
Abstract
Mutations in the PARK2 gene coding for parkin cause autosomal recessive juvenile parkinsonism (AR-JP), a familial form of Parkinson’s disease (PD). Parkin functions as an E3 ubiquitin ligase, and loss of this ubiquitin ligase activity appears to be the mechanism underlying pathogenesis of AR-JP. Recently, the spectrum of genetic, clinical, and pathological findings on AR-JP has been significantly expanded. Moreover, a considerable number of parkin interactors and/or substrates have been identified and characterized, and animal models of parkin deficiency have been generated. In this review, we provide an overview of the most relevant findings and discuss their implications for the pathogenesis of AR-JP and sporadic PD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder and occurs sporadically in the vast majority of cases, i.e., without a positive family history. As a rule, no exogenous cause for PD can be identified. Sporadic cases with no known cause are classified as idiopathic Parkinson’s syndrome (IPS; Lang and Lozano 1998). Recently, rare monogenetic forms of PD have significantly improved our understanding of the pathogenesis of the far more common IPS (Dawson and Dawson 2003a). For instance, autosomal recessive juvenile parkinsonism (AR-JP) as a clinically defined entity has been studied for decades, primarily in Japan (Yamamura et al. 1973, 1993; Ishikawa and Tsuji 1996). The discovery of the responsible gene has led to tremendous insight into the pathogenesis of both AR-JP and IPS (Giasson and Lee 2001). Prior to the identification of the gene, the defining characteristics of AR-JP were: (1) the existence of affected family members within the same generation, often born to consanguineous parents; (2) an early onset (<40 years); (3) a parkinsonian syndrome with a number of atypical clinical features; (4) the absence of Lewy body (LB) pathology in the brain. However, the full variety and complexity of this familial form of PD was only appreciated once its cause was defined at the molecular level, thereby allowing for the accurate diagnosis, even in cases not fitting these criteria (Abbas et al. 1999; Klein et al. 2000). Here, we review the genetics and the expanded spectrum of the clinical and pathological findings of AR-JP and the molecular biology of parkin.
Genetics
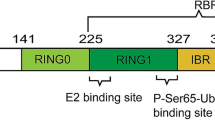
In 1997, the gene locus (PARK2) of AR-JP was mapped to chromosome 6q25.2–27 (Matsumine et al. 1997). Shortly thereafter, the gene was identified, and the encoded protein was named parkin (Kitada et al. 1998). The PARK2 gene contains 12 exons that are separated by extended intronic regions. As a consequence, the gene spans more than 1.53 Mb, ranking among the largest genes in the human genome (Kitada et al. 1998; West et al. 2001). The open reading frame codes for a protein of 465 amino acids with an apparent molecular weight of 52 kDa, which, in contrast to its very large genomic structure, places parkin close to the average length of proteins in the human proteome, as calculated by the EMBL-EBI (http://www.ebi.ac.uk/proteome/). Parkin is an evolutionary conserved gene product, with orthologs in Caenorhabditis elegans, Drosophila melanogaster, mouse, rat, and other species (Horowitz et al. 1999; Culetto and Sattelle 2000; Kitada et al. 2000; Bae et al. 2003). It has a modular structure, containing an N-terminal ubiquitin-like (UBL) domain, a central linker region, and a C-terminal cassette of two RING domains, separated by an In-Between-RING (IBR) domain (Fig. 1). Interestingly, the parkin promoter has been found to function as a bidirectional promoter, regulating not only transcription of parkin, but also transcription of a gene upstream of and antisense to PARK2 (West et al. 2003). This gene spans 0.6 Mb, contains five exons, and has been named parkin co-regulated gene (PACRG). The (patho-) physiological relevance of the corresponding gene product is not known.

Modular structure of parkin and missense mutations associated with AR-JP. Parkin contains a UBL domain, a central linker region, and a C-terminal cassette of two RING domains (R1, R2), separated by an In-Between-RING (IBR) domain. Locations of reported familial-associated disease-causing missense mutations are indicated. Disease-causing deletions, insertions, frameshifts, and nonsense point mutations are not depicted. Note that an association with AR-JP has not unequivocally been shown for all mutations. Some have only been found in isolated cases and some only as heterozygous mutations, and so a clear distinction between mutation and rare polymorphism is not always possible. For detailed references for all reported mutations, see the recent review by Mata and colleagues (2004)
The parkin mutations that initially led to the identification of the gene were large homozygous deletions of one or five exons, respectively (Kitada et al. 1998). Since then, a multitude of mutations has been identified: deletions of single or multiple exons, duplications or triplications of exons, frameshift mutations, point mutations, which can be subdivided into missense (resulting in the replacement of one amino acid residue by another) and nonsense mutations (resulting in a stop codon), and splice site (i.e., intronic) mutations (Abbas et al. 1999; Lucking et al. 2000; West et al. 2002). The majority of point mutations localize to the RING-IBR-RING domain in the C-terminal half of parkin and, in particular, to the first RING domain (R1), implying essential functional relevance for this region of the protein (Fig. 1). However, there is no true mutational hotspot, as mutations have been found in all the domains of parkin.
Parkin mutations associated with AR-JP occur as either homozygous or compound heterozygous mutations, i.e., with different mutations on both alleles. To find both alleles affected by some mutation that interferes with the expression or function of a protein is, of course, to be expected in a patient suffering from an autosomal-recessive disease. However, several cases have been published in which, despite extensive screening, only one of the alleles appears to be mutated (Farrer et al. 2001; West et al. 2002). Given the size and complexity of the genomic structure of the PARK2 gene, a disease-associated mutation in the seemingly unaffected allele may have been missed in each of these cases; however, it appears more likely that loss of one copy of the parkin gene constitutes a risk factor for PD. This notion postulates haploinsufficiency of parkin as a causative factor with reduced penetrance. Indeed, the analysis of two families by Farrer and co-workers (2001) has shown the association of a heterozygous 40-bp deletion in exon 3 with a parkinsonian syndrome, passed on in a dominant fashion with reduced penetrance, thereby supporting such a model of haploinsufficiency. Parkin is also S-nitrosylated in vitro and in vivo, and S-nitrosylation inhibits parkin’s E3 ligase activity (see below) and its protective function (Chung et al. 2004). Thus, a heterozygous parkin mutation coupled with nitrosative stress could lead to the manifestation of haploinsufficiency, accounting for the observation of disease-associated heterozygous mutations. A toxic gain of function or a dominant-negative effect of parkin carrying certain missense mutations is another possible explanation. However, the finding that truncated proteins are not even expressed at detectable levels in the brains of patients with deletions of exons 3 or 4 (Shimura et al. 1999, 2001) strengthens the concept of loss-of-function mutations as the predominant pathomechanism in AR-JP.
Whereas overall, the monogenetic forms of PD are very rare, among those genes identified to date, mutations in parkin appear to be the most common cause of familial forms of PD. In a large European study, including only patients with an age at onset ≤45 years (or with an affected sibling showing such an early onset), parkin mutations have been identified in up to 50% of familial cases (i.e., cases with at least one affected sibling), and in 18% of sporadic (or isolated) cases (Lucking et al. 2000). In patients without family history, 44% with an age at onset ≤30, but only 3% with an age at onset >30, were found to be associated with parkin mutations. Other reports confirm this order of magnitude, with considerable variation most likely being attributable, at least in part, to the variable inclusion criteria of the different studies (Abbas et al. 1999; Kann et al. 2002).
Clinical manifestation
In addition to the recessive pattern of inheritance, AR-JP had traditionally been characterized by the typical symptoms of PD (bradykinesia, rigidity and resting tremor) associated with: (1) early onset, typically before the age of 40; (2) foot dystonia at onset; (3) hyperreflexia of the lower limbs; (4) diurnal fluctuations with a marked sleep benefit; (5) good response to Levodopa-therapy; (6) early onset of Levodopa-induced dyskinesias; (7) slow disease progression (Ishikawa and Tsuji 1996). However, the results of more recent studies, in which the diagnosis of AR-JP has been based on molecular genetics, suggest that patients with parkin mutations cannot be identified based on clinical findings (Klein et al. 2000; Lucking et al. 2000). “AR-JP-specific” symptoms such as early dystonia and hyperreflexia, although more frequent in AR-JP than in IPS, turn out to be less frequent than previously reported. As a result, many cases of AR-JP appear to be clinically indistinguishable from IPS (Abbas et al. 1999), and, in the same study (including only familial cases), the reported average age at onset was 38 years, adding a question mark to the term “juvenile” in AR-JP; indeed, 40% of patients had an age at onset >40 years, putting them somewhat close to the typical manifestation of IPS. Remarkably, not only is there a wide variation of the age at onset, ranging from 7–58 years, but even within single families with identical parkin mutations, variation of up to 20 years in the age of onset has been observed (Lucking et al. 2000), indicating that there are strong modulating factors, either genetic or environmental. Moreover, there appears to be no correlation of certain types of mutations with distinctive clinical features. For example, truncating mutations do not result in an earlier onset and/or more severe phenotype than missense mutations, although one might expect the latter to interfere less with parkin’s enzymatic activity than the former. Further analysis of pedigrees spanning several generations and a thorough characterization of the effect of missense mutations on the molecular properties of parkin should prove most helpful in addressing the question as to whether different mutations result in distinct pathogenetic and/or pathological and clinical features.
Pathology
Histopathologically, AR-JP is characterized by loss of pigmented neurons in the substantia nigra pars compacta (SNpc) and in the locus coeruleus (LC), and by the absence of LBs (Takahashi et al. 1994; Mori et al. 1998; Hayashi et al. 2000; van de Warrenburg et al. 2001). LBs are cytoplasmic proteinaceous inclusion bodies that are considered the pathological hallmark of IPS (Lang and Lozano 1998). They are strongly immunoreactive for α-synuclein and ubiquitin and for a number of other proteins, but fibrillized α-synuclein appears to be the primary structural component (Goedert 2001). Absence of LBs has long been recognized as one of the defining features of AR-JP (Yamamura et al. 1993; Mori et al. 1998). However, a recent report of LB pathology in a case of parkin-associated PD has cast doubts on this notion (Farrer et al. 2001). Curiously, this particular patient carried two compound heterozygous mutations (a 40-bp deletion in exon 3, and an R275W missense mutation on the other allele), each of which appears to be sufficient to cause a parkinsonian syndrome by itself. Since the R275W mutation only decreases the E3 ligase function of parkin (see below) without abolishing it (Chung et al. 2001), the presence of LBs in this patient might be “the exception that proves the rule” (Dawson and Dawson 2003b). The data are still inconclusive, and further pathological studies are required to clarify whether mutations in parkin lead to the absence of LBs.
Conflicting data also exist regarding the presence of parkin in LBs in IPS pathology. Initially, LBs were reported to be immunoreactive for parkin (Shimura et al. 1999, 2001). However, lack of highly specific parkin antibodies has long been recognized as a serious obstacle to in vivo studies on parkin. Recently, a whole panel of monoclonal parkin antibodies has been thoroughly characterized (Pawlyk et al. 2003), providing a tool that should prove most helpful to clarifying aspects of parkin biology related to its regional and subcellular localization. These antibodies have, however, shown that parkin is absent from LBs.
Another distinctive pathological finding in AR-JP is the more restricted distribution of pathology compared with IPS. Although SNpc and LC catecholaminergic neurons are the most recognized cell populations undergoing neurodegeneration in IPS, other nuclei (such as the dorsal motor nucleus of the vagus, the anterior olfactory nucleus, and the nucleus basalis Meynert) are affected more severely and earlier in the course of the disease (Braak et al. 2003). In late stages, histopathological analysis of PD brains reveals widespread LB pathology, with involvement of multiple neurotransmitter and functional systems, including the neocortex. Although pathological evidence is somewhat limited so far, SNpc and LC appear to be affected by neurodegeneration in an exclusive fashion in AR-JP, with varying degrees of astrocytic gliosis (Hayashi et al. 2000; Gouider-Khouja et al. 2003). It remains to be seen whether these findings stand up to a critical and more systematic analysis of a larger number of autopsy samples.
Parkin functions as an E3 ligase
Like many other proteins containing a RING domain, parkin has been found to function as an E3 ligase (Shimura et al. 2000; Zhang et al. 2000b). E3 ligases are part of the cellular machinery that tags proteins with ubiquitin, thereby targeting them for degradation by the proteasome. The ubiquitin–proteasome system (UPS) plays a major role in many vital cellular processes, and its dysfunction has been implicated in the pathogenesis of neurodegenerative disorders (Ciechanover and Brundin 2003; Giasson and Lee 2003; Moore et al. 2003). Since the UPS has recently been reviewed extensively (Berke and Paulson 2003; Pickart 2004), we will only provide a short overview.
Ubiquitin is a 76-amino-acid protein that becomes covalently linked with its C-terminus to the lysine side-chain of a substrate. This posttranslational modification of proteins is called ubiquitination and occurs through sequential steps catalyzed by ubiquitin-activating (E1), conjugating (E2), and ligase (E3) enzymes (Fig. 2). In subsequent cycles of the same process, additional ubiquitin molecules are linked onto the previously ligated ubiquitin (most commonly to the lysine residue at position 48), resulting in the formation of a polyubiquitin chain. This polyubiquitin chain is the signal recognized by the 26S proteasome, a very large multisubunit protein complex with proteolytic activity localized on the inside of the barrel-shaped core complex (also called the 20S proteasome). The 19S cap, located on both ends of the central 20S complex, recognizes the polyubiquitin chain, which is removed and cleaved into monomers by deubiquitinating enzymes, such as ubiquitin C-terminal hydrolase L1 (UCH-L1). It then unfolds the targeted protein and feeds it into the inner chamber of the 20S core complex. There, it is proteolytically cleaved into small peptides that are then released into the cytosol (Fig. 2). The process of protein degradation via 26S proteasome complex is ATP-dependent.

The ubiquitin–proteasome system (UPS). Simplified scheme of the sequential activity of the various components involved in targeted degradation of proteins by the UPS (Ub ubiquitin)
The UPS is crucially involved in two tasks. One is the accurate timely regulation of the level of short-lived proteins that play a key role in processes such as cell-cycle progression, signal transduction, and metabolism. The other task is protein-quality control. More than 30% of newly synthesized proteins have been reported to be rapidly degraded without proper folding or possibly because of improper folding (Schubert et al. 2000). In addition, a variety of external stress factors can result in the misfolding of previously functional proteins, requiring either chaperone-mediated refolding activity or degradation of the misfolded proteins. The finding that ubiquitin itself is one of the so-called heat-shock proteins underlines the importance of the UPS in the cellular stress response (Tanaka et al. 2001).
The picture is further complicated by the observation that, instead of the formation of polyubiquitin chains, certain substrates can be tagged by one or two ubiquitin molecules only (Fig. 2). This mono-ubiquitination or di-ubiquitination does not target a protein for degradation by the proteasome; rather, in the case of membrane-bound proteins, it constitutes a signal for endocytosis, ultimately leading to lysosomal proteolysis of the protein. Recently, mono-ubiquitination/di-ubiquitination has been implicated as a more generally employed signaling mechanism for intracellular trafficking (Aguilar and Wendland 2003).
E3 ligases confer substrate specificity to the ubiquitination machinery and serve as scaffolding proteins. They interact with a cognate E2 and with the specific substrate in order to catalyze (or facilitate) the transfer of ubiquitin from the E2 onto the substrate. In the case of parkin, the ubiquitin-conjugating enzymes Ubc-H7 and Ubc-H8 are the collaborating E2s (Imai et al. 2000; Shimura et al. 2000; Zhang et al. 2000b). Additionally, parkin interacts with endoplasmic-reticulum-associated E2s UBC6 and UBC7 (Imai et al. 2001). The E2 enzymes interact with the RING-IBR-RING domain of parkin, and more specifically, at least in the case of Ubc-H8, with the second (or C-terminal) RING domain (R2; Zhang et al. 2000b). Cell culture experiments and in vitro-studies unequivocally show that parkin does indeed function as an E3 ligase (Imai et al. 2000; Shimura et al. 2000; Zhang et al. 2000b). Mutations associated with AR-JP impair the interaction of parkin with E2s and reduce or abolish its ubiquitin ligase activity. Interestingly, parkin has been found to undergo self-ubiquitination, possibly promoting its own degradation and thereby regulating its own expression level (Zhang et al. 2000b).
The family of RING-type E3 ligases can be further subdivided into single-subunit RING E3s and multisubunit RING E3s (Pickart 2001). In the latter, a small RING domain-containing protein typically plays a key organizational role as a scaffolding molecule recruiting the various components (including the E2) to the complex, without being catalytically active as ubiquitin ligase itself. SCF (Skp1-Cullin-F-box protein) E3s are a typical example of this kind of E3 ligase complex (Tyers and Jorgensen 2000). Given its size and modular structure, parkin has commonly been classified as a single-subunit E3 (Pickart 2001). However, parkin has recently been reported to interact with two known components of SCF complexes (hSel-10 and Cullin-1), which potentiate parkin’s ubiquitin ligase activity (Staropoli et al. 2003). Furthermore, parkin forms a complex with Hsp70 and CHIP, enhancing its E3 enzymatic activity and its ability to inhibit cell death induced by unfolded protein stress (Imai et al. 2002). It is unclear at this point whether parkin preferentially functions as a part of one of these complexes under physiological conditions or whether it is recruited into one or other the only under certain pathological conditions. It remains to be seen how these reports of entirely different parkin-containing protein complexes can be reconciled, and why there appears to be no overlap of the groups of parkin interactors identified with different approaches.
The quest for parkin substrates
Parkin functions as an E3 ligase, presumably targeting specific substrate proteins for degradation by the proteasome, and loss of parkin (or loss of parkin function through missense mutations) causes AR-JP. Taken together, these observations suggest a model of AR-JP pathogenesis in which, because of the absence of parkin, some protein cannot be properly degraded and, through its accumulation, causes dysfunction and eventually the death of susceptible neurons. Identification of parkin substrates has therefore been a major focus of many laboratories working on parkin, and several putative substrates have been reported (Table 1).
The first substrate to be identified was CDCrel-1 (cell division control-related protein 1; Zhang et al. 2000b). It belongs to a family of GTPases called septins and is predominantly expressed in the nervous system, where it is associated with synaptic vesicles (Beites et al. 1999). Although it has been implicated in neurotransmitter release, CDCrel-1 knockout mice do not have a severe phenotype; indeed, changes in the expression pattern of other members of the septin family appear to be the only abnormality (Peng et al. 2002). Intriguingly, overexpression of CDCrel-1 in SNpc neurons by adeno-associated virus-mediated gene transfer induces dopamine-dependent neurodegeneration, providing evidence for a potential role of CDCrel-1 in the pathogenesis of AR-JP (Dong et al. 2003). Such a role is further supported by the recent finding that CDCrel-1 accumulates in the brains of AR-JP patients (Choi et al. 2003).
Synphilin-1 was originally identified as an interactor of α-synuclein involved in the formation of inclusion bodies in cultured cells (Engelender et al. 1999). Its physiological function is unknown. Synphilin-1 is a component of LBs in PD and related synucleinopathies (Wakabayashi et al. 2000, 2002). Parkin interacts with and ubiquitinates synphilin-1 and promotes the ubiquitination of inclusion bodies when overexpressed with α-synuclein and synphilin-1 in cell culture (Chung et al. 2001). Therefore, synphilin-1 links parkin and α-synuclein in a common biochemical pathway with potential significance for the formation of LBs and the pathogenesis of both IPS and AR-JP. A missense mutation (R621C) in the synphilin-gene has been identified in two (out of more than 300) patients with sporadic PD, but not in more than 700 control samples (Marx et al. 2003), supporting an important role for this protein in PD pathogenesis. Interestingly, when overexpressed in cultured cells, the mutant synphilin-1 (compared with the wildtype protein) shows a decreased propensity to form aggregates, accompanied by an increased susceptibility to apoptotic stimuli (Marx et al. 2003).
The finding that α-synuclein is the major component of LBs, which are, on the other hand, absent in parkin-associated PD, has fuelled the idea that parkin activity is required for the formation of LBs. Therefore, α-synuclein has become an intensely studied candidate substrate of parkin. Whereas the abundant unmodified form of α-synuclein does not interact with parkin (Chung et al. 2001), Shimura and colleagues have identified a rare O-glycosylated form of α-synuclein (αSp22) that interacts with and is ubiquitinated by parkin (Shimura et al. 2001). Familial-linked parkin mutants fail to bind αSp22, and, importantly, αSp22 appears to accumulate in AR-JP brains. Curiously though, most laboratories have been unable to detect this glycosylated form of α-synuclein. The relevance of αSp22 for the pathogenesis of PD/AR-JP thus awaits further clarification.
Parkin-associated endothelin receptor-like receptor (Pael-R) is a putative G-protein-coupled transmembrane protein with homology to the endothelin receptor type B (Imai et al. 2001). In the brain, it is primarily expressed in oligodendrocytes, but also in a few distinct subpopulations of neurons, including SNpc catecholaminergic neurons. When overexpressed in cultured cells, Pael-R tends to become unfolded and insoluble, inducing the unfolded protein response and eventually leading to cell death. Parkin ubiquitinates this insoluble form of Pael-R, promoting its degradation, which results in the suppression of unfolded-protein-induced cell death. Under these conditions, parkin apparently acts as part of the endoplasmic-reticulum-associated protein degradation machinery, utilizing the endoplasmic-reticulum-associated E2 enzymes, Ubc6 and Ubc7. Two further observations strongly support the important pathogenetic role of Pael-R: the insoluble form of Pael-R accumulates in AR-JP brains (Imai et al. 2001), and panneuronal expression of Pael-R in Drosophila causes age-dependent selective degeneration of dopaminergic neurons (Yang et al. 2003).
Recently, a number of additional putative parkin substrates have been identified. Co-immunoprecipitation with and in vivo ubiquitination by parkin have been shown for all of the following substrates. α/β-Tubulin shows decreased stability in cells overexpressing parkin, and parkin colocalizes with microtubules (Ren et al. 2003). Steady-state levels of synaptotagmin XI expression are also decreased in the presence of parkin, and LBs are immunoreactive for this member of a large family of calcium-binding proteins (Huynh et al. 2003). Overexpression of p38, a component of the mammalian aminoacyl-tRNA synthetase complex, results in aggresome-like inclusion body formation and/or cell death, depending on the cell type. Furthermore, p38 is present in LBs (Corti et al. 2003). SEPT5_v2/CDCrel-2, another member of the septin family and a close homolog to CDCrel-1, has been reported as a parkin substrate (Choi et al. 2003). Cyclin E specifically interacts with parkin in the context of complex formation of parkin with hSel-10 and Cullin-1 (Staropoli et al. 2003). In primary neuronal cell cultures, knockdown of parkin increases the kainate-induced accumulation of cyclin E and concomitant cell death, whereas overexpression of parkin has the opposite effect in this model of excitotoxicity. Brain lysates of one AR-JP patient and two PD patients were shown to contain increased levels of cyclin E compared with control or Alzheimer’s disease brains (Staropoli et al. 2003).
Typically, one expects an E3 ligase to be highly specific for one or possibly a small number of substrates. Thus, the large number of putative parkin substrates is somewhat surprising. However, recruitment of parkin to different protein complexes under different conditions, as discussed above, might result in distinct patterns of specificity, thereby accounting for the unexpected variety of parkin interactors and substrates.
Animal models
Since there is strong evidence supporting the loss of function of parkin as a mechanism underlying AR-JP, the generation of parkin knockout animals should be an attractive way to model aspects of human disease in a mammalian organism. Three parkin-deficient mice have been reported so far (Goldberg et al. 2003; Itier et al. 2003; von Coelln et al. 2004). The first two reports describe parkin knockout mice generated by targeted disruption of exon 3. These mice show subtle behavioral deficits, some dysfunction of dopamine and glutamate neurotransmission, and abnormalities in dopamine metabolism, but no loss of SNpc or LC catecholaminergic neurons. In contrast, we have generated parkin null mice by targeted disruption of exon 7 and have found loss of noradrenergic LC neurons and a marked reduction of the norepinephrine-dependent startle response (von Coelln et al. 2004). Longevity and gross motor function are not compromised in either exon 7 and exon 3 parkin-deficient mice, and so far, none of the putative parkin substrates has been reported to accumulate in their brains. Rather, proteomic analysis of the exon-3-deleted mice revealed a decrease of several proteins involved in mitochondrial oxidative phosphorylation and oxidative stress (Palacino et al. 2004). Proteomic analysis in the exon-7-deleted mice may reveal the accumulation of authentic parkin substrates, as these mice have loss of LC neurons, whereas exon-3-deleted mice have relatively subtle abnormalities. The mild phenotype in parkin knockout mice compared with that in the human disease may be attributable to (1) a redundancy in the mouse E3 ligase enzyme family that is lacking in humans, although this seems somewhat unlikely for an evolutionary conserved protein; (2) the relatively short lifespan of mice, which may not live sufficiently long enough to accumulate toxic amounts of some potentially noxious parkin substrate; or (3) the loss of parkin activity requiring another insult, such as a toxic stimulus or cellular stressor to induce a PD-like syndrome.
In a Drosophila model, parkin null mutants exhibit a reduced lifespan, apoptotic cell death of the indirect flight muscles, and male sterility (Greene et al. 2003). Muscle degeneration and defective spermatogenesis can be attributed to mitochondrial dysfunction. The significance of these defects is uncertain, since no signs of myopathy or impaired fertility have been reported in AR-JP patients. They do, however, warrant a more thorough analysis of parkin’s contribution to mitochondrial function.
Protective effects of parkin in different model systems
Studies exploring the idea that certain toxic stimuli or stressors might be necessary to uncover parkin’s potential as a survival-promoting factor for catecholaminergic neurons have yielded some exciting insights. Parkin protects against kainate-induced excitotoxicity and cyclin E accumulation (Staropoli et al. 2003). In cell culture (including primary mesencephalic cultures), overexpression of parkin protects against proteasome inhibition and toxicity induced by overexpression of A53T-α-synuclein, a mutant form of α-synuclein associated with autosomal-dominant PD (Petrucelli et al. 2002). Conversely, knockdown of parkin in the same system renders cells more susceptible to proteasome inhibition. It is surprising that parkin protects against proteasome-inhibition-induced toxicity, since parkin targets proteins for degradation through the proteasome. Certain proteins accumulating in the presence of proteasome inhibitors could possibly be transformed by parkin into less toxic forms. Ubiquitination-dependent sequestration into aggresomes or inclusion bodies might account for parkin’s protective role in the setting of proteasomal inhibition. This model, however, remains highly speculative. Alternatively, since chemical inhibitors of the proteasome do not suppress 100% of its activity, but rather leave some residual proteolytic function intact, the overexpression of parkin might result in the preferential degradation of toxic parkin substrates, even in the presence of proteasome inhibitors. In Drosophila, neuronal expression of α-synuclein or Pael-R results in the degeneration of dopaminergic neurons (Feany and Bender 2000; Yang et al. 2003). Coexpression of parkin suppresses both α-synuclein-induced and Pael-R-induced toxicity, whereas interference with endogenous Drosophila parkin expression augments toxicity (Yang et al. 2003). Parkin’s potential to provide protection against such a variety of toxic insults has led to speculations about it being “a multipurpose neuroprotective agent” (Feany and Pallanck 2003). It remains to be seen whether all these effects can be attributed to parkin’s E3 ligase activity, or whether there is some additional/alternative parkin function.
Models of pathogenesis
It is an ongoing controversy whether, in IPS, the sequestration of aggregated α-synuclein represents an attempt of the cell to protect itself from some toxic form of α-synuclein, or whether LBs actually trigger cell dysfunction and death by obstructing proper intracellular trafficking and by trapping other vital cellular components (Giasson and Lee 2003). In other words, are LBs beneficial or noxious to the cell? Accordingly, the absence of LBs in AR-JP can be interpreted in two different ways: (1) parkin function may be necessary for the formation of LBs to detoxify deleterious α-synuclein species, and its absence breaches the neuron’s line of defence, resulting in early onset disease; (2) the mechanism of neurodegeneration in AR-JP may be entirely different from IPS, possibly unrelated to α-synuclein. Related to this puzzle, a major paradox of the parkin field is why the loss of a protein that is presumably involved in the degradation of other proteins does not promote detectable protein accumulation, but rather results in a PD-like disorder without the characteristic proteinaceous inclusion bodies.
Two hypothetical models arise from these conflicting views: a “unifying” and a “distinguishing” model (Fig. 3). According to the former, the occurrence of a toxic stimulus, possibly repeatedly over a long period of time, leads to the increased generation of toxic metabolites, e.g., unfolded/misfolded proteins. Excessive oxidative stress is an interesting candidate for such an initial toxic stimulus, given the accumulating evidence for oxidative damage in PD (Zhang et al. 2000a). Parkin-mediated ubiquitination of these toxic metabolites then represents an attempt by the affected cell to detoxify them by proteasomal degradation. However, if the proteasome itself is impaired at the same time, either by the aggregation of misfolded proteins or by the direct damage inflicted by the initial toxic stimulus, then the ubiquitinated species accumulate. As a last line of defence, they are therefore sequestered into aggresomes, which over time develop into LBs. In the absence of parkin, the toxic metabolites cannot be ubiquitinated, resulting in a more aggressive disease process, and the absence of LBs. One major flaw in this model is that recent evidence strongly argues against ubiquitination as a requirement for inclusion body formation (Sampathu et al. 2003); indeed, a significant percentage of inclusion bodies in synucleinopathies such as PD are synuclein-positive, but ubiquitin-negative (Spillantini et al. 1998a, 1998b; Gomez-Tortosa et al. 2000). On the other hand, parkin has been shown to undergo S-nitrosylation in vitro and in vivo rendering it inactive (Chung et al. 2004). In brain tissue from patients with sporadic PD, parkin is indeed S-nitrosylated. Thus, parkin inactivation by this specific posttranslational modification might be an important contributing factor to the pathogenesis of IPS, consistent with the “unifying model.”

Models of the pathogenesis of IPS and parkin-associated AR-JP. a The “unifying model” postulates a common pathogenetic mechanism for both forms of PD, with a central role for parkin in the formation of protein aggregates and Lewy bodies. b According to the “distinguishing model,” AR-JP is a distinct disease entity, its pathogenesis being unrelated to protein aggregation or α-synuclein-induced toxicity. Conversely, in this model, parkin does not play an (essential) role in the pathogenesis of IPS
The “distinguishing model” predicts an increased level of some parkin substrate (“substrate X,” Fig. 3) in the absence of its cognate E3 ligase, inducing neurodegeneration without ever becoming insoluble or forming inclusion bodies. Cyclin E as a possible promoter of apoptotic cell death represents an attractive candidate for such a molecule that conceivably could do its fatal job without ever accumulating to a histologically detectable extent. The pathogenesis of IPS, according to this model, is entirely unrelated to parkin, most likely with α-synuclein as the key player. In this case, the question remains whether LBs are beneficial or noxious, but without relevance for the pathogenesis of AR-JP.
In the next few years, a more detailed analysis of the various animal models and a further dissection of the molecular pathways of parkin and its multiple interactors will most likely provide answers to some of these mysteries and may lead to new strategies for therapeutic intervention.
References
Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Bohme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A (1999) A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum Mol Genet 8:567–574
Aguilar RC, Wendland B (2003) Ubiquitin: not just for proteasomes anymore. Curr Opin Cell Biol 15:184–190
Bae YJ, Park KS, Kang SJ (2003) Genomic organization and expression of parkin in Drosophila melanogaster. Exp Mol Med 35:393–402
Beites CL, Xie H, Bowser R, Trimble WS (1999) The septin CDCrel-1 binds syntaxin and inhibits exocytosis. Nat Neurosci 2:434–439
Berke SJ, Paulson HL (2003) Protein aggregation and the ubiquitin proteasome pathway: gaining the UPPer hand on neurodegeneration. Curr Opin Genet Dev 13:253–261
Braak H, Del Tredici K, Rub U, Vos RA de, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Choi P, Snyder H, Petrucelli L, Theisler C, Chong M, Zhang Y, Lim K, Chung KK, Kehoe K, D’Adamio L, Lee JM, Cochran E, Bowser R, Dawson TM, Wolozin B (2003) SEPT5_v2 is a parkin-binding protein. Brain Res Mol Brain Res 117:179–189
Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM (2001) Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med 7:1144–1150
Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM (2004) S-Nitrosylation of parkin regulates ubiquitination and compromises parkins protective function. Science 304:1328–1330
Ciechanover A, Brundin P (2003) The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40:427–446
Corti O, Hampe C, Koutnikova H, Darios F, Jacquier S, Prigent A, Robinson JC, Pradier L, Ruberg M, Mirande M, Hirsch E, Rooney T, Fournier A, Brice A (2003) The p38 subunit of the aminoacyl-tRNA synthetase complex is a parkin substrate: linking protein biosynthesis and neurodegeneration. Hum Mol Genet 12:1427–1437
Culetto E, Sattelle DB (2000) A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum Mol Genet 9:869–877
Dawson TM, Dawson VL (2003a) Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302:819–822
Dawson TM, Dawson VL (2003b) Rare genetic mutations shed light on the pathogenesis of Parkinson disease. J Clin Invest 111:145–151
Dong Z, Ferger B, Paterna JC, Vogel D, Furler S, Osinde M, Feldon J, Bueler H (2003) Dopamine-dependent neurodegeneration in rats induced by viral vector-mediated overexpression of the parkin target protein, CDCrel-1. Proc Natl Acad Sci USA 100:12438–12443
Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, Margolis RL, Troncoso JC, Lanahan AA, Worley PF, Dawson VL, Dawson TM, Ross CA (1999) Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet 22:110–114
Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, Singleton A, Tanner C, Hardy J, Langston JW (2001) Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 50:293–300
Feany MB, Bender WW (2000) A Drosophila model of Parkinson’s disease. Nature 404:394–398
Feany MB, Pallanck LJ (2003) Parkin: a multipurpose neuroprotective agent? Neuron 38:13–16
Giasson BI, Lee VM (2001) Parkin and the molecular pathways of Parkinson’s disease. Neuron 31:885–888
Giasson BI, Lee VM (2003) Are ubiquitination pathways central to Parkinson’s disease? Cell 114:1–8
Goedert M (2001) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2:492–501
Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635
Gomez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT (2000) Alpha-synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol (Berl) 99:352–357
Gouider-Khouja N, Larnaout A, Amouri R, Sfar S, Belal S, Ben Hamida C, Ben Hamida M, Hattori N, Mizuno Y, Hentati F (2003) Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism Relat Disord 9:247–251
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100:4078–4083
Hayashi S, Wakabayashi K, Ishikawa A, Nagai H, Saito M, Maruyama M, Takahashi T, Ozawa T, Tsuji S, Takahashi H (2000) An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord 15:884–888
Horowitz JM, Myers J, Stachowiak MK, Torres G (1999) Identification and distribution of parkin in rat brain. Neuroreport 10:3393–3397
Huynh DP, Scoles DR, Nguyen D, Pulst SM (2003) The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum Mol Genet 12:2587–2597
Imai Y, Soda M, Takahashi R (2000) Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem 275:35661–35664
Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell 105:891–902
Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, Takahashi R (2002) CHIP is associated with parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol Cell 10:55–67
Ishikawa A, Tsuji S (1996) Clinical analysis of 17 patients in 12 Japanese families with autosomal-recessive type juvenile parkinsonism. Neurology 47:160–166
Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, et al (2003) Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet 12:2277–2291
Kann M, Jacobs H, Mohrmann K, Schumacher K, Hedrich K, Garrels J, Wiegers K, Schwinger E, Pramstaller PP, Breakefield XO, Ozelius LJ, Vieregge P, Klein C (2002) Role of parkin mutations in 111 community-based patients with early-onset parkinsonism. Ann Neurol 51:621–625
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Kitada T, Asakawa S, Minoshima S, Mizuno Y, Shimizu N (2000) Molecular cloning, gene expression, and identification of a splicing variant of the mouse parkin gene. Mamm Genome 11:417–421
Klein C, Pramstaller PP, Kis B, Page CC, Kann M, Leung J, Woodward H, Castellan CC, Scherer M, Vieregge P, Breakefield XO, Kramer PL, Ozelius LJ (2000) Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann Neurol 48:65–71
Lang AE, Lozano AM (1998) Parkinson’s disease. First of two parts. N Engl J Med 339:1044–1053
Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A (2000) Association between early-onset Parkinson’s disease and mutations in the parkin gene. French Parkinson’s Disease Genetics Study Group. N Engl J Med 342:1560–1567
Marx FP, Holzmann C, Strauss KM, Li L, Eberhardt O, Gerhardt E, Cookson MR, Hernandez D, Farrer MJ, Kachergus J, Engelender S, Ross CA, Berger K, Schols L, Schulz JB, Riess O, Kruger R (2003) Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson’s disease. Hum Mol Genet 12:1223–1231
Mata IF, Lockhart PJ, Farrer MJ (2004) Parkin genetics: one model for Parkinson’s disease. Hum Mol Genet 13 (Suppl 1):R127–R133
Matsumine H, Saito M, Shimoda-Matsubayashi S, Tanaka H, Ishikawa A, Nakagawa-Hattori Y, Yokochi M, Kobayashi T, Igarashi S, Takano H, Sanpei K, Koike R, Mori H, Kondo T, Mizutani Y, Schaffer AA, Yamamura Y, Nakamura S, Kuzuhara S, Tsuji S, Mizuno Y (1997) Localization of a gene for an autosomal recessive form of juvenile parkinsonism to chromosome 6q25.2–27. Am J Hum Genet 60:588–596
Moore DJ, Dawson VL, Dawson TM (2003) Role for the ubiquitin–proteasome system in Parkinson’s disease and other neurodegenerative brain amyloidoses. Neuromol Med 4:95–108
Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y (1998) Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 51:890–892
Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279:18614–18622
Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM (2003) Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem 278:48120–48128
Peng XR, Jia Z, Zhang Y, Ware J, Trimble WS (2002) The septin CDCrel-1 is dispensable for normal development and neurotransmitter release. Mol Cell Biol 22:378–387
Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR (2002) Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36:1007–1019
Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70:503–533
Pickart CM (2004) Back to the future with ubiquitin. Cell 116:181–190
Ren Y, Zhao J, Feng J (2003) Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J Neurosci 23:3316–3324
Sampathu DM, Giasson BI, Pawlyk AC, Trojanowski JQ, Lee VM (2003) Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am J Pathol 163:91–100
Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404:770–774
Shimura H, Hattori N, Kubo S, Yoshikawa M, Kitada T, Matsumine H, Asakawa S, Minoshima S, Yamamura Y, Shimizu N, Mizuno Y (1999) Immunohistochemical and subcellular localization of parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann Neurol 45:668–672
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25:302–305
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ (2001) Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 293:263–269
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998a) Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251:205–208
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998b) Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A (2003) Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron 37:735–749
Takahashi H, Ohama E, Suzuki S, Horikawa Y, Ishikawa A, Morita T, Tsuji S, Ikuta F (1994) Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology 44:437–441
Tanaka K, Suzuki T, Chiba T, Shimura H, Hattori N, Mizuno Y (2001) Parkin is linked to the ubiquitin pathway. J Mol Med 79:482–494
Tyers M, Jorgensen P (2000) Proteolysis and the cell cycle: with this RING I do thee destroy. Curr Opin Genet Dev 10:54–64
von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM (2004) Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci USA 101:10744–10749
Wakabayashi K, Engelender S, Yoshimoto M, Tsuji S, Ross CA, Takahashi H (2000) Synphilin-1 is present in Lewy bodies in Parkinson’s disease. Ann Neurol 47:521–523
Wakabayashi K, Engelender S, Tanaka Y, Yoshimoto M, Mori F, Tsuji S, Ross CA, Takahashi H (2002) Immunocytochemical localization of synphilin-1, an alpha-synuclein-associated protein, in neurodegenerative disorders. Acta Neuropathol (Berl) 103:209–214
Warrenburg BP van de, Lammens M, Lucking CB, Denefle P, Wesseling P, Booij J, Praamstra P, Quinn N, Brice A, Horstink MW (2001) Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 56:555–557
West A, Farrer M, Petrucelli L, Cookson M, Lockhart P, Hardy J (2001) Identification and characterization of the human parkin gene promoter. J Neurochem 78:1146–1152
West A, Periquet M, Lincoln S, Lucking CB, Nicholl D, Bonifati V, Rawal N, Gasser T, Lohmann E, Deleuze JF, Maraganore D, Levey A, Wood N, Durr A, Hardy J, Brice A, Farrer M (2002) Complex relationship between parkin mutations and Parkinson disease. Am J Med Genet 114:584–591
West AB, Lockhart PJ, O’Farell C, Farrer MJ (2003) Identification of a novel gene linked to parkin via a bi-directional promoter. J Mol Biol 326:11–19
Yamamura Y, Sobue I, Ando K, Iida M, Yanagi T (1973) Paralysis agitans of early onset with marked diurnal fluctuation of symptoms. Neurology 23:239–244
Yamamura Y, Arihiro K, Kohriyama T, Nakamura S (1993) Early-onset parkinsonism with diurnal fluctuation—clinical and pathological studies. Rinsho Shinkeigaku 33:491–496
Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B (2003) Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron 37:911–924
Zhang Y, Dawson VL, Dawson TM (2000a) Oxidative stress and genetics in the pathogenesis of Parkinson’s disease. Neurobiol Dis 7:240–250
Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM (2000b) Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA 97:13354–13359
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
von Coelln, R., Dawson, V.L. & Dawson, T.M. Parkin-associated Parkinson’s disease. Cell Tissue Res 318, 175–184 (2004). https://doi.org/10.1007/s00441-004-0924-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-004-0924-4