-dependent B-Raf and ERK activation (Yamaguchi et al. 2006). The study has also demonstrated that increases in [Ca2+]i restore the normal anti-mitogenic response to cAMP in renal cells derived from either human ADPKD or ARPKD kidneys. These data suggest that the PKD proteins (PC1, PC2, and fibrocystin) play a role in maintaining Ca2+ homeostasis. Mutations in any of the PKD proteins may lead to reduce [Ca2+]i and activate the cAMP mitogenic pathway.
Epidermal growth factor receptor axis-mediated proliferation
Evidence from a number of laboratories has demonstrated a significant role for the epideraml growth factor receptor (EGFR) axis in promoting epithelial hyperplasia in cystic epithelia, with resultant renal cyst formation and progressive enlargement in both murine and human ADPKD and ARPKD (Du and Wilson 1995; Gattone et al. 1996; Lowden et al. 1994; Lu et al. 1999; Nauta et al. 1995; Neufield et al. 1992; Orellana et al. 1995; Pugh et al. 1995; Richards et al. 1998; Sweeney and Avner 1998; Sweeney et al. 2003; Torres 2004). Renal cystic epithelia demonstrate both quantitative (overexpression) and qualitative (mislocalization) expression of one or more members of the ErbB family of receptors as shown in Fig. 4. In addition, evidence from rodent models (including the orthologous polycystic kidney rat; PCK rat) suggests that similar abnormalities of the EGFR axis may mediate biliary epithelial hyperplasia and biliary ductal ectasia (Nauta et al. 1995; Sato et al. 2005; Sweeney et al. 2000, 2003). EGF has an important role in the expansion of renal cysts. Cystic epithelial cells from patients with ARPKD or ADPKD are unusually susceptible to the proliferative stimulus of EGF. Moreover, cyst fluid of patients contains mitogenic concentrations of EGFR ligands, which are secreted into the lumen of cysts in amounts that can induce cellular proliferation (Klingel et al. 1992; Lakshmanan and Fisher 1993; Neufield et al. 1992; Rohatgi et al. 2004; Sullivan et al. 1998; Sweeney and Avner 1996; Ye et al. 1992). In all animal models studied to date, abnormalities in the expression and localization of members of the EGFR axis have been reported. Cyst-causing genes on different chromosomes, irrespective of whether they are orthologous with human PKD genes, result in cystic epithelia with EGFR-axis abnormalities. This includes the previously noted Kif3A cystic model that has a tissue-specific inactivation of Kif3A in renal tubular epithelial cells. As previously noted, Kif3A inactivation results in cystic lesions lined by epithelia that demonstrate pronounced apical expression of EGFR.

Immunohistochemical localization of ErbB family receptors. a Basolateral staining of EGFR (ErbB1) in a normal murine kidney section. b–d Cystin (CPK) deficiency, B-cell progenitor kinase (BPK) deficiency, and human ARPKD, respectively, demonstrate increased expression and apical mislocalization of EGFR (ErbB1) in lectin-identified collecting tubules. Note normal basolateral localization in non-cystic tubules within sections in b, c. e, f PKD and human ARPKD, respectively, demonstrate high expression and apical localization of ErbB2. Bars 0.1 mM
A recently developed murine model of ADPKD that produces reduced levels of full-length Pkd1-encoded protein, PC-1, develops renal cystic lesions suggesting that reduced Pkd1 expression is sufficient for renal cyst formation in ADPKD. This model demonstrates up-regulation of heparin-binding epidermal growth factor-like growth factor accompanied by increased protein levels and activity (phosphorylation) of EGFR (Jiang et al. 2006).
These data indicate that abnormalities in the EGFR axis are a common cellular phenotype downstream from a number of different primary gene mutations. Despite interesting speculations, the precise mechanisms of the striking relationship remain unknown (Dell et al. 2004; Du and Wilson 1995; Grantham 2000; Lu et al. 1999; Sweeney et al. 2003; Wilson 2004b).
The overexpression and abnormally located EGFRs on the apical (luminal) surface of cyst-lining epithelia in human ARPKD and murine models have been shown specifically to bind EGF/TGF-α with high affinity, to autophosphorylate, and to generate mitogenic signals in response to EGF and/or cyst fluid (Sweeney and Avner 1996, 1998). This creates a sustained cycle of autocrine-paracrine stimulation of proliferation in cysts similar to that seen in many forms of cancer.
Secretion
ADPKD cysts have a fundamental structural difference from ARPKD cysts. ADPKD cysts rapidly close off from urinary flow and continue to expand by transepithelial secretion. As stated above, ARPKD cysts remain open with respect to the entire nephron, maintaining both afferent and efferent tubular connections. However, secretion is still a necessary element of cyst formation in ARPKD, and the difference in cyst structure suggests a different secretory mechanism.
In ADPKD, the weight of the evidence indicates that Cl− is secreted via a cAMP-mediated co-transport mechanism in the basolateral membrane and the cystic fibrosis transmembrane conductance regulator (CFTR) in the apical membrane, leading to expansion of cysts, especially after they have detached from the nephron of origin (Belibi et al. 2004; Sullivan and Grantham 1996). This does not appear to be the case in ARPKD. By using a genetic complementation approach, the BPK mouse (deficient in B-cell progenitor kinase) has been crossed with a CFTR knockout mouse (Clarke et al. 1992). The results demonstrate that the absence of CFTR does not alter the course of renal or biliary cyst development or growth (Nakanishi et al. 2001).
ARPKD cystic epithelia have been shown to exhibit net fluid secretion through a decrease in principal cell sodium absorption. This is attributable to an EGF-mediated decrease in the alpha-subunit of the epithelial Na channel (Veizis et al. 2004; Veizis and Cotton 2005). However, conflicting data has been reported (Rohatgi et al. 2003).
Biliary epithelia in ARPKD have been shown to be hyper-proliferative in response to EGF (Nauta et al. 1995; Sato et al. 2005). However, the effect of EGF hypersentivity in biliary epithelia on secretory mechanisms is unclear.
Translational implications
A critical test of the veracity of proposed pathophysiology is the effectiveness of targeted therapy on modulating disease. To date, no specific therapies that limit renal cyst development and progressive enlargement or biliary ductal ectasia in ARPKD are in clinical use (Davis et al. 2001). However, the studies noted above suggest that therapeutic targeting of the cAMP and EGFR-axis abnormalities documented in cystic epithelia may translate into effective therapies for the proliferation and secretory abnormalities of ARPKD (and, by analogy, ADPKD).
To date, decreasing cAMP through vasopressin V2 receptor (VPV2R) antagonism in renal CTs has demonstrated efficacy in animal models of PKD orthologous to human diseases (ARPKD → PCK rat: adolescent nephronophthisis → pcy mouse; ADPKD → pkd2ws25/− mouse; Gattone et al. 2003; Torres 2005; Torres et al. 2004). However, because of the restricted expression of the VPV2R on renal principal cells, such approaches are limited to the renal lesions of nephronophthisis and may provide only partial benefit in such multisystem diseases as ADPKD, and particularly ARPKD.
Therapeutic targeting of the EGFR axis has promise in ADPKD, particularly in the dual renal and biliary cell abnormalities in ARPKD. To date, genetic complementation to decrease EGFR (Richards et al. 1998), the inhibition of EGFR, c-ErbB2, and c-ErbB4 through small molecule covalent inhibitors (Dell et al. 2001; Sweeney et al. 2000, 2003), and the inhibition of EGFR ligand availability, alone and in combination with EGFR tyrosine kinase activity inhibitors (Dell et al. 2001; Sweeney et al. 2003) have demonstrated dramatic efficacy in ameliorating renal CT abnormalities, biliary ductal ectasia, and periportal fibrosis in a variety of rodent ARPKD models. Similarly, therapeutic targeting of substrates downstream from the EGFR, such as MAPK/MEK5/ERK5/MEK1/2 are effective in ameliorating biliary dysgenesis of the PCK rat, in vitro (Sato et al. 2005).
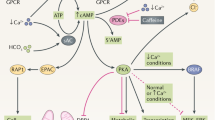
A particularly promising approach appears to be the specific targeting of downstream intermediates of both the cAMP- and EGFR-axis-mediated pathways. As shown in Fig. 5, cSrc (pp66Src) is one such component that interacts with critical pathophysiological pathways and exists at multiple locations in proximity to multermeric polycystic protein complexes. Preliminary studies targeting cSrc with a specific inhibitor (WY-606; Boschelli et al. 2005a,b) have been particularly effective in ameliorating both renal and biliary epithelial abnormalities in ARPKD models (Gunay-Aygun et al. 2006).

Signaling pathways involved in PKD pathogenesis. Both cAMP- and EGFR-axis-mediated pathways have been shown to be involved in the development and progression of PKD. Representation of the way that these pathways may intersect, and the manner in which c-Src (pp66Src) may act as a downstream intermediate of both the cAMP- and EGFR-axis-mediated pathways. Proteins such as c-Src that interact with critical pathophysiological pathways and exist at multiple locations in proximity to multermeric polycystic protein complexes may provide attractive therapeutic targets (ENac epithelial Na channel, CFTR cystic fibrosis transmembrane conductance regulator, PKA protein kinase A, MMP’s matrix metalloproteinases, HB-EGF heparin-binding epidermal growth factor-like growth factor, AC adenylate cyclase, MEK MAP/ERK kinase)
Concluding remarks
The delineation of the molecular and cellular pathophysiology of ARPKD has led to a new era of therapeutic innovation through targeting specific abnormalities of cystic epithelia. Such innovations bring great hope to patients with RCD, and particularly children devastated by the dual organ pathophysiology of ARPKD.
References
Adeva M, El-Youssef M, Rosetti S, Kamath PS, Kubly V, Consugar MB, Milliner DM, King BF, Torres VE, Harris PC (2006) Clinical and molecular characterizations defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine 85:1–21
Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM Jr, Grantham JJ (2004) Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int 66:964–973
Bergmann C, Senderek J, Sedlacek B, Pegiazoglou I, Puglia P, Eggermann T, Rudnik-Schoneborn S, Furu L, Onuchic LF, De Baca M, Germino GG, Guay-Woodford L, Somlo S, Moser M, Buttner R, Zerres K (2003) Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J Am Soc Nephrol 14:76–89
Bergmann C, Senderek J, Kupper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schoneborn S, Kirfel J, Furu L, Onuchic LF, Rossetti S, Harris PC, Somlo S, Guay-Woodford L, Germino GG, Moser M, Buttner R, Zerres K (2004a) PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat 23:453–463
Bergmann C, Senderek J, Schneider F, Dornia C, Kupper F, Eggermann T, Rudnik-Schoneborn S, Kirfel J, Moser M, Buttner R, Zerres K (2004b) PKHD1 mutations in families requesting prenatal diagnosis for autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat 23:487–495
Bergmann C, Kupper F, Dornia C, Schneider F, Senderek J, Zerres K (2005a) Algorithm for efficient PKHD1 mutation screening in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat 25:225–231
Bergmann C, Kupper F, Schmitt CP, Vester U, Neuhaus TJ, Senderek J, Zerres K (2005b) Multi-exon deletions of the PKHD1 gene cause autosomal recessive polycystic kidney disease (ARPKD). J Med Genet 42:e63
Bergmann C, Senderek J, Windelen E, Kupper F, Middeldorf I, Schneider F, Dornia C, Rudnik-Schoneborn S, Konrad M, Schmitt CP, Seeman T, Neuhaus TJ, Vester U, Kirfel J, Buttner R, Zerres K (2005c) Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int 67:829–848
Bernstein J, Slovis TL (1992) Polycystic diseases of the kidney. In: Edelmann C (ed) Pediatric kidney diseases. Little, Brown, Boston, pp 1139–1157
Blyth H, Ockenden BG (1971) Polycystic disease of kidneys and liver presenting in childhood. J Med Genet 8:257–284
Boschelli DH, Wu B, Barrios Sosa AC, Chen JJ, Golas JM, Boschelli F (2005a) Inhibition of Src kinase activity by 7-[(2,4-dichloro-5-methoxyphenyl)amino]-2-heteroaryl-thieno[3,2-b]pyridine-6-carbonitriles. Bioorg Med Chem Lett 15:4681–4684
Boschelli DH, Wu B, Barrios Sosa AC, Durutlic H, Chen JJ, Wang Y, Golas JM, Lucas J, Boschelli F (2005b) Synthesis and Src kinase inhibitory activity of 2-phenyl- and 2-thienyl-7-phenylaminothieno[3,2-b]pyridine-6-carbonitriles. J Med Chem 48:3891–3902
Clarke LL, Grubb BR, Gabriel SE, Smithies O, Koller BH, Boucher RC (1992) Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science 257:1125–1128
Cole BR, Conley SB, Stapleton FB (1987) Polycystic kidney disease in the first year of life. J Pediatr 111:693–699
Davis ID, MacRae Dell K, Sweeney WE, Avner ED (2001) Can progression of autosomal dominant or autosomal recessive polycystic kidney disease be prevented? Semin Nephrol 21:430–440
Davis ID, Ho M, Hupertz V, Avner ED (2003) Survival of childhood polycystic kidney disease following renal transplantation: the impact of advanced hepatobiliary disease. Pediatr Transplant 7:364–369
Dell K, Avner E (2003) Autosomal recessive polycystic kidney disease gene reviews; genetic disease online reviews at gene tests-gene clinics. University of Washington, Seattle
Dell KM, Nemo R, Sweeney WE, Levin JI, Frost P, Avner ED (2001) A novel inhibitor of tumor necrosis factor-alpha converting enzyme ameliorates polycystic kidney disease. Kidney Int 60:1240–1248
Dell K, McDonald R, Watkins SL, Avner ED (2004) Polycystic kidney disease. In: Avner ED, Harmon WE, Niaudet P (eds) Pediatric nephrology. Lippincott Williams & Wilkins, Philadelphia, pp 675–699
Desmet VJ (1992) Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation”. Hepatology 16:1069–1083
Du J, Wilson PD (1995) Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Physiol 269:C487–C495
Furu L, Onuchic LF, Gharavi A, Hou X, Esquivel EL, Nagasawa Y, Bergmann C, Senderek J, Avner E, Zerres K, Germino GG, Guay-Woodford LM, Somlo S (2003) Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J Am Soc Nephrol 14:2004–2014
Gabow PA (1993) Autosomal dominant polycystic kidney disease. N Engl J Med 329:332–342
Gattone VH 2nd, Kuenstler KA, Lindemann GW, Lu X, Cowley BD Jr, Rankin CA, Calvet JP (1996) Renal expression of a transforming growth factor-alpha transgene accelerates the progression of inherited, slowly progressive polycystic kidney disease in the mouse. J Lab Clin Med 127:214–222
Gattone VH 2nd, Wang X, Harris PC, Torres VE (2003) Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9:1323–1326
Grantham JJ (1996) The etiology, pathogenesis, and treatment of autosomal dominant polycystic kidney disease: recent advances. Am J Kidney Dis 28:788–803
Grantham JJ (1997a) Mechanisms of progression in autosomal dominant polycystic kidney disease. Kidney Int Suppl 63:S93–S97
Grantham JJ (1997b) Renal cell proliferation and the two faces of cyclic adenosine monophosphate. J Lab Clin Med 130:460–469
Grantham JJ (2000) Time to treat polycystic kidney diseases like the neoplastic disorders that they are. Kidney Int 57:339–340
Grantham JJ, Geiser JL, Evan AP (1987) Cyst formation and growth in autosomal dominant polcystic kidney disease. Kidney Int 31:1145–1152
Grantham JJ, Ye M, Davidow C, Holub B, Sharma M (1995) Evidence for a potent lipid secretagogue in the cyst fluids of patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 6:1242–1249
Guan KL, Figueroa C, Brtva TR, Zhu T, Taylor J, Barber TD, Vojtek AB (2000) Negative regulation of the serine/threonine kinase B-Raf by Akt. J Biol Chem 275:27354–27359
Guay-Woodford L (1996) Autosomal recessive polycystic kidney disease. In: Watson M, Torres VH (eds) Polycystic kidney disease. Oxford University Press, New York
Guay-Woodford LM (2003) Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol 285:F1034–F1049
Guay-Woodford LM, Desmond RA (2003) Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics 111:1072–1080
Guay-Woodford LM, Muecher G, Hopkins SD, Avner ED, Germino GG, Guillot AP, Herrin J, Holleman R, Irons DA, Primack W, Thomson PD, Waldo FB, Lunt PW, Zerres K (1995) The severe perinatal form of autosomal recessive polycystic kidney disease maps to chromosome 6p21.1–p12: implications for genetic counseling. Am J Hum Genet 56:1101–1107
Gunay-Aygun M, Avner ED, Bacallao RL, et al (2006) Autosomal recessive polycystic kidney disease NIH symposium summary statement. J Pediatr (in press)
Harris PC, Rossetti S (2004) Molecular genetics of autosomal recessive polycystic kidney disease. Mol Genet Metab 81:75–85
Hiesberger T, Bai Y, Shao X, McNally BT, Sinclair AM, Tian X, Somlo S, Igarashi P (2004) Mutation of hepatocyte nuclear factor-1{beta} inhibits Pkhd1 gene expression and produces renal cysts in mice. J Clin Invest 113:814–825
Hildebrandt F, Otto E (2005) Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet 6:928–940
Hogan MC, Griffin MD, Rossetti S, Torres VE, Ward CJ, Harris PC (2003) PKHDL1, a homolog of the autosomal recessive polycystic kidney disease gene, encodes a receptor with inducible T lymphocyte expression. Hum Mol Genet 12:685–698
Igarashi P, Somlo S (2002) Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol 13:2384–2398
Igarashi P, Shao X, McNally BT, Hiesberger T (2005) Roles of HNF-1beta in kidney development and congenital cystic diseases. Kidney Int 68:1944–1947
Jafar TH, Stark PC, Schmid CH, Strandgaard S, Kamper AL, Maschio G, Becker G, Perrone RD, Levey AS (2005) The effect of angiotensin-converting-enzyme inhibitors on progression of advanced polycystic kidney disease. Kidney Int 67:265–271
Jiang ST, Chiou YY, Wang E, Lin HK, Lin YT, Chi YC, Wang CK, Tang MJ, Li H (2006) Defining a link with autosomal-dominant polycystic kidney disease in mice with congenitally low expression of pkd1. Am J Pathol 168:205–220
Jorgensen MJ (1977) The ductal plate malformation. Acta Pathol Microbiol Scand Suppl 257:1–87
Kaariainen H, Koskimies O, Norio R (1988) Dominant and recessive polycystic kidney disease in children: evaluation of clinical features and laboratory data. Pediatr Nephrol 2:296–302
Kaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM (1989) Autosomal recessive polycystic kidney disease. Pediatr Nephrol 3:43–49
Klingel R, Dippold W, Storkel S, Meyer zum Buschenfelde KH, Kohler H (1992) Expression of differentiation antigens and growth-related genes in normal kidney, autosomal dominant polycystic kidney disease, and renal cell carcinoma. Am J Kidney Dis 19:22–30
Lakshmanan J, Fisher DA (1993) An inborn error in epidermal growth factor prohormone metabolism in a mouse model of autosomal recessive polycystic kidney disease. Biochem Biophys Res Commun 196:892–901
Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P (2003) Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA 100:5286–5291
Losekoot M, Haarloo C, Ruivenkamp C, White SJ, Breuning MH, Peters DJ (2005) Analysis of missense variants in the PKHD1-gene in patients with autosomal recessive polycystic kidney disease (ARPKD). Hum Genet 118:185–206
Lowden DA, Lindemann GW, Merlino G, Barash BD, Calvet JP, Gattone VH 2nd (1994) Renal cysts in transgenic mice expressing transforming growth factor-alpha. J Lab Clin Med 124:386–394
Lu W, Fan X, Babakhanlou H, Law T, Rifal N, Harris PC, Perez-Atayde AR, Renneke HG, Zhou J (1999) Late onset of renal and hepatic cysts in Pkd1-targeted heterozygotes. Nat Gen 21:160–161
MacRae Dell K, Nemo R, Sweeney WE Jr, Avner ED (2004) EGF-related growth factors in the pathogenesis of murine ARPKD. Kidney Int 65:2018–2029
Masyuk TV, Huang BQ, Ward CJ, Masyuk AI, Yuan D, Splinter PL, Punyashthiti R, Ritman EL, Torres VE, Harris PC, LaRusso NF (2003) Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology 125:1303–1310
Menezes LF, Cai Y, Nagasawa Y, Silva AM, Watkins ML, Da Silva AM, Somlo S, Guay-Woodford LM, Germino GG, Onuchic LF (2004) Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int 66:1345–1355
Murcia NS, Sweeney WE, Avner ED (1999) New insights into the molecular pathophysiology of polycystic kidney disease. Kidney Int 55:1187–1197
Nagano J, Kitamura K, Hujer KM, Ward CJ, Bram RJ, Hopfer U, Tomita K, Huang C, Miller RT (2005) Fibrocystin interacts with CAML, a protein involved in Ca(2+) signaling. Biochem Biophys Res Commun 338:880–889
Nagasawa Y, Matthiesen S, Onuchic LF, Hou X, Bergmann C, Esquivel E, Senderek J, Ren Z, Zeltner R, Furu L, Avner E, Moser M, Somlo S, Guay-Woodford L, Buttner R, Zerres K, Germino GG (2002) Identification and characterization of Pkhd1, the mouse orthologue of the human ARPKD gene. J Am Soc Nephrol 13:2246–2258
Nakanishi K, Sweeney WE Jr, Macrae Dell K, Cotton CU, Avner ED (2001) Role of CFTR in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 12:719–725
Nauli SM, Zhou J (2004) Polycystins and mechanosensation in renal and nodal cilia. Bioessays 26:844–856
Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33:129–137
Nauta J, Sweeney WE, Rutledge JC, Avner ED (1995) Biliary epithelial cells from mice with congenital polycystic kidney disease are hyperresponsive to epidermal growth factor. Pediatr Res 37:755–763
Neufield TK, Douglass D, Grant M, Ye M, Silva F, Nadasdy T, Grantham JJ (1992) In vitro formation and expansion of cysts derived from human renal cortex epithelial cells. Kidney Int 41:1222–1236
Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schoneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, Germino GG (2002) PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet 70:1305–1317
Orellana SA, Sweeney WE, Neff CD, Avner ED (1995) Epidermal growth factor receptor expression is abnormal in murine polycystic kidney. Kidney Int 47:490–499
Osathanondh V, Potter EL (1964a) Pathogenesis of polycystic kidneys. Historical survey. Arch Pathol 77:459–465
Osathanondh V, Potter EL (1964b) Pathogenesis of polycystic kidneys. Survey of results of microdissection. Arch Pathol 77:510–512
Osathanondh V, Potter EL (1964c) Pathogenesis of polycystic kidneys. Type 1 due to hyperplasia of interstitial portions of collecting tubules. Arch Pathol 77:466–473
Pazour GJ (2004) Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol 15:2528–2536
Pazour GJ, Rosenbaum JL (2002) Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol 12:551–555
Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol 151:709–718
Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB (2002) Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol 12:R378–R380
Phillips CL, Miller KJ, Filson AJ, Nurnberger J, Clendenon JL, Cook GW, Dunn KW, Overbeek PA, Gattone VH 2nd, Bacallao RL (2004) Renal cysts of inv/inv mice resemble early infantile nephronophthisis. J Am Soc Nephrol 15:1744–1755
Praetorius HA, Spring KR (2001) Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol 184:71–79
Praetorius HA, Spring KR (2003a) Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol 191:69–76
Praetorius HA, Spring KR (2003b) The renal cell primary cilium functions as a flow sensor. Curr Opin Nephrol Hypertens 12:517–520
Praetorius HA, Spring KR (2005) A physiological view of the primary cilium. Annu Rev Physiol 67:515–529
Praetorius HA, Frokiaer J, Nielsen S, Spring KR (2003) Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J Membr Biol 191:193–200
Praetorius HA, Praetorius J, Nielsen S, Frokiaer J, Spring KR (2004) Beta1-integrins in the primary cilium of MDCK cells potentiate fibronectin-induced Ca2+ signaling. Am J Physiol Renal Physiol 287:F969–F978
Pugh JL, Sweeney WE Jr, Avner ED (1995) Tyrosine kinase activity of the EGF receptor in murine metanephric organ culture. Kidney Int 47:774–781
Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP, Avner ED (1998) Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. J Clin Invest 101:935–939
Rohatgi R, Greenberg A, Burrow CR, Wilson PD, Satlin LM (2003) Na transport in autosomal recessive polycystic kidney disease (ARPKD) cyst lining epithelial cells. J Am Soc Nephrol 14:827–836
Rohatgi R, Zavilowitz B, Vergara M, Woda C, Kim P, Satlin LM (2004) Cyst fluid composition in human autosomal recessive polycystic kidney disease. Pediatr Nephrol 20:552–553
Rossetti S, Torra R, Coto E, Consugar M, Kubly V, Malaga S, Navarro M, El-Youssef M, Torres VE, Harris PC (2003) A complete mutation screen of PKHD1 in autosomal-recessive polycystic kidney disease (ARPKD) pedigrees. Kidney Int 64:391–403
Roy S, Dillon MJ, Trompeter RS, Barratt TM (1997) Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol 11:302–306
Sato Y, Harada K, Kizawa K, Sanzen T, Furubo S, Yasoshima M, Ozaki S, Ishibashi M, Nakanuma Y (2005) Activation of the MEK5/ERK5 cascade is responsible for biliary dysgenesis in a rat model of Caroli’s disease. Am J Pathol 166:49–60
Sharp AM, Messiaen LM, Page G, Antignac C, Gubler MC, Onuchic LF, Somlo S, Germino GG, Guay-Woodford LM (2005) Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J Med Genet 42:336–349
Sullivan LP, Grantham JJ (1996) Mechanisms of fluid secretion by polycystic epithelia. Kidney Int 49:1586–1591
Sullivan LP, Wallace DP, Grantham JJ (1998) Chloride and fluid secretion in polycystic kidney disease. J Am Soc Nephrol 9:903–916
Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, Hopkins N (2004) A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development 131:4085–4093
Sweeney WE, Avner ED (1996) BPK cyst fluid contains EGF and TGF-a like peptides which are motogenic and phosphorylate apical EGFR. J Am Soc Nephrol 7:1606
Sweeney WE Jr, Avner ED (1998) Functional activity of epidermal growth factor receptors in autosomal recessive polycystic kidney disease. Am J Physiol 275:F387–F394
Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED (2000) Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int 57:33–40
Sweeney WE Jr, Hamahira K, Sweeney J, Garcia-Gatrell M, Frost P, Avner ED (2003) Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int 64:1310–1319
Torres VE (2004) Therapies to slow polycystic kidney disease. Nephron Exp Nephrol 98:e1–e7
Torres VE (2005) Vasopressin antagonists in polycystic kidney disease. Kidney Int 68:2405–2418
Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH (2004) Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med 10:363–364
Veizis IE, Cotton CU (2005) Abnormal EGF-dependent regulation of sodium absorption in ARPKD collecting duct cells. Am J Physiol Renal Physiol 288:F474–F482
Veizis EI, Carlin CR, Cotton CU (2004) Decreased amiloride-sensitive Na+ absorption in collecting duct principal cells isolated from BPK ARPKD mice. Am J Physiol Renal Physiol 286:F244–F254
Wallace DP, Christensen M, Reif G, Belibi F, Thrasher B, Herrell D, Grantham JJ (2002) Electrolyte and fluid secretion by cultured human inner medullary collecting duct cells. Am J Physiol Renal Physiol 283:F1337–F1350
Wang S, Luo Y, Wilson PD, Witman GB, Zhou J (2004) The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. J Am Soc Nephrol 15:592–602
Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC (2002) The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 30:259–269
Ward CJ, Yuan D, Masyuk TV, Wang X, Punyashthiti R, Whelan S, Bacallao R, Torra R, LaRusso NF, Torres VE, Harris PC (2003) Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet 12:2703–2710
Welling LW, Grantham JJ (1991) Cystic and developmental diseases of the kidney. In: Brenner BMR, Rector FC (eds) The kidney. Saunders, Philadelphia, pp 1657–1694
Wilson PD (2004a) Polycystic kidney disease. N Engl J Med 350:151–164
Wilson PD (2004b) Polycystic kidney disease: new understanding in the pathogenesis. Int J Biochem Cell Biol 36:1868–1873
Witzgall R (2005) New developments in the field of cystic kidney diseases. Curr Mol Med 5:455–465
Xiong H, Chen Y, Yi Y, Tsuchiya K, Moeckel G, Cheung J, Liang D, Tham K, Xu X, Chen XZ, Pei Y, Zhao ZJ, Wu G (2002) A novel gene encoding a TIG multiple domain protein is a positional candidate for autosomal recessive polycystic kidney disease. Genomics 80:96–104
Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, Rome LA, Sullivan LP, Grantham JJ (2000) cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int 57:1460–1471
Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ (2003) Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int 63:1983–1994
Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP (2004) Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 279:40419–40430
Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP (2006) Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol 17:178–187
Ye M, Grant M, Sharma M, Elzinga L, Swan S, Torres VE, Grantham JJ (1992) Cyst fluid from human autosomal dominant polycystic kidneys promotes cyst formation and expansion by renal epithelial cells in vitro. J Am Soc Nephrol 3:984–994
Yoder BK, Hou X, Guay-Woodford LM (2002) The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 13:2508–2516
Zerres K, Rudnik-Schoneborn S, Steinkamm C, Mucher G (1996) Autosomal recessive polycystic kidney disease. Nephrol Dial Transplant 11 (Suppl 6):29–33
Zerres K, Mücher G, Becker J, Steinkamm C, Rudnik-Schöneborn S, Heikkilä P, Rapola J, Salonen R, Germino GG, Onuchic L, Somlo S, Avner ED, Harman LA, Stockwin JM, Guay-Woodford LM (1998) Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet 76:137–144
Zerres K, Senderek J, Rudnik-Schoneborn S, Eggermann T, Kunze J, Mononen T, Kaariainen H, Kirfel J, Moser M, Buettner R, Bergmann C (2004) New options for prenatal diagnosis in autosomal recessive polycystic kidney disease by mutation analysis of the PKHD1 gene. Clin Genet 66:53–57
Zhang BH, Guan KL (2000) Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J 19:5429–5439
Zhang MZ, Mai W, Li C, Cho SY, Hao C, Moeckel G, Zhao R, Kim I, Wang J, Xiong H, Wang H, Sato Y, Wu Y, Nakanuma Y, Lilova M, Pei Y, Harris RC, Li S, Coffey RJ, Sun L, Wu D, Chen XZ, Breyer MD, Zhao ZJ, McKanna JA, Wu G (2004) PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci USA 101:2311–2316
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors are supported by the National Institutes of Health (grant no. 1-P50-DK57306), the PKD Foundation (grant no. 76a2r), and the Children’s Research Institute, Children’s Hospital of Wisconsin.
Rights and permissions
About this article
Cite this article
Sweeney, W.E., Avner, E.D. Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD). Cell Tissue Res 326, 671–685 (2006). https://doi.org/10.1007/s00441-006-0226-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-006-0226-0