
Abstract
We present a comprehensive clinically oriented workflow for large-insert genome sequencing (liGS)-based nucleotide level resolution and interpretation of de novo (dn) apparently balanced chromosomal abnormalities (BCA) in prenatal diagnosis (PND). Retrospective or concomitant with conventional PND and liGS, molecular and newly developed clinically inspired bioinformatic tools (TAD-GConTool and CNV-ConTool) are applied to analyze and assess the functional and phenotypic outcome of dn structural variants (dnSVs). Retrospective analysis of four phenotype-associated dnSVs identified during conventional PND precisely reveal the genomic elements disrupted by the translocation breakpoints. Identification of autosomal dominant disease due to the disruption of ANKS1B and WDR26 by t(12;17)(q23.1;q21.33)dn and t(1;3)(q24.11;p25.3)dn breakpoints, respectively, substantiated the proposed workflow. We then applied this workflow to two ongoing prenatal cases with apparently balanced dnBCAs: 46,XX,t(16;17)(q24;q21.3)dn referred for increased risk on combined first trimester screening and 46,XY,t(2;19)(p13;q13.1)dn referred due to a previous trisomy 21 pregnancy. Translocation breakpoints in the t(16;17) involve ANKRD11 and WNT3 and disruption of ANKRD11 resulted in KBG syndrome confirmed in postnatal follow-up. Breakpoints in the t(2;19) are within ATP6V1B1 and the 3′ UTR of CEP89, and are not interpreted to cause disease. Genotype–phenotype correlation confirms the causative role of WDR26 in the Skraban-Deardorff and 1q41q42 microdeletion phenocopy syndromes, and that disruption of ANKS1B causes ANKS1B haploinsufficiency syndrome. In sum, we show that an liGS-based approach can be realized in PND care providing additional information concerning clinical outcomes of dnBCAs in patients with such rearrangements.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A causal relationship between a balanced chromosomal abnormality (BCA) and a congenital anomaly is predicted in up to 40% of cases presenting a clinical phenotype-associated BCA (Redin et al. 2017). Recognition of de novo (dn) BCAs leading to disorders constitutes a formidable challenge in prenatal diagnosis (PND). Conventional low-resolution karyotyping remains the standard approach for assigning rearrangement breakpoints of cytogenetically visible dnBCAs in the prenatal setting. Breakpoints of some BCAs have been localized though molecular cytogenomic approaches including FISH, but high-resolution chromosomal microarrays (CMA) are generally insensitive to BCAs (David et al. 2003). More recently, massively parallel sequencing-based methods have been used facilitating nucleotide level resolution of BCAs (Chen et al. 2008; Talkowski et al. 2011).
Long-insert genome sequencing (liGS), with high physical coverage and low sequence depth, has been applied within an actionable timeframe of a PND, for precise identification of BCA breakpoints (Talkowski et al. 2012; Ordulu et al. 2016). BCAs must also be evaluated in the context of copy-number variation (CNV) burden, and the relevance and expanding knowledge of topologically associated domains (TADs) in mechanisms of disease (Dixon et al. 2012; Lupiáñez et al. 2015).
In the present study, we apply the liGS approach for identification of structural variant (SV) breakpoints in four retrospectively analyzed dnBCAs identified during conventional PND and in two ongoing PNDs with dnBCAs. Two bioinformatic tools to assist prediction of the phenotypic outcome of SVs and CNVs in the routine clinical setting were developed including evaluation of the local genomic landscape in which these dnBCAs occurred. Finally, we consider the predictability of the phenotypic outcome of these dnBCAs identified during PND.
Materials and methods
Patients, karyotyping and CMA
Two fetal and four adolescent probands with dnBCAs identified by fetal karyotyping during a conventional PND protocol and their family members were analyzed. Karyotyping and CMA are described in Supplementary Material and Methods.
liGS library preparation, sequencing, bioinformatic analysis and resolution
liGS library preparation, sequencing, and bioinformatic analysis of sequencing data were carried out essentially as described by Talkowski et al. (2011) and Collins et al. (2017). Briefly, after aligning read-pairs against the reference genome, chimeric and improper read-pairs were selected, categorized, clustered and filtered against a so-called blacklist, a list of genomic regions with systematic short-read mappability biases, with an overlap cut-off ≥ 30% (Collins et al. 2017). Based on cluster analysis, different types of balanced and unbalanced SVs such as translocations, insertions (ins), inversions (inv), complex (cx) SV, deletions (del) and tandem duplications (dup) can be identified by liGS. The resolution of liGS is equivalent to the median insert size plus twice the S.D., i.e., ~ 4.5 kb. SVs identified in 689 participants with autism spectrum disorder were used as an SV reference dataset (SVref dataset; Collins et al. 2017). Deletions and tandem duplications identified by depth-of-coverage and improper cluster analysis were cross validated (Klambauer et al. 2012; Collins et al. 2016) and analyzed using our CNV-ConTool.
As long as read-pair clusters do not overlap low-complexity regions, our clinically oriented pipeline includes all translocations, ins, del and dup above 30 kb, and cx SV above 10 kb. A more detailed description is available in Supplementary Material.
Identification of cluster-specific split-reads, CNVs and bioinformatic tools
For the identification of cluster-specific split-reads encompassing BCA or SV breakpoints, a custom Python algorithm was developed and applied. This process uses read-pairs with one of the reads mapped within a breakpoint cluster and the respective paired read unmapped. Detailed description of this algorithm is available in Supplementary Material.
To assist prediction of the phenotypic outcome of SV and CNVs, two bioinformatic tools were developed. TAD-Gene Content Tool (TAD-GConTool) using TAD data from Dixon et al. (2012) and Moore et al. (2015), identifies breakpoint spanning and flanking TADs and retrieves a series of protein-coding and non-coding RNA genes and genomic elements localized within the TADs, as well as associated structural and functional information. Additionally, this tool has the ability to construct the sequence-based nomenclature of the SVs according to the International System for Human Cytogenomic Nomenclature (ISCN) 2016. This tool will be updated in concert with revisions to ISCN 2016, anticipated to be ISCN 2020. CNV-Content Tool (CNV-ConTool) was developed to search for overlap between patient-specific CNVs and those from public databases. This second tool also retrieves data on genes affected by these CNVs. Detailed descriptions of both bioinformatic tools are available in Supplementary Material and Methods.
Both TAD-GConTool and CNV-ConTool can be accessed online at http://dgrctools-insa.min-saude.pt. Source codes are available at https://github.com/DGRC-PT/.
Amplification of junction fragments
Amplification conditions for junction and control fragments of BCA and proband-specific CNVs are summarized in Supplementary Table 1.
Lymphoblastoid cell lines (LCLs), RNA extractions and expression studies
Establishment of LCLs from peripheral blood lymphocytes, extraction of RNAs from LCLs, peripheral blood and amniocytes, and quality assessment and quantification of RNAs were performed essentially as described previously (David et al. 2003).
Genome-wide assessment of gene expression levels in LCL or amniocytes of the probands and controls were performed using the Affymetrix Human Transcriptome Array 2.0 (HTA 2.0, ThermoFisher Scientific). Sample and array processing and data analysis were carried out according to the manufacturer’s instructions and are detailed in the Supplementary Material and Methods.
Variant interpretation and disease prediction
Variants produced by liGS were interpreted according to ACMG sequence based variant criteria (Richards et al. 2015) or CNV criteria (Riggs et al. 2019). For disease prediction (Table 1) bespoke criteria were developed to guide clinicians in the interpretation of sequencing results and ACMG variant classification, as follows:
Disease causing a structural variant resulting in loss-of-function (LoF) of an annotated gene transcript causing an autosomal dominant (AD) clinically relevant or major developmental disorder, where LoF is a known mechanism of the disease;
Disease plausible a structural variant resulting in disruption of an annotated gene transcript intolerant to LoF variants but not yet associated with human disorders, affected gene reported with an important biological function, or convergent genomic and biological evidence (GWAS, gene expression, phenotypic data and other) supporting at least partial involvement of the disrupted gene in the patient clinical phenotype.
Non-disease causing a structural variant resulting in no disruption of protein-coding genes within the bpTADs, no human pathology reported to be associated with genomic elements localized within the bpTADs or no statistically significant GWAS data and/or data supporting at least partial overlap between the genetic traits associated with the affected genomic region and the patient phenotype; and
Low potential of disease a structural variant resulting in LoF of an annotated gene transcript solely causing autosomal recessive (AR) disorders or not associated with an AD clinically relevant or major developmental disorder causing gene localized within the breakpoint topological associating domains (bpTADs);
Results
Patient medical histories
A 39 year-old female presented with an elevated risk for aneuploidy following 1st trimester combined tests with increased nuchal translucency (4.1 mm, > 95th percentile). Chorionic villus sampling (CVS) was performed at 14 weeks gestation for karyotyping of the fetus (designated DGRC0016). Neither parent had any relevant family medical history.
Ultrasound examination at 20 weeks revealed hypoplastic nasal bone and atrioventricular septal defect (AVSD) with ventricular septal defect (VSD) confirmed by fetal echocardiography. Besides AVSD and fetal growth restriction (5th centile), no other fetal anomalies were observed on subsequent evaluations.
Postnatal echocardiogram confirmed the reported AVSD with small VSD but without hemodynamic compromise. At 20 months of age, DGCR0016 presented with developmental delay, most evident in the postural control and locomotor areas, growth restriction and the characteristic facial gestalt to fulfill the criteria for a clinical diagnosis of KBG syndrome (KBGS) (Supplementary Table 2) (Low et al. 2016).
The 40 year-old mother of the second prenatal proband, DGCR0019, had a history of previous pregnancy termination due to trisomy 21 and was referred for amniocentesis at 17 weeks of gestation due to maternal anxiety. Postnatal medical examination of the newborn was phenotypically normal. Besides slightly hypohidrotic skin noticed at four months of age, no other health problem was noted.
Clinical phenotypes of retrospectively analyzed probands are described in Supplementary Results (DGRC0006-t(8;14), DGRC0013-inv(13), DGRC0025-t(12;17), and DGRC0030-t(1;3)) and summarized in Table 1. Clinical features of probands DGRC0006 and DGRC0013 do not match a specific genetic diagnosis, whereas DGRC0025 (Supplementary Fig. 1 and Supplementary Table 3) and DGRC0030 (Supplementary Fig. 2 and Supplementary Table 4) present clinical phenotypes matching a recently reported ANKS1B haploinsufficiency syndrome (Carbonell et al. 2019) and Skraban-Deardorff syndrome (SKDEAS OMIM #617616) (Skraban et al. 2017), respectively.
Conventional prenatal diagnosis
Cytogenetic analysis of the CVS of DGRC0016 revealed a de novo apparently balanced reciprocal translocation, 46,XX,t(16;17)(q24;q21.3)dn (Fig. 1a, b). CMA identified an 810 kb de novo deletion at 8q24.21 interpreted to be a variant of uncertain significance based on a total score of -0.15 (1A, 2H, 3A, 4I and 5A criteria) obtained from the ACMG CNV interpretation guidelines (Riggs et al. 2019). Karyotyping of the amniotic fluid cells of DGRC0019 revealed a de novo apparently balanced reciprocal translocation, 46,XY,t(2;19)(p13;q13.1)dn (Fig. 2a, b). The balanced nature of the translocations was confirmed by CMA and breakpoints mapped on average with a 7 Mb resolution by karyotyping.

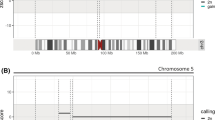
Ideograms, partial karyotype and liGS-based localization of the t(16;17)(q24.3;q21.3)dn breakpoints at genomic and gene levels. a, b Ideograms and GTL-banded normal and derivative metaphase chromosomes. Chromosome 17 ideogram is shaded in yellow. Beside the derivative ideograms the karyotype and liGS-based resolution of the breakpoints are specified. Filled diamonds or arrows indicate chromosome breakpoints. Karyotype resolution indicates the size of the identified disrupted chromosome band. c, d Localization of the t(16;17)(q24.3;q21.3)dn breakpoints at genomic and gene level based on the translocation-specific chimeric cluster. Black and blue arrowheads depict chimeric reads aligned to chromosomes 16 and 17, respectively. Below, gene structure of the disrupted genes, reference transcript numbers and the translational initiation codons (ATG) are indicated. A split-read between positions chr17:46,781,986 and chr16:89,401,732 identified at the der(17) breakpoint is shown by a double arrowhead

Ideograms, partial karyotype and liGS-based localization of the t(2;19)(p13.3;q13.11)dn breakpoints at genomic and gene levels. a, b Ideograms and GTL-banded normal and derivative metaphase chromosomes. Chromosome 19 ideogram is shaded in yellow. Beside the derivative ideograms the karyotype and liGS-based resolution of the breakpoints are specified. Filled diamonds or arrows indicate chromosome breakpoints. Karyotype resolution indicates the size of the disrupted chromosome band established by metaphase analysis. c, d Localization of the t(2;19)(p13.3;q13.11)dn breakpoints at genomic and gene levels based on the translocation-specific chimeric cluster. Black and blue arrowheads depict chimeric reads aligned to chromosomes 2 and 19, respectively. Below, gene structure of the disrupted ATP6B1, reference transcript number and the translational initiation codon are indicated. Additionally, the genomic position of the 19q13.1 breakpoint within the 3′UTR of CEP89 is shown
Detection of SVs from liGS data
Two prenatal and four retrospective probands were sequenced using Illumina short-read (25 bp) sequencing of liGS libraries. Metrics for the libraries are summarized in Supplementary Table 5. Physical coverage was between 42 to 88-fold whereas sequence depth was about one-fold. Chimeric and improper read-pairs ranged between approximately 4–8%.
SVs were identified at liGS resolution of ~ 4.5 kb, but clinical reported at resolution of ≥ 30 kb. A summary of identified chimeric read-pair clusters denoting translocations, ins, inv and cx SV are shown in Supplementary Table 6. At clinical resolution, on average three fully resolved, novel or non-polymorphic (< 1%) SVs were discovered, whereas at liGS resolution, an average of five additional novel or non-polymorphic SVs were identified.
Likewise, a summary of identified del and dup is shown in Supplementary Table 7. At a clinical resolution of ≥ 30 kb, an average of 18 del and dup were identified per proband, but only four del and six dup are considered novel or non-polymorphic (< 1%) based on the SVref dataset (Collins et al. 2017). At liGS resolution, after filtering, an additional 14 del and 15 dup were detected per patient.
Identification of breakpoints at nucleotide resolution
liGS of the fetal DNA sample DGRC0016 identified the 16q breakpoint within a 70 bp region (chr16:89,401,663–89,401,732) at 16q24.3, and the 17q breakpoint was delimited to a 2,300 bp region (chr17:46,781,986–46,784,286) at 17q21.31 (Fig. 1). A split-read was found at the der(17) breakpoint. Sequencing of the second fetal DNA sample DGRC0019 identified the 2p breakpoint within a 485 bp fragment (chr2:70,941,289–70,941,773) at 2p13.3, and the 19q breakpoint was mapped within a 132 bp sequence (chr19:32,878,469–32,878,600) at 19q13.11. Junction fragments for both cases were amplified and Sanger sequenced (Supplementary Table 1 and Supplementary Figs. 3 and 4).
The karyotype of DGRC0016 was revised to t(16;17)(16pter → 16q24.3::17q21.31 → 17qter;17pter → 17q21.31::16q24.3 → 16qter)dn, and according to next-gen cytogenetics nomenclature (Ordulu et al. 2014) is described as 46,XX,t(16;17)(q24;q21.3)dn.seq[GRCh38] t(16;17)(16pter → 16q24.3(89,401,715)::17q21.31(46,784,035) → 17qter;17pter → 17p21.31(46,781,998::16q24.3(89,401,718) → 16qter)dn. The translocation was reclassified as unbalanced due to the 2,036 bp deletion identified at the 17q21.31 breakpoint (Supplementary Fig. 3).
The karyotype of DGRC0019 was revised to t(2;19)(19qter → 19q13.11::2p13.3 → 2qter; 19pter- > 19q13.11::2p13.3 → 2pter)dn, and according to next-gen cytogenetics nomenclature is described as 46,XY,t(2;19)(p13;q13.1)dn.seq[GRCh38] t(2;19)(19qter(-) → 19q13.11(32,878,515)::2p13.3( +)(70,941,507) → 2qter;19pter → 19q13.11( +)(32,878,512)::CATA::2p13.3(-)(70,941,502) → 2pter)dn.
Characterization of breakpoint regions
In DGRC0016, the 16q24.3 breakpoint at position chr16:89,401,715 disrupts IVS3 of ANKRD11 (Ankrd11 repeat domain 11, OMIM *611192), whereas the 17q21.31 breakpoint at position chr17:46,781,998 disrupts IVS1 of WNT3 (Wnt family member 3, OMIM *165330) (Fig. 1). Haploinsufficiency of ANKRD11 causes AD KBGS (OMIM #148050) (Sirmaci et al. 2011).
Homozygous pathogenic variants in WNT3 are associated (but not yet independently confirmed) with recessive tetra-amelia syndrome-1 (TETAMS1, OMIM #165330), a severe malformation syndrome that includes complete absence of all four limbs and other severe anomalies (Niemann et al. 2004). As both ANKRD11 and WNT3 are transcribed on the negative strand, the translocation results in two chimeric genes (Supplementary Fig. 5). Although the chimeric gene at the der(16) breakpoint lacks ANKRD11 exons 1–3, it has an intact ANKRD11 open-reading frame downstream of WNT3 exon 1, translational initiation codon and WNT3 5′ regulatory region.
Regarding the gene content of the 16q24.3 breakpoint-spanning TAD (brTAD) in human embryonic stem cells (hESC) (Dixon et al. 2012), the mitochondrial metalloprotease protein coding gene, SPG7 (paraplegin matrix AAA peptidase subunit) associated with AR/AD adult-onset spastic paraplegia 7 (SPG7, OMIM #607259) is localized 89 kb from the breakpoint (Supplementary Fig. 3 and Supplementary Table 8) (Sánchez-Ferrero et al. 2013). Concerning the 17q21.31 in the brTAD (Fig. 3), in addition to the disrupted WNT3, the myosin light chain 4 gene (MYL4) is localized 425 kb distal to the breakpoint and is etiologic in dominant atrial fibrillation, familial, 18 (ATFB18, OMIM #617280) with an age of onset of 35 years (Orr et al. 2016). Further distal in the brTAD is ITGB3 or platelet glycoprotein IIIa, which has been reported to cause AR or AD platelet-related mild bleeding disorders (BDPLT16, OMIM #187800).

available at https://decipher.sanger.ac.uk/ and their LoF intolerance, expressed as oe-ratio of LoF variants stated below the genes (http://gnomad.broadinstitute.org/). ANKRD11 causing KBGS is marked with a hash mark (#). Data for IMR90 fibroblasts, hESCs and LCL GM12878 are according to Dixon et al. (2012) and Moore et al. (2015), respectively
TADs spanning translocation breakpoints of t(16;17) in LCL GM12878, IMR90, and hESC, and chimeric TADs from derivative chromosomes. a 16q24 breakpoint region. b 17q21.3 breakpoint region. Chromosome 16 TADs are depicted in black or gray, whereas those of chromosome 17 in blue or light blue. Below the TAD tracks, CCCTC-binding factor (CTCF) sites from the analyzed region are shown according to the Chip-seq track in IMR90 fibroblasts (IMR90 CTCF IgG-rab ChIP-seq Signal from ENCODE/SYDH; ENCODE Project Consortium). Horizontal lines with folded gray arrowheads indicate the position of genes in sense and antisense orientations. Genes are color-coded according to their haploinsufficiency index (HI)
In DGRC0019, the 2p13.3 breakpoint at position chr2:70,941,502 disrupts IVS1 of ATP6V1B1 (ATPase H + transporting V1 subunit B1, OMIM *192132), whereas the 19q13.11 breakpoint at position chr19:32,878,515 is located within the 3′ UTR of the CEP89 transcript NM_032816 (centrosomal protein 89, OMIM *615470) (Fig. 2). The disrupted ATPase is a component of the vacuolar ATPases, a multi-subunit enzyme that mediates acidification of eukaryotic intracellular organelles. Pathogenic variants within this gene are reported to cause an AR distal renal tubular acidosis with progressive nerve deafness (OMIM #267300) (Karet et al. 1999). A homozygous deletion comprising CEP89 and SLC7A9 has been reported in a patient with isolated mitochondrial complex IV deficiency, intellectual disability and multisystemic problems (van Bon et al. 2013). SLC7A9, causing cystinuria (OMIN #220,100) with AR and AD inheritance with incomplete penetrance, was identified within the hESC and LCL GM12878 brTADs 8.75 kb proximal from the 19q13.11 breakpoint (Supplementary Fig. 6d and Supplementary Tables 9 and 10) (Rao et al. 2014; Leclerc et al. 2002).
In DGRC0006, the 8q12.3 breakpoint disrupts IVS1 of a large intergenic non-coding (Linc) RNA LINC01414 or RP11-32K4.1 with a brain-specific expression pattern and unreported biological function (Supplementary Fig. 7). The 14q31.2 breakpoint is in a large gene poor region.
In DGRC0013, IVS1 of FLT1 (Fms related tyrosine kinase 1, OMIM *165070) is disrupted (Supplementary Fig. 8). FLT1 is a tyrosine kinase receptor for vascular endothelial growth factors (VEGF) with important roles in angiogenesis and vasculogenesis. Although this receptor has been implicated in development and homeostasis of many organs, it is not yet associated with a human disorder (Tjwa et al. 2003). Flt1 knockout mice models show increased angiogenesis, left ventricle wall thickening and enlargement of the left ventricle cavity, only the last of which is consistent with the DGRC0013 phenotype (Fong et al. 1995; Mei et al. 2015). However, it is not unsurprising that a disruption of a single allele in FLT1 is not totally representative of the loss-of function phenotype in the knockout mouse. No Flt1 knockout mice study showed abnormalities of the tricuspid valve as did DGRC0013, but the repression of VEGF was described as part of the mechanism for heart valve morphogenesis (Chang et al. 2004). Moreover, the enhanced expression of FLT1 in atrioventricular valves, per FANTOM CAT browser, correlates with the reported valve abnormality in the patient (Hon et al. 2017).
In DGCR0025, ANKS1B (Ankrd11 repeat domain 11, OMIM *611192) IVS9 is disrupted (Supplementary Fig. 9). ANKS1B is a tyrosine kinase effector of activity-dependent post-synaptic signaling and a component of the postsynaptic density complex (Jordan et al. 2007). ANKS1B shows an enriched brain-specific expression pattern. Recently, monogenic heterozygous microdeletions in ANKS1B have been reported to cause a spectrum of neurodevelopmental phenotypes (Carbonell et al. 2019).
Finally, in DGRC0030 the breakpoints disrupt exon 12 of WDR26 (WD repeat-containing protein 26; OMIM *617424) and IVS1 of ATP2B2 (ATPase plasma membrane Ca2 + transporting 2; OMIM *108733) (Supplementary Fig. 10). Pathogenic variants in these genes are reported to cause AD SKDEAS and AD non-syndromic sensorineural hearing impairment, respectively (Skraban et al. 2017; Smits et al. 2019).
Genomic imbalances
The median size of del and dup at clinical resolution is 64 and 49 kb, respectively. Two deletions, 53.512 kb at 3p24.1 (27,354,680–27,408,191) and 836.049 kb at 8q24.21 (129,061,233–129,897,281), identified in DGRC0016 (Supplementary Figs. 11, 12 and Supplementary Table 11) were not found in public CNV databases. The deletion at 3p24.1, classified as a VUS with a total score of − 0.45 (1A, 3A, 4 J(− 0.30), 5C(− 0.15)) according to ACMG CNV criteria (Riggs et al. 2019), is present in the proband’s phenotypically normal mother and brother, and, therefore, unlikely to contribute to an abnormal phenotype. As for the 836.047 kb de novo deletion, none of the affected genes has been considered to cause a reported phenotype and the deletion is interpreted as VUS according to ACMG CNV criteria (Riggs et al. 2019). Moreover, although several genetic traits have been associated by GWAS with the affected genomic region, none of these represent developmental disorders (Supplementary Table 12). Posteriorly, this SV was considered as unrelated to the patient's reported clinical features. Regarding DGRC0019, with the exception of a 12,033 bp deletion within the olfactory receptor family 5 subfamily B pseudogene region (chr11q12.1:58,336,732–58,348,764), no other proband-specific alteration was detected (Supplementary Table 13).
Proband-specific del and dup identified in the retrospectively analyzed probands are summarized in Supplementary Table 14, and inv, ins and cx SV in Supplementary Table 15. Most likely, none of these SVs has a pathogenic implication.
Expression studies
From the disrupted genes in prenatal probands, only ANKRD11 and CEP89 are ubiquitously expressed in LCLs. WNT3 shows skin enriched expression, whereas ATP6V1B1 has kidney, lung and skin enhanced expression (Supplementary Figs. 13 and 14). Expression array profiling of the t(16;17) proband’s LCLs shows that due to low sensitivity of this HTA 2.0 array, the whole gene expression level of ANKRD11 is roughly the same as that in controls (7.77 vs. 7.85, SD 0.09) whereas that of WNT3 is increased (5.86 vs. 4.6, SD 0.01) (Supplementary Table 16). The increased WNT3 exon 5 signal intensity (33.58) may explain the observed whole gene elevated WNT3 expression (Supplementary Fig. 15). Expression levels of the remaining genes from both brTAD were roughly similar to controls (Supplementary Table 16).
HTA 2.0 expression data of cultured human amniocytes are not available in the literature. Therefore, expression data of the t(2;19) proband’s cultured amniocytes were compared to LCLs as control. Noticeable altered expression above the threshold of the microarray was not observed at the level of gene, exon or exon splicing (data not shown).
Predictability of the phenotypic outcome of dnBCA
The pathogenicity of an SV should be assessed separately for each breakpoint and jointly as a single alteration. In the absence of established guidelines or criteria for classification of SV, we based our classification on ACMG criteria for sequence variants (Richards et al. 2015). Variant classification and clinical interpretation of BCAs is summarized in Table 1.
During PND of DGRC0016, the t(16;17) rearrangement at 16q24.3 was classified as PM6 (ACMG criterion PM6—assumed de novo, but without confirmation of paternity and maternity) and the absence of ANKRD11 exons 1–3 in the der(16) as PSV1 (Table 1). Therefore, the 16q24.3 rearrangement was interpreted as a likely pathogenic variant, most likely leading to a KBG syndrome-like phenotype. Postnatally, it was further classified as PP4 (PP4—patient’s phenotype and family history highly specific for a disease with a single gene etiology). Moreover, although KBG syndrome is typically milder and less frequently diagnosed in females, the patient’s clinical features meet the diagnostic criteria for KBG (Richards et al. 2015) (Supplementary Table 2) and, therefore, the ACMG interpretation was upgraded to pathogenic (Table 1).
During PND of DGRC0019, the t(2;19) rearrangement at 2p13.3 was also classified as PM6 and according to our interpretation criteria (Table 1) was predicted to have a “Low potential of disease.”
For DGRC0006, none of the affected genes or identified genomic alterations is associated with pathologies nor show overlap with the patient’s phenotype. Furthermore, GWAS data do not reach genome-wide statistical significance (Supplementary Table 17). Therefore, based on our criteria we consider this variant as “Non-disease causing” (Table 1).
In DGRC0013 and DGRC0025, dnBCA breakpoints directly disrupt genes with a low ratio of observed/expected (oe) number of LoF variants indicating a strong LoF intolerance (Table 1 and Supplementary Tables 18 and 19) but neither are curated in ClinGen. Both SVs (i.e., involving FLT1 and ANKS1B) can only be scored to PM6, but based on our criteria are predicted to be “Disease plausible” (Table 1). Of note, the clinical phenotype of DGRC0025 matches a recently reported ANKS1B haploinsufficiency syndrome (Carbonell et al. 2019).
Finally, in DGRC0030, the SV disrupting the disease gene WDR26 was classified as PVS1, PM6 and PP4 corresponding to pathogenic by ACMG criteria (Richards et al. 2015). Thus, for the WDR26 variant our interpretation was “Disease causing” and the proband’s clinical phenotype coincides with that of age-matched patients with SKDEAS (Skraban et al. 2017). ATP2B2 is not curated as a dominant disease locus (Supplementary Table 20), but the ATP2B2 variant is classified as “Disease plausible” (Table 1).
TAD analysis
It is now clearly established in the literature that disruption of TADs and the creation of neo-TADs are dominant mechanisms of SVs (Lupiáñez et al. 2015; Franke et al. 2016). The main source of knowledge of TAD maps are generated by chromosome conformation capture (Hi-C) data. Details of TADs involved in DGRC0016 are presented in Fig. 3. None of the t(16;17) breakpoints disrupt an interaction loop (data not shown) (Rao et al. 2014). Hi-C contact heatmaps of the t(2;19) breakpoint regions for LCLs and IMR90 are shown in Supplementary Fig. 6. Although the 2p13.3 breakpoint disrupts two interaction loops (Supplementary Fig. 6a, b) (Rao et al. 2014), none of the involved genes shows LoF sensitivity.
Discussion
Genome sequencing data of two ongoing fetal and four retrospective samples with dnBCA identified during conventional PND were analyzed by liGS, followed by comprehensive structural analyses of candidate genes from the disrupted bpTADs and prediction of the phenotypic outcome. Moreover, to facilitate implementation of this analysis, two new bioinformatic tools applicable in the clinical setting have been developed. Using this information and the developed bioinformatic tools, we propose an analytical workflow for identification and interpretation of de novo SVs in their genomic landscape (Fig. 4).

Proposed workflow for nucleotide level resolution and interpretation of de novo structural rearrangements in their genomic landscape in prenatal diagnosis. a In the case of BCAs associated with fetal anomalies or de novo SVs, concomitantly with the conventional PND protocol, liGS should be performed for nucleotide level resolution of the rearrangement breakpoints in their genomic landscape. Inherited BCAs are referred to a clinical geneticist to lay out the follow-up required, including potential inclusion of liGS. Foreseeably, upon improvement of liGS-based methods, all prenatally identified non-polymorphic SV may be analyzed by such an approach. b Long-insert-based genome sequencing such as mate-pair, with short or medium reads, from 2 × 25 to 150 bp, can be used. Increasing the sequence coverage enables identification of SNV and indels from the same sequencing data. c The proposed bioinformatic workflow: (i) Sequence data decoded in different types of read-pairs, (ii) Clusters denoting different types of balanced and unbalanced SVs (translocations, ins, inv, cx SV, del and dup) are identified at the liGS resolution, (iii) Identification of cluster-specific split-reads, (iv) Genomic regions comprising deletions and tandem duplications revealed by the two procedures merged, (v) Potentially pathogenic candidate genes, genomic loci and CNVs revealed by bioinformatic tools TAD-GConTool and CNV-ConTool, and (vi) Analysis of the disrupted or dysregulated genes and CNVs, orthogonal confirmation, validation, and preparation of report by a certified medical geneticist
In DGCR0016, translocation breakpoints disrupt a single allele of ANKRD11 and of WNT3, wherein haploinsufficiency of ANKRD11 causes AD KBGS. KBGS was first reported by Herrmann et al. (1975) in three unrelated families with the surnames initials being K, B and G. The common phenotypic characteristics of this multiple congenital anomaly comprises, among others, a characteristic facial appearance (including protruding ears and hypertelorism), hand anomalies, neurologic involvement, and postnatal short stature (Skjei et al. 2007), which are consistent with the phenotype observed in the patient and fits KBGS diagnostic criteria (Low et al. 2016).
In DGCR0019, translocation breakpoints disrupt genes tolerant to LoF variants, ATP6V1B1 and CEP89. No gene causing AD or developmental disorder was identified within the bpTADs. The predicted outcome was confirmed by absence of a postnatal clinical phenotype. Nevertheless, longer term follow-up would be warranted to exclude any later onset of a disorder that might be associated as recently demonstrated for prenatally detected dnBCAs (Halgren et al. 2018) or natural history of individuals with postnatal dnBCAs (Currall et al. 2018).
Of the four retrospectively analyzed dnBCAs, similarly to the aforementioned DGCR0016, disruption of WDR26 predictably will lead to SKDEAS. The patient's clinical phenotype highlights that these phenocopies, SKDEAS and 1q41q42 deletion syndrome, are primarily caused by disruption of WDR26.
Although the breakpoints of dnBCAs inv(13) and t(12;17) disrupt genes not yet curated in ClinGen, the fact that these are significantly LoF intolerant genes involved in several biological processes, reinforced by convergent evidence, led us to predict that they are “Disease plausible”. Furthermore, DGRC0025 clinical phenotype overlaps ANKS1B haploinsufficiency syndrome.
Finally, for the postnatal phenotype of DGRC0006, the t(8;14) variant is predicted as “Non-disease causing.” Although pathogenic cx SVs smaller than our clinical resolution cutoff have been reported (Sanchis-Juan et al. 2018), at the higher resolution of liGS no additional presumably pathogenic SV was identified in DGRC0006. Exome sequencing (ES) has not been performed, and other non-genetic factors unrelated to the translocation may be responsible for the phenotype (e.g., environmental or multifactorial factors).
Short-read sequencing by either genome sequencing (GS) or ES has been applied in the prenatal setting. ES in fetuses with structural anomalies was recently elucidated in a large-scale study (Lord et al. 2019; Petrovski et al. 2019) revealing a genetic etiology in about 10% of affected fetuses. However, short-read sequencing is not optimal for the identification of SVs. The physical coverage of GS is relatively low, whereas ES is high but will miss breakpoints localized within non-coding sequence. The long-insert size of the liGS libraries, intended for the identification of BCAs and CNVs, and low read size results in high physical but low sequence coverage.
The lack of transcriptome data on gestational age- and sex-matched first trimester cells from CVS and amniotic fluid is a current limitation for the introduction of gene expression analysis in the clinical prenatal setting. Clearly, the future of prenatal diagnosis for SVs will require generation of gene expression data by RNA-Seq linked to Hi-C of CVS cells and amniocytes, as is available now in public databases for adult tissues. Presently, the interpretation of current PND of dnBCAs could be limited to disruption of major dominant genes leading to Mendelian disorders as occurred for ANKRD11, WDR26 and ANKS1B. Nonetheless, cytogenetics laboratories should be attentive to take into consideration the architectural features of genomes to address fully the disease potential of a SVs (Lupiáñez et al. 2015).
In comparison to karyotyping and CMA analysis, we demonstrate the benefits of an liGS-based approach and our clinically inspired pipeline for identification of dnBCA breakpoints and interpretation of the genomic landscape on which these occurred in the prenatal setting. We show the predictability of the clinical outcome of these BCAs and plan to provide updated bioinformatic tools to facilitate data analysis and a workflow for implementation of genome sequencing in the diagnostic prenatal setting.
References
Carbonell AU, Cho CH, Tindi JO et al (2019) Haploinsufficiency in the ANKS1B gene encoding AIDA-1 leads to a neurodevelopmental syndrome. Nat Commun 10:3529
Chang CP, Neilson JR, Bayle JH et al (2004) A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell 118:649–663
Chen W, Kalscheuer V, Tzschach A et al (2008) Mapping translocation breakpoints by next-generation sequencing. Genome Res 18:1143–1149
Collins RL, Stone MR, Brand H et al (2016) CNView: a visualization and annotation tool for copy number variation from whole-genome sequencing. bioRxiv. https://doi.org/10.1101/049536
Collins RL, Brand H, Redin CE et al (2017) Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol 18:36
Currall BB, Chen M, Sallari RC et al (2018) Loss of LDAH associated with prostate cancer and hearing loss. Hum Mol Genet 27:4194–4203
David D, Cardoso J, Marques B et al (2003) Molecular characterization of a familial translocation implicates disruption of HDAC9 and possible position effect on TGFB2 in the pathogenesis of Peters’ anomaly. Genomics 81:489–503
Dixon JR, Selvaraj S, Yue F et al (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380
Fong GH, Rossant J, Breitman ML (1995) Role of the FLT1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376:66–69
Franke M, Ibrahim DM, Andrey G et al (2016) Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538:265–269
Halgren C, Nielsen NM, Nazaryan-Petersen L et al (2018) Risks and recommendations in prenatally detected de novo balanced chromosomal rearrangements from assessment of long-term outcomes. Am J Hum Genet 102:1090–1103
Herrmann J, Pallister PD, Tiddy W, Opitz JM (1975) The KBG syndrome: a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig Artic 11:7–18
Hon CC, Ramilowski JA, Harshbarger J et al (2017) An atlas of human long non-coding RNAs with accurate 5' ends. Nature 543:199–204
Jordan BA, Fernholz BD, Khatri L, Ziff EB (2007) Activity-dependent AIDA-1 nuclear signaling regulates nucleolar numbers and protein synthesis in neurons. Nat Neurosci 10:427–435
Karet FE, Finberg KE, Nelson RD et al (1999) Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21:84–90
Klambauer G, Schwarzbauer K, Mayr A et al (2012) cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucl Acids Res 40:e69
Leclerc D, Boutros M, Suh D et al (2002) SLC7A9 mutations in all three cystinuria subtypes. Kidney Int 62:1550–1559
Lord J, McMullan DJ, Eberhardt RY et al (2019) Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 393:747–757
Low K, Ashraf T, Canham N et al (2016) Clinical and genetic aspects of KBG syndrome. Am J Med Genet A 170:2835–2846
Lupiáñez DG, Kraft K, Heinrich V et al (2015) Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161:1012–1025
Mei L, Huang Y, Lin J et al (2015) Increased cardiac remodeling in cardiac-specific Flt-1 receptor knockout mice with pressure overload. Cell Tissue Res 362:389–398
Moore BL, Aitken S, Semple CA (2015) Integrative modeling reveals the principles of multi-scale chromatin boundary formation in human nuclear organization. Genome Biol 16:110
Niemann S, Zhao C, Pascu F et al (2004) Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am J Hum Genet 74:558–563
Ordulu Z, Wong KE, Currall BB et al (2014) Describing sequencing results of structural chromosome rearrangements with a suggested next-generation cytogenetic nomenclature. Am J Hum Genet 94:695–709
Ordulu Z, Kammin T, Brand H et al (2016) Structural chromosomal rearrangements require nucleotide-level resolution: lessons from next-generation sequencing in prenatal diagnosis. Am J Hum Genet 99:1015–1033
Orr N, Arnaout R, Gula LJ et al (2016) A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun 7:11303
Petrovski S, Aggarwal V, Giordano JL et al (2019) Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet 393:758–767
Rao SSP, Huntley MH, Durand NC et al (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159:1665–1680
Redin C, Brand H, Collins RL et al (2017) The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet 49:36–45
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Riggs ER, Andersen EF, Cherry AM et al (2019) Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. https://doi.org/10.1038/s41436-019-0686-8
Sánchez-Ferrero E, Coto E, Beetz C et al (2013) SPG7 mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p. A510V. Clin Genet 83:257–262
Sanchis-Juan A, Stephens J, French CE et al (2018) Complex structural variants in Mendelian disorders: identification and breakpoint resolution using short- and long-read genome sequencing. Genome Med 10:95
Sirmaci A, Spiliopoulos M, Brancati F et al (2011) Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet 89:289–294
Skjei KL, Martin MM, Slavotinek AM (2007) KGB syndrome: report of twins, neurological characteristics, and delineation of diagnostic criteria. Am J Med Genet 143A:292–300
Skraban CM, Wells CF, Markose P et al (2017) WDR26 haploinsufficiency causes a recognizable syndrome of intellectual disability, seizures, abnormal gait, and distinctive facial features. Am J Hum Genet 101:139–148
Smits JJ, Oostrik J, Beynon AJ et al (2019) De novo and inherited loss-of-function variants of ATP2B2 are associated with rapidly progressive hearing impairment. Hum Genet 138:61–72
Talkowski ME, Ernst C, Heilbut A et al (2011) Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet 88:469–481
Talkowski ME, Ordulu Z, Pillalamarri V et al (2012) Clinical diagnosis by whole-genome sequencing of a prenatal sample. N Engl J Med 367:2226–2232
Tjwa M, Luttun A, Autiero M, Carmeliet P (2003) VEGF and PlGF: two pleiotropic growth factors with distinct roles in development and homeostasis. Cell Tissue Res 314:5–14
van Bon BW, Oortveld MA, Nijtmans LG et al (2013) CEP89 is required for mitochondrial metabolism and neuronal function in man and fly. Hum Mol Genet 22:3138–3151
Acknowledgements
We dedicate this article to Maria Guida Boavida who founded the Human Genetics Laboratory as the precursor of the current Department of Human Genetics at the National Health Institute Doutor Ricardo Jorge, Lisbon, Portugal. She and her colleagues performed the first chromosomal prenatal diagnosis from cultured amniocytes in Portugal in July 1977 (no. 77180). However, the first translocation in PND, a paternally inherited Robertsonian t(13;14) (no. 80321), was only identified in June 1980. Almost 40 years later, we report application of an liGS-based approach for the identification of dnBCAs breakpoints in PND. First, we are grateful to the family members for their involvement in this study. We thank Dr. Michael Talkowski and members of his lab for library preparation and sequencing data generation. We thank Jader Cruz for echographic diagnosis of fetal heart defects, to Hildeberto Correia, Ana Cristina Alves and Bárbara Marques for conventional prenatal diagnosis, and to Márcia Rodrigues, Sofia Nunes, Teresa Kay, and Rui Gonçalves for their contribution to the project. C.C.M. is supported (GM061354) by the National Institutes of Health (USA) and the NIHR Manchester Biomedical Research Centre, UK. This research was supported by national funds through FCT—Fundação para a Ciência e a Tecnologia, Research Grant HMSP-ICT/0016/2013 of the Harvard Medical School—Portugal Program in Translational Research and Information.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Informed consent
This study was approved by the Ethics Committee of the National Institute of Health Doutor Ricardo Jorge and was carried out according to the Principles of the Declaration of Helsinki of the World Medical Association. Samples were obtained after written informed consent of the participants or their legal representatives. Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
David, D., Freixo, J.P., Fino, J. et al. Comprehensive clinically oriented workflow for nucleotide level resolution and interpretation in prenatal diagnosis of de novo apparently balanced chromosomal translocations in their genomic landscape. Hum Genet 139, 531–543 (2020). https://doi.org/10.1007/s00439-020-02121-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02121-x