
Abstract
Genome-wide association studies have reported 56 independently associated colorectal cancer (CRC) risk variants, most of which are non-coding and believed to exert their effects by modulating gene expression. The computational method PrediXcan uses cis-regulatory variant predictors to impute expression and perform gene-level association tests in GWAS without directly measured transcriptomes. In this study, we used reference datasets from colon (n = 169) and whole blood (n = 922) transcriptomes to test CRC association with genetically determined expression levels in a genome-wide analysis of 12,186 cases and 14,718 controls. Three novel associations were discovered from colon transverse models at FDR ≤ 0.2 and further evaluated in an independent replication including 32,825 cases and 39,933 controls. After adjusting for multiple comparisons, we found statistically significant associations using colon transcriptome models with TRIM4 (discovery P = 2.2 × 10− 4, replication P = 0.01), and PYGL (discovery P = 2.3 × 10− 4, replication P = 6.7 × 10− 4). Interestingly, both genes encode proteins that influence redox homeostasis and are related to cellular metabolic reprogramming in tumors, implicating a novel CRC pathway linked to cell growth and proliferation. Defining CRC risk regions as one megabase up- and downstream of one of the 56 independent risk variants, we defined 44 non-overlapping CRC-risk regions. Among these risk regions, we identified genes associated with CRC (P < 0.05) in 34/44 CRC-risk regions. Importantly, CRC association was found for two genes in the previously reported 2q25 locus, CXCR1 and CXCR2, which are potential cancer therapeutic targets. These findings provide strong candidate genes to prioritize for subsequent laboratory follow-up of GWAS loci. This study is the first to implement PrediXcan in a large colorectal cancer study and findings highlight the utility of integrating transcriptome data in GWAS for discovery of, and biological insight into, risk loci.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is estimated that genetic variants explain 12–35% of the heritability in colorectal cancer (CRC) risk (Lichtenstein et al. 2000; Czene et al. 2002; Jiao et al. 2014). To date, Genome-Wide Association Studies (GWAS) have identified 56 independent common risk variants that are robustly associated with CRC (Peters et al. 2015; Schumacher et al. 2015; Orlando et al. 2016). However, the functional relevance of most discovered CRC-risk variants (89%) remains unclear. The biological mechanisms linking CRC-associated risk variants with target genes have only been validated in the laboratory for six regions [8q24 MYC (Pomerantz et al. 2009), 8q23.3 EIF3H (Pittman et al. 2010), 11q23.1 COLCA1 and COLCA2 (Biancolella et al. 2014), 15q13.3 GREM1 (Lewis et al. 2014), 16q22.1 CDH1 (Shin et al. 2004), and 18q21.1 SMAD7 (Fortini et al. 2014)]. Given that most of the associated loci do not include coding variants, a large portion of CRC genetic risk is thought to be explained by regulatory variation that modulates the expression of target genes. This hypothesis is supported by the observation that CRC risk variants are enriched in colon expression quantitative trait loci (eQTLs) (Hulur et al. 2015) and active regulatory regions of colorectal enhancers (Bien et al. 2017). Together, this evidence highlights the value of studying transcriptional regulation in relation to CRC risk.
Large-scale efforts are underway to map regulatory elements across tissues and cell types. Many transcriptome studies have been conducted where genotype and expression levels are jointly assayed for many individuals, enabling the discovery of tissue-specific eQTLs. For instance, the Genotype-Tissue Expression (GTEx) Project (GTEx Consortium 2013) is building a biospecimen repository to comprehensively map tissue-specific eQTLs across human tissues, which currently includes transcriptomes from 169 colon transverse samples. These data provide a remarkable new resource for understanding function in non-coding regions that can be used to inform GWAS.
We employed the computational method, PrediXcan (Gamazon et al. 2015), to perform a CRC transcriptome-wide association study using reference datasets to ‘impute’ unobserved expression levels into GWAS datasets. Variant prediction models were developed using colon transverse transcriptomes (n = 169) from GTEx (GTEx Consortium 2013) and a larger whole blood transcriptome panel (n = 922) from the depression genes and networks (DGN) (Battle et al. 2014). We included whole blood as a previous analysis demonstrated that gene regulatory elements of immune cell types from peripheral blood are enriched for variants with more significant CRC association P (Bien et al. 2017). Further, laboratory follow-up of the CRC GWAS locus 11q23 implicates two genes, COLCA1 and COLCA2, which are co-expressed in immune cell types and correlate with inflammatory processes (Peltekova et al. 2014). In addition to novel discovery, the PrediXcan approach can aid in prioritization of candidate target genes in non-coding GWAS loci and thereby inform testable hypotheses for laboratory follow-up. Therefore, as a secondary analysis we investigated the association of imputed gene expression with CRC in the 44 genetic regions harboring one or more of the 56 independent variants (r2 < 0.2) that are associated with CRC in previous GWAS (P ≤ 5 × 10− 8) and were replicated in an independent dataset.
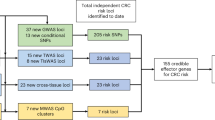
We aimed to discover novel loci associated with CRC, and refine established regulatory risk loci by reducing the list of putative gene targets. Employing PrediXcan, we tested genetically regulated gene expression for association with CRC in a two-stage approach. In the discovery stage, up to 8277 gene sets were tested in 12,186 cases and 14,718 controls from the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO) and the Colon Cancer Family Registry (CCFR). This discovery set was also used to identify potential target genes in the 44 genetic regions harboring 56 known CRC risk variants. We attempted replication of three novel genes that were not positioned within 1 Mb of the 56 previously reported risk variants and with false discovery rate (FDR) ≤ 0.2 for CRC risk in a large and independent study of 32,825 cases and 39,933 controls from the Colorectal Transdisciplinary (CORECT) consortium, UK Biobank, and additional CRC GWAS (Fig. 1).

Schematic illustration of the study design training data was comprised of joint observations of imputed variant genotypes and tissue-specific gene expression from reference datasets (DGN and GTEx). Elastic net regularization was used to train genetic variant predictors of gene expression and downloaded from PredictDB.org. Models for colon transverse tissues and whole blood were used for imputation of expression into independent GWAS datasets for Colorectal Cancer (CRC). Imputed gene expression was then tested for association with case (ca.)–control (co.) status in the discovery stage. Novel gene associations with a false discovery rate (FDR) = 0.2 were assessed in an independent CRC GWAS dataset. As a secondary analysis, the association of genetically determined expression of genes in 44 GWAS-associated risk regions was examined
Results
Imputation of genetically regulated gene expression
Gene expression levels were imputed using previously published multi-variant models built using elastic net regularization (variant weight gene models V6 available online from PredictDB.org). For each tissue and gene, a quality metric referred to as predictive R2 was provided as the correlation between the observed and predicted expression from the multi-variant model based on a tenfold cross validation. After restricting to protein coding genes with a predictive R2 > 0.01 (≥ 10% correlation between predicted and observed expression), the discovery analysis tested the association of imputed expression for 4850 genes using colon transverse models and 8277 genes using whole blood models. On average, colon transverse models used 22 variants (SD = 19) per gene with a range of 1–173 variants. The number of variants in whole blood models were slightly larger on average with a mean of 34 variants (SD = 24) per gene, ranging from 1 to 213 variants. We report CRC association results and predictive R2 for imputed expression of each gene with P ≤ 0.05 in either colon transcriptome or whole blood analysis (Online Resource 2 Table S2).
Discovery of new CRC susceptibility genes
In total, multivariate logistic regression was used to test the association of CRC with genetically impute gene expression for 4850 genes from colon transverse models and 8277 genes from whole blood models. We employed PrediXcan in 12,186 cases and 14,718 controls from 16 GWAS studies. Replication was attempted for associations meeting an FDR = 0.2 threshold in the discovery phase if they were in a novel CRC region using an independent GWAS dataset comprised of 32,825 cases and 39,933 controls from the CORECT consortium, UK Biobank, and additional GWAS as described in Online Resource 1. In the discovery phase, colon transcriptome models identified CRC association with imputed genetically regulated gene expression in three putative novel regions. Two out of three genes tested in the replication dataset were significant after adjusting for multiple comparisons (α = 0.05/3 = 0.017) (Online Resource Fig S1, Table 1). In addition to being more than 1 Mb away from previously identified risk variants, we confirmed that none of the variant predictors used to impute gene expression for these three genes were in LD (r2 ≤ 0.1) with previously published CRC-risk variants. In the 7q22.1 locus, increased expression of TRIM4 was associated with reduced CRC risk with an odds ratio (OR) of 0.94 [95% confidence interval (CI) 0.91–0.97, discovery P = 2.2 × 10− 4]. Reduced CRC risk was also statistically associated with increased genetically regulated gene expression of TRIM4 in the independent replication dataset (P = 0.01). The second novel locus, 14q22.1, was also found to be inversely associated, where increased genetically regulated gene expression of PYGL was associated with decreased CRC risk, showing an OR of 0.90 (95% CI 0.85–0.96) in the discovery dataset (discovery P = 2.3 × 10− 4) as well as in the replication dataset (P = 7.9 × 10− 4). Imputed genetically regulated gene expression for SLC22A31 was associated with increased CRC risk in the discovery phase (P = 1.3 × 10− 4), but did not replicate in the independent dataset. We found no associations in novel regions using whole blood variant models that reached FDR = 0.2 in the discovery phase.
Colon Transverse PrediXcan analyses were repeated for TRIM4 and PYGL in the discovery dataset stratifying cases by proximal (n = 4454 cases), distal (n = 3580 cases), and rectal (n = 2936 cases) cancer sites. We excluded 1216 cases from the stratified analysis because the colon cancer site was unspecified. We found that for both genes the effects and p values were similar between the three sites. For TRIM4, the CRC association with genetically imputed gene expression had an OR of 0.94 (95% CI 0.90–0.98, P = 3 × 10− 3) in proximal colon cases compared to an OR of 0.95 (95% CI 0.90–1.0, P = 5 × 10− 2) in distal colon cases and an OR of 0.93 (95% CI 0.88–0.98, P = 2 × 10− 2) in rectal cases. There was no significant difference in the effect estimates between these cancer sites for TRIM4 (Q-test for heterogeneity P = 1.0). Similarly, for PYGL, the CRC association with genetically regulated gene expression had an OR of 0.89 (95% CI 0.82–0.97, P = 3 × 10− 3) in proximal colon cases compared to an OR of 0.91 (95% CI 0.83–1.0, P = 2 × 10− 2) in distal colon cases and an OR of 0.86 (95% CI 0.77–0.95, P = 5 × 10− 4) in rectal cases with no significant difference in effects (Q test for heterogeneity P = 0.98).
We further investigated the replicated CRC-associated PrediXcan genes by summarizing the single-variant CRC association results for variants that were included in the prediction models, referred to hereafter as ‘variant predictors’ (Online Resources 3–6 Fig S2). In TRIM4, the association was mostly driven by one LD block with 62 correlated genetic variant predictors used to impute genetically regulated gene expression in colon tissue models. Among the variant predictors of TRIM4, rs2527886 was most significantly associated with CRC (P = 1.8 × 10− 4). Bioinformatic follow-up of the TRIM4 locus showed that in the genomic region containing variants correlated with rs2527886, there were six enhancers with strong Chromatin Immunoprecipitation Sequencing (ChIP-seq) H3K27ac signal in either normal colorectal crypt cells or a CRC cell line (Online Resource 1 Fig S3). Using peak signal from H3K27ac activity to define enhancer regions, two enhancers were gained in ten or more CRC cell lines compared to normal colorectal crypt cells, referred to as recurrent variant enhancer loci (VEL) (Akhtar-Zaidi et al. 2012). Rs2527886 is positioned within one of these VEL. Peak ChIP-seq binding region for CTCF suggests that the VEL harboring rs2527886 may be in physical contact with the TRIM4 promoter. In the same VEL, one of the LD variants, rs2525548 (LD r2 = 0.99), is positioned within transcription factor binding sites for RUNX3, FOX, NR3C1, and BATF (Online Resource 1 Fig S3). In the PYGL locus, rs12589665 is the variant predictor with the strongest marginal association with CRC (P = 3.2 × 10− 4). We identified 7 enhancers in the region spanning the variants in LD with rs12589665, and three variants in LD with the lead predictor variant were positioned in VEL. Two of these variants, rs72685325 (r2 = 0.62) and rs72685323 (r2 = 0.53), were positioned within binding sites for 7 transcription factors (Online Resource 1 Fig S3).
A series of exploratory analyses were conducted to assess whether the observed inflation in association signals (λ = 1.1) was the result of bias in our data or modeling error. Results suggest that inflation was not driven by genes with low predictive R2 values (Online Resource 1 Fig S4), other potential confounding factors common to GWAS like genotyping batch effects (Online Resource 1 Fig S5) or cryptic population structure (Online Resource 1 Fig S6–S7), or due to inflated Z statistics by modeling genes with little variability in expression (Online Resource 1 Fig. S8–S11). Observed inflation was slightly reduced, but still elevated when looking at the marginal association results for the variant predictors (λ = 1.07; Online Resource 1 Fig S12) and when excluding genes with high predicted co-expression (λ = 1.07; Online Resource 1 Fig S13). Collectively, this exploration suggests that the observed inflation is less likely to be the result of modeling or analytical error and more likely reflects the polygenicity of CRC.
Refinement of known CRC GWAS-risk regions
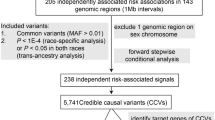
We first assembled a list of 56 previously reported independent (r2 ≤ 0.2) CRC GWAS risk variants and defined a distance-based region surrounding each variant as the chromosomal position of the first reported (index) variant ± 1 Mb (Online Resource 1 Table S3). We then combined overlapping risk regions by taking the minimum and maximum chromosomal positions of all regions that overlapped, resulting in a total of 44 CRC risk regions harboring 1–4 independent CRC-risk variants. In these 44 regions, there was an average of 20 (SD ± 17) protein-coding genes per region annotated by the Consensus Coding Sequence Database (CCDS). The average number of protein-coding genes per region with imputed genetically regulated gene expression in the tissue-specific models was reduced to an average of 10 (SD ± 8) genes in colon transverse, and 14 (SD ± 11) genes in whole blood. Further, in these regions we found that of the total number of genes with genetically regulated gene expression across the two models, an average 45% of the genes overlapped. We found that 34/44 (77%) of CRC-risk regions overlapped the transcription start site of a gene associated with CRC at a P < 0.05. Comparing the number of genes with a P < 0.05 to the total number of CCDS genes within 1 Mb of an index variant resulted in an average reduction of 82% per region (Table 2).
We further investigated the regions that did not show evidence of gene association and found that GWAS reported risk variants in 3/10 of these regions were a coding variant or were in LD with a coding variant (3q26-MYNN and LRRC34, 10q24.32-WBP1L, 14q22.2-BMP4). Additionally, 2/10 of the risk variants were originally discovered in East Asian populations and risk SNPs had weaker association in our study (10q22.3-rs704017 P = 1 × 10− 4 and 10q24.32-rs4919687 P = 1 × 10− 2). Another 2/10 GWAS risk variant did not replicate in our study (4q31.1-rs60745952 P = 0.8 and 16p13.2-rs79900961 P = 0.26). In the remaining 3/10 regions, we found that the index variants did not reach genome-wide significance, reflecting power limitations in our discovery dataset (4q32.2-rs35509282 P = 6 × 10− 3, 16q24.1-rs16941835 P = 4 × 10− 3, and 20p12.3-rs961253 P = 4 × 10− 5).
Among the 34 regions containing associated genes, we found that the most significant gene association in the PrediXcan analysis was often the strongest candidate based on either known CRC etiology and gene function or results from previous laboratory follow-up (e.g. COLCA2, LAMC1, POLD3, SMAD7, TGFB1). In addition to confirming suspected genes, new candidates were also identified. For example, CXCR1 (P = 8 × 10− 5) and CXCR2 (P = 9 × 10− 5) were among the strongest associations. Notably, these genes are biologically relevant targets given that they encode cytokine receptors known to be implicated in a variety of cancers.
Discussion
In this study, we employed the PrediXcan in 12,186 cases and 14,718 controls. Genetic variant predictors of gene expression from both colon transverse and whole blood transcriptomes were used to test the association of CRC risk with imputed gene expression. We replicated novel associations of TRIM4 and PYGL in a large independent study of over 70,000 participants. In addition, we identified strong gene targets in several known GWAS loci, including genes that were previously not reported as putative candidates.
The two novel gene associations discovered in colon transverse models implicate genes involved with hypoxia-induced metabolic reprogramming, which is a hallmark of tumorigenesis in solid tumors. TRIM4 is a member of a superfamily of ubiquitin E3 ligases comprised of over 70 genes notably defined by a highly conserved N-terminal RING finger domain. This family of proteins has been implicated in a number of oncogenic or tumor suppressor activities that involve pathways related to CRC (Myc, Ras, etc.) (Sato et al. 2012; Chen et al. 2012; Zaman et al. 2013; Tocchini et al. 2014; Zhou et al. 2014; Zhan et al. 2015), and recently have been implicated in inflammatory and immune related activities (Eames et al. 2012; Versteeg et al. 2014). Somatic alterations in other TRIM genes have been associated with a large number of cancers including colon (Glebov et al. 2006; Noguchi et al. 2011; Hatakeyama 2011). While TRIM4 has not previously been implicated in cancer risk, the strong homology across gene members of this family and their implications in cancer and immunity make this gene an interesting candidate. Moreover, a recent study suggests that expression of TRIM4 plays a role in sensitizing cells to oxidative stress-induced death and regulation of reactive oxygen species (ROS) levels (H2O2) through ubiquitination of the redox regulator peroxide reductase (Tomar et al. 2015). Regulation of ROS levels and the cellular antioxidant system has previously been implicated in the pathophysiology of many diseases including inflammation and tumorigenesis (López-Lázaro 2007; Holmdahl et al. 2013). ROS are associated with cell cycle, proliferation, differentiation and migration and are elevated in colon as well as other cancers (Vaquero et al. 2004; Kumar et al. 2008; Afanas’ev 2011; Lin et al. 2017). Notably, many of the established environmental risk factors for colon cancer implicate oxidative stress pathways, including high alcohol consumption, smoking, increased consumption of red and processed meats (Stevens et al. 1988; Bird et al. 1996), or decreased consumption of fruits and vegetables (La Vecchia et al. 2013). In future laboratory analysis, it would be interesting to investigate whether the association of increased TRIM4 expression with decreased CRC risk is mechanistically acting through the regulation of ROS and cell growth.
Under the hypoxic conditions of the tumor microenvironment, constant reprogramming of glycogen metabolism is essential for providing the energy requirements necessary for cell growth and proliferation. PYGL (the second novel finding) encodes the key enzyme involved in glycogen degradation, releases glucose-1-phosphate so that it can enter the pentose phosphate pathway, which is important for generating NADPH, nucleotides, amino acids, and lipids required for continued cell proliferation (Favaro et al. 2012). It has previously been shown that depletion of PYGL leads to oxidative stress (increased ROS levels), and subsequent P53-induced growth arrest in cancer cells (Favaro et al. 2012). Of note, small molecule inhibitors of PYGL are currently under investigation for the treatment of diabetes (Praly and Vidal 2010). However, while decreased expression of PYGL in the tumor may result in tumor senescence, our results suggest that decreased PYGL expression is associated with increased risk of CRC. Like the dynamic role of expression for genes involved in the TGF-beta pathway, these conflicting observations between cancer risk and effects of early versus late induction of PYGL on cancer survival are likely reflecting the importance of context and fluctuating nutrient and oxygen availability within the tumor microenvironment.
Importantly, we found that the PrediXcan analysis identified new candidate genes in known GWAS loci that had previously gone undetected. For instance, in the recently identified 2q35 locus (Orlando et al. 2016), the authors originally reported the two closest genes, PNKD and TMBIM1, as potential targets for the putative regulatory locus marked by the index variant, rs992157. The authors reported eQTL evidence showing that rs992157 was associated with expression of nearby genes PNKD and TMBIM1 in lymphoblastoid cells, but not colorectal adenocarcinoma cells. In our PrediXcan analysis, expression of two other genes in this region, CXCR1 and CXCR2, were among the most strongly associated genes in the entire analysis, while the associations for PNKD (P = 6 × 10− 3) and TMBIM1 (P = 0.01) showed weaker associations. Our study added independent evidence for an association of the locus with CRC given that the index variant was only borderline significantly associated in previous analysis and identify two promising targets, CXCR1 and CXCR2. These genes are of note due to their chemotherapeutic properties. Specifically, the CXCR inhibitor, Reparixin, is currently under investigation for progression free survival of metastatic triple negative breast cancer in a stage 2 clinical trial (NCT02370238). Interestingly, expression of CXCR1 and CXCR2 has been shown to be elevated in colon tumor epithelium relative to normal adjacent tissue (P < 0.001). While there is still much to be learned, it is possible that this drug could also be useful for the treatment of CRC (Dabkeviciene et al. 2015).
This study had many strengths, most notably the use of reference transcriptome data to perform gene-level association testing in several large GWAS studies to both uncover novel associations and identify likely functional gene targets in known loci. By integrating reference transcriptome data, this study focused on genes that are expressed in CRC-relevant tissues. Furthermore, this method provided biologically relevant sets to aggregate variants, thereby improving statistical power by reducing the burden of multiple comparisons. In addition, our study was quite large, being comprised of nearly 100,000 participants across the discovery and replication datasets.
Our study had several limitations. For many genes, the predictive R2 for genetic variant models was relatively low, indicating that a small proportion of the variance in gene expression was explained by these models. In a recent publication, Su et al. (2018) demonstrated through extensive simulations that while there is an attenuation of true signal as a results of this, the diminishment in power was less than anticipated and more importantly this does not increase type I error. Predictive performance values were relatively strong in the models used for PYGL (R2 = 0.26) TRIM4. (R2 = 0.51) corresponding to 51% and 71% correlation between predicted and observed expression, respectively. In general, larger sample sizes for the reference panel will be needed to achieve better prediction models, particularly for rarer variants. While PYGL and TRIM4 were discovered using the colon tissue model, the whole blood model also showed evidence of association. This finding was not surprising in light of the recent GTEx paper demonstrating that many GWAS loci implicate shared eQTLs (GTEx Consortium et al. 2017). It should also be noted that variant predictors could implicate enhancers influencing the expression of multiple genes and because this study only evaluates genetically influenced expression levels, there is uncertainty that the associated gene is the causally related gene. As such, laboratory follow-up remains a critical extension of these findings; however, this laborious work can now be more targeted based on results from this analysis.
The loci identified using GWAS are most often located in non-coding regions and provide little biological insight. In contrast, the PrediXcan method directly tests putative target genes providing strong hypotheses for subsequent laboratory follow-up. The CXCR1 and CXCR2 findings are of interest given their therapeutic potential. As such, these findings provide preliminary support for new molecular targets that could potentially repurpose a putative cancer therapeutic agent and highlight the utility of integrating functional data for discovery of, and biological insight into risk loci.
Future analyses would be improved by increasing the number of transcriptomes. Similarly, larger GWAS sample sizes, or imputation of other molecular phenotypes (ChIP-seq, DNase-Seq, etc.) as data become available could be fruitful in the identification of important enhancer(s) or other regulatory elements that could influence the expression of one or more genes.
In conclusion, we identified two novel loci through the association of genetically predicted gene expression for TRIM4 and PYGL with CRC risk and identified strong target genes in known loci. The CXCR1 and CXCR2 findings highlight the advantage of using gene-based methods to identify stronger candidate genes and potentially expedite clinically relevant discovery. Further functional studies are required to confirm our findings and understand their biologic implications. This, in turn, could provide further insight into CRC etiology and potentially new therapeutic targets.
Materials and methods
Description of study cohorts
The discovery phase was comprised of 26,904 participants (12,186 CRC cases and 14,718 controls) of European ancestral heritage across 16 studies (described in methods and materials of Online Resource 1). Details of genotyping, QC and single-variant GWAS have been previously reported (Peters et al. 2013; Schumacher et al. 2015). The replication phase included a total of 32,825 cases and 39,933 controls. In addition to previously published CRC GWAS studies from CORECT (Schumacher et al. 2015) we included UK Biobank (application number 8614) and new CRC GWAS from additional GWAS. A nested case–control dataset from the UK Biobank resource was constructed defining cases as subjects with primary invasive CRC diagnosed, or who died from CRC according to ICD9 (1530–1534, 1536–1541) or ICD10 (C180, C182–C189, C19, C20) codes. Control selection was done in a time-forward manner, selecting one control for each case, first from the risk set at the time of the case’s event, and then multiple passes were made to match second, third and fourth controls. For prevalent cases, each case was matched with four controls that exactly matched the following matching criteria: year at enrollment, race/ethnicity, and sex. In total, 5356 cases and 21,407 matched controls were included from UK Biobank in the replication analysis. For the site-stratified analysis, “proximal” colon cancer was defined as hepatic flexure, transverse colon, cecum and ascending colon (ICD9 1530,1531,1534,1536), “distal” colon cancer was defined as descending colon, sigmoid colon, and splenic flexure (ICD9 1532,1533,1537) and “rectal” was defined as rectosigmoid junction, and rectum (ICD9 1540,1541).
Studies, sample selection and matching are described in Online Resource 1, which provides details on sample numbers, and demographic characteristics of study participants. All participants provided written informed consent, and each study was approved by the relevant research ethics committee or institutional review board.
Whole-genome sequencing reference genotype imputation panel
We performed low-pass whole-genome sequencing of 2192 samples (details in Online Resource 1) at the University of Washington Sequencing Center (Seattle, WA, USA). A detailed description is provided in the Online Resource 1. In brief, after sample QC and removal of samples with estimated DNA contamination > 3% (16), duplicated samples (5) or related individuals (1), sex discrepancies (0), and samples with low concordance with genome-wide variant array data (11), there were a total of 1439 CRC cases and 720 controls of European ancestry available for subsequent imputation. These data were used as a reference imputation panel for the discovery and replication GWAS datasets.
GWAS genotype data and quality control
In brief, genotyped variants were excluded based on call rate (< 98%), lack of Hardy–Weinberg Equilibrium in controls (HWE, P < 1 × 10− 4), and low minor allele frequency (MAF < 0.05). We imputed the autosomal variants of all studies to an internal imputation reference panel derived from whole genome sequencing (described above). We employed a two-stage imputation strategy (Howie et al. 2012) where entire chromosomes were first pre-phased using SHAPEIT2 (Delaneau et al. 2013), followed by imputation using minimac3 (Das et al. 2016). Only variants with an imputation quality R2 > 0.3 were included for subsequent analyses.
Imputation of genetically regulated gene expression in study cohort
Jointly measured genome variant data and transcriptome data sets were used by Gamazon et al. to develop additive models of gene expression levels. The weights for the estimation were downloaded from the publicly available database (http://hakyimlab.org/predictdb/). We used these models to estimate genetically regulated expression of genes in colon transverse, and whole blood. These estimates represent multi-variant prediction of tissue-specific gene expression levels.
In-depth details of the reference cohort, datasets, and model building have previously been described (Gamazon et al. 2015). To summarize, jointly measured genome-wide genotype data and RNA-seq data were obtained from two different projects: (1) the DGN cohort (Battle et al. 2014) (whole blood, n = 922) and (2) GTEx (GTEx Consortium 2015) (transverse colon, n = 169), predominantly of European ancestry. Gamazon et al. used approximately 650,000 variants with MAF > 0.05 to impute non-genotyped dosages using the 1000G Phase 1 v3 reference panel variants with MAF > 0.05 and imputation R2 > 0.8 was retained for subsequent model building. In each tissue, Gamazon et al. normalized gene expression by adjusting for sex, the top 3 principal components (derived from genotype data) and the top 15 PEER factors (to quantify hidden experimental confounders). These genomic and transcriptomic data sets were used to train additive models of gene expression levels with elastic net regularization (Gamazon et al. 2015). The model can be written as
where Yg is the expression trait of gene g, wk,g is the effect size of genetic marker k for g, Xk is the number of reference variant alleles of marker k and ε is the contribution of other factors influencing gene expression. The effect sizes (wk,g) in Eq. (1) were estimated using the elastic net penalized approach. The summation in Eq. 1 is referred to as the genetically determined component of gene expression. The variant models (weights, w_k,g) were downloaded from the publicly available database (http://hakyimlab.org/predictdb/).
The heritability of gene expression was used to estimate how well the variant models predict gene expression levels. The narrow-sense heritability for each gene was calculated by Gamazon et al. (2015), using a variance-component model with a genetic relationship matrix (GRM) estimated from genotype data, as implemented in GCTA (Yang et al. 2011). The proportion of the variance in gene expression explained by these local variants was calculated using a mixed-effects model (Torres et al. 2014; Gamazon et al. 2015). This heritability was highly correlated with the predictive R2 (The cross-validated R2 value found when training the model). Only genes with R2 ≥ 0.01(≥ 10% correlation between predicted and observed expression) were tested for association with CRC. Furthermore, this analysis focused on the component of heritability driven by variants in the vicinity (1 Mb) of each gene (cis-variants) because the component based on distal variants could not be estimated with enough accuracy to make meaningful inferences.
Genotypes were treated as continuous variables (dosages). Using the variant weights provided by Gamazon et al. we estimated the genetically regulated gene expression (GReX) of each gene g
where wk is the single-variant coefficient derived by regressing the gene expression trait Y on variant Xk using the reference transcriptome data. To address linkage disequilibrium among variant predictors, Gamazon et al. (2015) used the variable selection method to select a sparser set of (less correlated) of predictors. Specifically, variant weights (wk) were derived using elastic net with the R package glmnet with α = 0.5. These weights are available from http://hakyimlab.org/predictdb/. Using Eq. 2, and the reference variant predictor weights (wk,g), the (unobserved) genetically determined expression of each gene g (GReX) was estimated in our GWAS sample. For both transcriptome models, separate analyses were performed for genetically based expression of genes (up to 2 tests per gene). Genes with predictive R2 > 0.01 were tested for association with CRC in our cohort (colon transverse n = 4850 genes, and whole blood n = 8277 genes).
Gene level tests of CRC association with imputed genetically regulated gene expression
Discovery phase
Statistical analyses of all data were conducted centrally at the GECCO coordinating center on individual-level data. Multivariate logistic regression models were adjusted for age, sex (when appropriate), center (when appropriate), and genotyping batch (ASTERISK) and the first four principal components to account for potential population substructure. Imputed genetically regulated gene expression (GREx), was treated as a continuous variable. All studies were analyzed together in a pooled dataset using logistic regression models to obtain odds ratios (ORs) and 95% confidence intervals (CIs). Quantile–quantile (Q–Q) plots were assessed to determine whether the distribution of the P was consistent with the null distribution (except for the extreme tail). All analyses were conducted using the R software (Version 3.0.1). Novelty of a gene finding was determined by taking all variant predictors of the gene and determining if they were in linkage disequilibrium (LD ≥ 0.2 in Phase 3 Thousand Genomes Europeans) with a previously reported GWAS index variant.
We identified suggestive findings in the discovery stage to be replicated in a second independent dataset. In the discovery stage we employed a false-discovery rate (FDR) threshold of 0.2 separately for colon transverse and whole blood models. FDR for each gene was calculated using the R statistical package p.adjust, which uses the method of Benjamini and Hochberg to calculate the expected proportion of false discoveries amongst the rejected hypotheses (Hochberg and Benjamini 1990). Genes meeting this threshold were carried forward for replication.
Replication phase
To replicate novel PrediXcan findings (n = 3 genes from colon transverse models) that had a FDR ≤ 0.2, we used the same GTEx colon transverse, elastic net prediction models (as we had done in the discovery GECCO-CCFR data) to impute genetically regulated gene expression in replication samples from (1) CORECT (pooled across consortium studies), (2) UK Biobank and (3) a pooled dataset of 5 independent GWAS datasets. Multivariate logistic regression was used to test the association of imputed genetically regulated gene expression with colorectal cancer risk in these three datasets and then meta-analyzed effects using inverse variance weighting of Z scores (details provided in Online Resource 1). A two-sided P value less than 0.05/(number of genes to be replicated) was considered statistically significant.
Definition of CRC risk regions and refinement of GWAS loci
The 56 previously reported CRC risk variants used in this analysis had an LD r2 ≤ 0.2 with other risk variants in our known list, or were otherwise previously reported to maintain statistical significance in regression models conditioning on other nearby risk variants (referred to hereon as ‘independent’ risk variants). For each of the 56 independent risk variants defined in Table S3, we further defined ‘risk regions’ as 1 megabase (Mb) upstream and 1 Mb downstream of each risk variant (2 Mb regions surrounding each risk variant). Overlapping 2 Mb risk regions were then combined into a single new risk region defined as the minimum and maximum chromosomal coordinates from one or more overlapping risk regions (the union of the overlapping regions). This resulted in a total of 44 regions harboring one or more risk variants (maximum of four independent risk variants). A list of transcription start sites (TSS) for genes that showed nominal association (P ≤ 0.05) between genetically regulated gene expression and CRC risk in colon transverse and whole blood models was then intersected with the list of 44 risk regions to identify a list a putative target genes regulated by non-coding GWAS risk variants.
Bioinformatic follow-up
Bioinformatic follow-up was performed for the TRIM4 and PYGL loci using the UCSC Genome Browser and publicly available functional data for CRC relevant tissues and cell-types from Roadmap, ENCODE, as well as previously published epigenomes (Akhtar-Zaidi et al. 2012). The TRIM4 and PYGL loci were defined as the genomic region containing all variants in LD (r2 ≥ 0.2 from Phase 3 Thousand Genomes Project) with the variant predictor having the strongest marginal CRC association (TRIM4-rs2527886 and PYGL-rs12589665). We then aligned the locus with refseq protein coding genes, epigenetic signals in normal crypts and CRC cell lines to identify recurrently gained and lost variant enhancer loci (VEL), and ChIP-seq transcription factor binding sites.
URLs
PrediXcan software, https://github.com/hakyimlab/PrediXcan; University of Michigan Imputation-Server, https://imputationserver.sph.umich.edu/start.html; GTEx Portal, http://www.gtexportal.org/; PredictDB, http://predictdb.org/.
Change history
28 June 2019
Every author has erroneously been assigned to the affiliation “62”. The affiliation 62 belongs to the author Graham Casey.
References
Afanas’ev I (2011) Reactive oxygen species signaling in cancer: comparison with aging. Aging Dis 2:219–230
Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O et al (2012) Epigenomic enhancer profiling defines a signature of colon cancer. Science 336:736–739. https://doi.org/10.1126/science.1217277
Al-Tassan NA, Whiffin N, Hosking FJ et al (2015) A new GWAS and meta-analysis with 1000 Genomes imputation identifies novel risk variants for colorectal cancer. Sci Rep 5:10442. https://doi.org/10.1038/srep10442
Battle A, Mostafavi S, Zhu X et al (2014) Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res 24:14–24. https://doi.org/10.1101/gr.155192.113
Biancolella M, Fortini BK, Tring S et al (2014) Identification and characterization of functional risk variants for colorectal cancer mapping to chromosome 11q23.1. Hum Mol Genet 23:2198–2209. https://doi.org/10.1093/hmg/ddt584
Bien SA, Auer PL, Harrison TA et al (2017) Enrichment of colorectal cancer associations in functional regions: insight for using epigenomics data in the analysis of whole genome sequence-imputed GWAS data. PLoS One 12:e0186518. https://doi.org/10.1371/journal.pone.0186518
Bird CL, Witte JS, Swendseid ME et al (1996) Plasma ferritin, iron intake, and the risk of colorectal polyps. Am J Epidemiol 144:34–41
Broderick P, Carvajal-Carmona L, Pittman AM et al (2007) A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet 39:1315–1317. https://doi.org/10.1038/ng.2007.18
Chen L, Chen D-T, Kurtyka C et al (2012) Tripartite motif containing 28 (Trim28) can regulate cell proliferation by bridging HDAC1/E2F interactions. J Biol Chem 287:40106–40118. https://doi.org/10.1074/jbc.M112.380865
Cui R, Okada Y, Jang SG, Ku JL, Park JG, Kamatani Y, Hosono N, Tsunoda T, Kumar V, Tanikawa C, Kamatani N, Yamada R, Kubo M, Nakamura Y, Matsuda K (2011) Common variant in 6q26-q27 is associated with distal colon cancer in an Asian population. Gut 60(6):799–805
Czene K, Lichtenstein P, Hemminki K (2002) Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int J Cancer 99:260–266. https://doi.org/10.1002/ijc.10332
Dabkeviciene D, Jonusiene V, Zitkute V et al (2015) The role of interleukin-8 (CXCL8) and CXCR2 in acquired chemoresistance of human colorectal carcinoma cells HCT116. Med Oncol 32:258. https://doi.org/10.1007/s12032-015-0703-y
Das S, Forer L, Schönherr S et al (2016) Next-generation genotype imputation service and methods. Nat Genet 48:1284–1287. https://doi.org/10.1038/ng.3656
Delaneau O, Howie B, Cox AJ et al (2013) Haplotype estimation using sequencing reads. Am J Hum Genet 93:687–696. https://doi.org/10.1016/j.ajhg.2013.09.002
Dunlop MG, Dobbins SE, Farrington SM et al (2012) Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat Genet 44:770–776. https://doi.org/10.1038/ng.2293
Eames HL, Saliba DG, Krausgruber T et al (2012) KAP1/TRIM28: an inhibitor of IRF5 function in inflammatory macrophages. Immunobiology 217:1315–1324. https://doi.org/10.1016/j.imbio.2012.07.026
Favaro E, Bensaad K, Chong MG et al (2012) Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab 16:751–764. https://doi.org/10.1016/j.cmet.2012.10.017
Fortini BK, Tring S, Plummer SJ, Edlund CK, Moreno V, Bresalier RS, Barry EL, Church TR, Figueiredo JC, Casey G (2014) Multiple functional risk variants in a SMAD7 enhancer implicate a colorectal cancer risk haplotype. PLoS One. https://doi.org/10.1371/journal.pone.0111914
Gamazon ER, Wheeler HE, Shah KP et al (2015) A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 47:1091–1098. https://doi.org/10.1038/ng.3367
Glebov OK, Rodriguez LM, Soballe P et al (2006) Gene expression patterns distinguish colonoscopically isolated human aberrant crypt foci from normal colonic mucosa. Cancer Epidemiol Biomark Prev 15:2253–2262. https://doi.org/10.1158/1055-9965.EPI-05-0694
GTEx Consortium (2013) The genotype-tissue expression (GTEx) project. Nat Genet 45:580–585. https://doi.org/10.1038/ng.2653
GTEx Consortium (2015) Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348:648–660. https://doi.org/10.1126/science.1262110
GTEx Consortium, Laboratory, Data Analysis & Coordinating Center (LDACC), Analysis Working Group, Statistical Methods Groups, Analysis Working Group et al (2017) Genetic effects on gene expression across human tissues. Nature 550:204–213. https://doi.org/10.1038/nature24277
Hatakeyama S (2011) TRIM proteins and cancer. Nat Rev Cancer 11:792–804. https://doi.org/10.1038/nrc3139
Hochberg Y, Benjamini Y (1990) More powerful procedures for multiple significance testing. Stat Med 9:811–818. https://doi.org/10.1002/sim.4780090710
Holmdahl R, Sareila O, Pizzolla A et al (2013) Hydrogen peroxide as an immunological transmitter regulating autoreactive T cells. Antioxid Redox Signal 18:1463–1474. https://doi.org/10.1089/ars.2012.4734
Houlston RS, Cheadle J, Dobbins SE et al (2010) Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet 42:973–977. https://doi.org/10.1038/ng.670
Howie B, Fuchsberger C, Stephens M et al (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44:955–959. https://doi.org/10.1038/ng.2354
Hulur I, Gamazon ER, Skol AD et al (2015) Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci. BMC Genom 16:138. https://doi.org/10.1186/s12864-015-1292-z
Jia W-H, Zhang B, Matsuo K et al (2013) Genome-wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nat Genet 45:191–196. https://doi.org/10.1038/ng.2505
Jiao S, Peters U, Berndt S et al (2014) Estimating the heritability of colorectal cancer. Hum Mol Genet 23:3898–3905. https://doi.org/10.1093/hmg/ddu087
Kinnersley B, Migliorini G, Broderick P, Whiffin N, Dobbins SE, Casey G, Hopper J, Sieber O, Lipton L, Kerr DJ, Dunlop MG, Tomlinson IP, Houlston RS, Colon Cancer Family Registry (2012) The TERT variant rs2736100 is associated with colorectal cancer risk. Br J Cancer 107:1001–1008
Kumar B, Koul S, Khandrika L et al (2008) Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res 68:1777–1785. https://doi.org/10.1158/0008-5472.CAN-07-5259
La Vecchia C, Decarli A, Serafini M et al (2013) Dietary total antioxidant capacity and colorectal cancer: a large case-control study in Italy. Int J Cancer 133:1447–1451. https://doi.org/10.1002/ijc.28133
Lewis A, Freeman-Mills L, de la Calle-Mustienes E et al (2014) A polymorphic enhancer near GREM1 influences bowel cancer risk through differential CDX2 and TCF7L2 binding. Cell Rep 8:983–990. https://doi.org/10.1016/j.celrep.2014.07.020
Lichtenstein P, Holm NV, Verkasalo PK et al (2000) Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 343:78–85. https://doi.org/10.1056/NEJM200007133430201
Lin S, Li Y, Zamyatnin AA et al (2017) Reactive oxygen species and colorectal cancer. J Cell Physiol 233:5119–5132. https://doi.org/10.1002/jcp.26356
López-Lázaro M (2007) Dual role of hydrogen peroxide in cancer: possible relevance to cancer chemoprevention and therapy. Cancer Lett 252:1–8. https://doi.org/10.1016/j.canlet.2006.10.029
Niell BL, Long JC, Rennert G, Gruber SB (2003) Genetic anthropology of the colorectal cancer-susceptibility allele APC I1307K: evidence of genetic drift within the Ashkenazim. Am J Hum Genet 73:1250–1260. https://doi.org/10.1086/379926
Noguchi K, Okumura F, Takahashi N et al (2011) TRIM40 promotes neddylation of IKKγ and is downregulated in gastrointestinal cancers. Carcinogenesis 32:995–1004. https://doi.org/10.1093/carcin/bgr068
Orlando G, Law PJ, Palin K et al (2016) Variation at 2q35 (PNKD and TMBIM1) influences colorectal cancer risk and identifies a pleiotropic effect with inflammatory bowel disease. Hum Mol Genet 25:2349–2359. https://doi.org/10.1093/hmg/ddw087
Peltekova VD, Lemire M, Qazi AM et al (2014) Identification of genes expressed by immune cells of the colon that are regulated by colorectal cancer-associated variants. Int J Cancer 134:2330–2341. https://doi.org/10.1002/ijc.28557
Peters U, Jiao S, Schumacher FR et al (2013) Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology 144:799–807.e24. https://doi.org/10.1053/j.gastro.2012.12.020
Peters U, Bien S, Zubair N (2015) Genetic architecture of colorectal cancer. Gut 64:1623–1636. https://doi.org/10.1136/gutjnl-2013-306705
Pittman AM, Naranjo S, Jalava SE et al (2010) Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of EIF3H. PLoS Genet 6:e1001126. https://doi.org/10.1371/journal.pgen.1001126
Pomerantz MM, Ahmadiyeh N, Jia L et al (2009) The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet 41:882–884. https://doi.org/10.1038/ng.403
Praly J-P, Vidal S (2010) Inhibition of glycogen phosphorylase in the context of type 2 diabetes, with focus on recent inhibitors bound at the active site. Mini Rev Med Chem 10:1102–1126
Sato T, Okumura F, Ariga T, Hatakeyama S (2012) TRIM6 interacts with Myc and maintains the pluripotency of mouse embryonic stem cells. J Cell Sci 125:1544–1555. https://doi.org/10.1242/jcs.095273
Schmit SL, Schumacher FR, Edlund CK et al (2016) Genome-wide association study of colorectal cancer in Hispanics. Carcinogenesis 37:547–556. https://doi.org/10.1093/carcin/bgw046
Schumacher FR, Schmit SL, Jiao S et al (2015) Corrigendum: genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nat Commun 6:8739. https://doi.org/10.1038/ncomms9739
Shin Y, Kim I-J, Kang HC et al (2004) A functional polymorphism (− 347 G → GA) in the E-cadherin gene is associated with colorectal cancer. Carcinogenesis 25:2173–2176. https://doi.org/10.1093/carcin/bgh223
Stevens RG, Jones DY, Micozzi MS, Taylor PR (1988) Body iron stores and the risk of cancer. N Engl J Med 319:1047–1052. https://doi.org/10.1056/NEJM198810203191603
Study COGENT, Houlston RS, Webb E et al (2008) Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet 40:1426–1435. https://doi.org/10.1038/ng.262
Su Y-R, Di C, Bien SA et al (2018) A mixed-effects model for powerful association tests in integrative functional genomics: an application to a large-scale genome-wide association study of colorectal cancer. Am J Hum Genet 102(5):904–919. https://doi.org/10.1016/j.ajhg.2018.03.019
Tenesa A, Farrington SM, Prendergast JGD et al (2008) Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet 40:631–637. https://doi.org/10.1038/ng.133
Tocchini C, Keusch JJ, Miller SB et al (2014) The TRIM-NHL protein LIN-41 controls the onset of developmental plasticity in Caenorhabditis elegans. PLoS Genet 10:e1004533. https://doi.org/10.1371/journal.pgen.1004533
Tomar D, Prajapati P, Lavie J et al (2015) TRIM4; a novel mitochondrial interacting RING E3 ligase, sensitizes the cells to hydrogen peroxide (H2O2) induced cell death. Free Radic Biol Med 89:1036–1048. https://doi.org/10.1016/j.freeradbiomed.2015.10.425
Tomlinson I, Webb E, Carvajal-Carmona L et al (2007) A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet 39:984–988. https://doi.org/10.1038/ng2085
Tomlinson IPM, Webb E, Carvajal-Carmona L et al (2008) A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet 40:623–630. https://doi.org/10.1038/ng.111
Torres JM, Gamazon ER, Parra EJ et al (2014) Cross-tissue and tissue-specific eQTLs: partitioning the heritability of a complex trait. Am J Hum Genet 95:521–534. https://doi.org/10.1016/j.ajhg.2014.10.001
Vaquero EC, Edderkaoui M, Pandol SJ et al (2004) Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem 279:34643–34654. https://doi.org/10.1074/jbc.M400078200
Versteeg GA, Benke S, García-Sastre A, Rajsbaum R (2014) InTRIMsic immunity: positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev 25:563–576. https://doi.org/10.1016/j.cytogfr.2014.08.001
Wang H, Schmit SL, Haiman CA et al (2017) Novel colon cancer susceptibility variants identified from a genome-wide association study in African Americans. Int J Cancer 140:2728–2733. https://doi.org/10.1002/ijc.30687
Whiffin N, Hosking FJ, Farrington SM et al (2014) Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum Mol Genet 23:4729–4737. https://doi.org/10.1093/hmg/ddu177
Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88:76–82. https://doi.org/10.1016/j.ajhg.2010.11.011
Zaman MM-U, Nomura T, Takagi T et al (2013) Ubiquitination-deubiquitination by the TRIM27-USP7 complex regulates tumor necrosis factor alpha-induced apoptosis. Mol Cell Biol 33:4971–4984. https://doi.org/10.1128/MCB.00465-13
Zeng C, Matsuda K, Jia W-H et al (2016) Identification of susceptibility loci and genes for colorectal cancer risk. Gastroenterology 150:1633–1645. https://doi.org/10.1053/j.gastro.2016.02.076
Zhan W, Han T, Zhang C et al (2015) TRIM59 promotes the proliferation and migration of non-small cell lung cancer cells by upregulating cell cycle related proteins. PLoS One 10:e0142596. https://doi.org/10.1371/journal.pone.0142596
Zhang B, Jia W-H, Matsuda K et al (2014) Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet 46:533–542. https://doi.org/10.1038/ng.2985
Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, Ferretti V, Laflamme P, Sundararajan S, Roumy S, Olivier JF, Robidoux F, Sladek R, Montpetit A, Campbell P, Bezieau S, O'Shea AM, Zogopoulos G, Cotterchio M, Newcomb P, McLaughlin J, Younghusband B, Green R, Green J, Porteous ME, Campbell H, Blanche H, Sahbatou M, Tubacher E, Bonaiti-Pellié C, Buecher B, Riboli E, Kury S, Chanock SJ, Potter J, Thomas G, Gallinger S, Hudson TJ, Dunlop MG (2007) Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet 39:989–994
Zhou Z, Ji Z, Wang Y et al (2014) TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 147:1043–1054. https://doi.org/10.1053/j.gastro.2014.07.021
Acknowledgements
ASTERISK: We are very grateful to Dr. Bruno Buecher without whom this project would not have existed. We also thank all those who agreed to participate in this study, including the patients and the healthy control persons, as well as all the physicians, technicians and students. CORECT: The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the CORECT Consortium, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government or the CORECT Consortium. We thank Alina Hoehn for her valuable contributions to table/figure generation and organization of this manuscript. We are incredibly grateful for the contributions of Dr. Brian Henderson and Dr. Roger Green over the course of this study and acknowledge them in memoriam. We are also grateful for support from Daniel and Maryann Fong. ColoCare: We thank the many investigators and staff who made this research possible in ColoCare Seattle and ColoCare Heidelberg. ColoCare was initiated and developed at the Fred Hutchinson Cancer Research Center by Drs. Ulrich and Grady. COLON and NQplus: the authors would like to thank the COLON and NQplus investigators at Wageningen University & Research and the involved clinicians in the participating hospitals. CCFR: The Colon CFR graciously thanks the generous contributions of their study participants, dedication of study staff, and financial support from the U.S. National Cancer Institute, without which this important registry would not exist. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the CCFR. CPS-II: The authors thank the CPS-II participants and Study Management Group for their invaluable contributions to this research. The authors would also like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, and cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program. DACHS: We thank all participants and cooperating clinicians, and Ute Handte-Daub, Utz Benscheid, Muhabbet Celik and Ursula Eilber for excellent technical assistance. Galeon: GALEON wishes to thank the Department of Surgery of University Hospital of Santiago (CHUS), Sara Miranda Ponte, Carmen M Redondo, and the staff of the Department of Pathology and Biobank of CHUS, Instituto de Investigación Sanitaria de Santiago (IDIS), Instituto de Investigación Sanitaria Galicia Sur (IISGS), SERGAS, Vigo, Spain, and Programa Grupos Emergentes, Cancer Genetics Unit, CHUVI Vigo Hospital, Instituto de Salud Carlos III, Spain. EPIC: We thank all participants and health care personnel in the Västerbotten Intervention Programme, as well as the Department of biobank research, Umeå University, and Biobanken norr, Västerbotten County Council. GECCO: The authors would like to thank all those at the GECCO Coordinating Center for helping bring together the data and people that made this project possible. The authors also acknowledge Deanna Stelling, Mark Thornquist, Greg Warnick, Carolyn Hutter, and team members at COMPASS (Comprehensive Center for the Advancement of Scientific Strategies) at the Fred Hutchinson Cancer Research Center for their work harmonizing the GECCO epidemiological data set. The authors acknowledge Dave Duggan and team members at TGEN (Translational Genomics Research Institute), the Broad Institute, and the Génome Québec Innovation Center for genotyping DNA samples of cases and controls, and for scientific input for GECCO. HPFS, NHS and PHS: We would like to acknowledge Patrice Soule and Hardeep Ranu of the Dana Farber Harvard Cancer Center High-Throughput Polymorphism Core who assisted in the genotyping for NHS, HPFS, and PHS under the supervision of Dr. Immaculata Devivo and Dr. David Hunter, Qin (Carolyn) Guo and Lixue Zhu who assisted in programming for NHS and HPFS, and Haiyan Zhang who assisted in programming for the PHS. We would like to thank the participants and staff of the Nurses’ Health Study and the Health Professionals Follow-Up Study, for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data. MCCS: This study was made possible by the contribution of many people, including the original investigators and the diligent team who recruited participants and continue to work on follow-up. We would also like to express our gratitude to the many thousands of Melbourne residents who took part in the study and provided blood samples. PLCO: The authors thank Drs. Christine Berg and Philip Prorok, Division of Cancer Prevention, National Cancer Institute, the Screening Center investigators and staff or the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, Mr. Tom Riley and staff, Information Management Services, Inc., Ms. Barbara O’Brien and staff, Westat, Inc., and Drs. Bill Kopp and staff, SAIC-Frederick. Most importantly, we acknowledge the study participants for their contributions to making this study possible. The statements contained herein are solely those of the authors and do not represent or imply concurrence or endorsement by NCI. PMH: The authors would like to thank the study participants and staff of the Hormones and Colon Cancer study. SEARCH: We acknowledge the contributions of Mitul Shah, Val Rhenius, Sue Irvine, Craig Luccarini, Patricia Harrington, Don Conroy, Rebecca Mayes, and Caroline Baynes. The Swedish low-risk colorectal cancer study: we thank Berith Wejderot and the Swedish low-risk colorectal cancer study group. UK Biobank: This research has been conducted using the UK Biobank Resource under Application Number 8614. WHI: The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf.
Funding
GECCO: This work was supported by the National Cancer Institute; National Institutes of Health; and the United States Department of Health and Human Services (U01 CA137088, R01 CA059045, U01 CA164930, R01 CA201407, R01 CA206279). Genotyping/Sequencing services were provided by the Center for Inherited Disease Research and is supported by a federal contract from the National Institutes of Health to The Johns Hopkins University (HHSN268201200008I). ASTERISK: a Hospital Clinical Research Program (PHRC-BRD09/C) from the University Hospital Center of Nantes and supported by the Regional Council of Pays de la Loire, the Groupement des Entreprises Françaises dans la Lutte contre le Cancer; the Association Anne de Bretagne Génétique; and the Ligue Régionale Contre le Cancer. ATBC: The ATBC Study is supported by the Intramural Research Program of the United States National Cancer Institute; National Institutes of Health; and by United States Public Health Service (HHSN261201500005C) from the National Cancer Institute, Department of Health and Human Services. COLO2&3: National Institutes of Health (R01 CA60987). CCFR: Illumina GWAS was supported by funding from the National Cancer Institute; and the National Institutes of Health (U01 CA122839, R01 CA143247). The Colon CFR/CORECT Affymetrix Axiom GWAS and OncoArray GWAS were supported by funding from the National Cancer Institute; and National Institutes of Health (U19 CA148107 to S.B.G.). The Colon CFR participant recruitment and collection of data and biospecimens used in this study were supported by the National Cancer Institute; and National Institutes of Health (UM1 CA167551) and through cooperative agreements between multiple Colon CFR centers: (U01 CA074778, U01/U24 CA097735 to Australasian Colorectal Cancer Family Registry, U01/U24 CA074799 to USC Consortium Colorectal Cancer Family Registry, U01/U24 CA074800 to Mayo Clinic Cooperative Family Registry for Colon Cancer Studies, U01/U24 CA074783 to Ontario Familial Colorectal Cancer Registry, U01/U24 CA074794 to Seattle Colorectal Cancer Family Registry, U01/U24 CA074806 to University of Hawaii Colorectal Cancer Family Registry). Additional support for case ascertainment was provided from the Surveillance, Epidemiology and End Results Program of the National Cancer Institute (N01-CN-67009, N01-PC-35142, HHSN2612013000121 to Fred Hutchinson Cancer Research Center), the Hawaii Department of Health (N01-PC-67001 and N01-PC-35137, HHSN26120100037C), and the California Department of Public Health (HHSN261201000035C to the University of Southern California), and the following state cancer registries: AZ, CO, MN, NC, NH, and by the Victoria Cancer Registry and Ontario Cancer Registry. CORECT: The CORECT Study was supported by the National Cancer Institute; National Institutes of Health; and the United States Department of Health and Human Services (U19 CA148107, R01 CA81488 to S.B.G., P30 CA014089, R01 CA197350 to S.B.G., P01 CA196569, R01 CA201407); and National Institutes of Environmental Health Sciences, National Institutes of Health (T32 ES013678). CPSII: The Cancer Prevention Study-II Nutrition Cohort is supported by the American Cancer Society. COLON: The COLON study is sponsored by Wereld Kanker Onderzoek Fonds, including funds from grant 2014/1179 as part of the World Cancer Research Fund International Regular Grant Programme, by Alpe d’Huzes and the Dutch Cancer Society (UM 2012-5653, UW 2013-5927, UW2015-7946), and by TRANSCAN (JTC2012-MetaboCCC, JTC2013-FOCUS). ColoCare: This work was supported by the National Institutes of Health (R01 CA189184, U01 CA206110, 2P30CA015704-40 to Gilliland]; the Matthias Lackas-Foundation; the German Consortium for Translational Cancer Research; and the EU TRANSCAN initiative. DACHS: This work is supported by the German Research Council [Deutsche Forschungsgemeinschaft, BR 1704/6-1, BR 1704/6-3, BR 1704/6-4 and CH 117/1-1); and the German Federal Ministry of Education and Research (01KH0404 and 01ER0814). DALS: This work is supported by the National Institutes of Health (R01 CA48998 to M. L. S.) EPIC: The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by Danish Cancer Society (Denmark); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF), Deutsche Krebshilfe, Deutsches Krebsforschungszentrum and Federal Ministry of Education and Research (Germany); the Hellenic Health Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); ERC-2009-AdG 232997 and Nordforsk, Nordic Centre of Excellence programme on Food, Nutrition and Health (Norway); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Swedish Cancer Society, Swedish Research Council and County Councils of Skåne and Västerbotten (Sweden); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom). ESTHER/VERDI: This work was supported by grants from the Baden-Württemberg Ministry of Science, Research and Arts, and the German Cancer Aid. HPFS: This work is supported by the National Institutes of Health (P01 CA055075, UM1 CA167552, R01 CA137178, R01 CA151993, R35 CA197735, K07 CA190673, P50 CA127003); NHS is supported by the National Institutes of Health (R01 CA137178, P01 CA087969, UM1 CA186107, R01 CA151993, R35 CA197735, K07 CA190673, and P50 CA127003); and PHS by the National Institutes of Health (R01 CA042182). MEC: This work is supported by the National Institutes of Health (R37 CA54281, P01 CA033619, R01 CA063464). MCCS: Cohort recruitment was supported by VicHealth and Cancer Council Victoria. GALEON: FIS Intrasalud (PI13/01136). The MCCS was further supported by Australian NHMRC grants (509348, 209057, 251553, 504711), and by infrastructure provided by the Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry and the Australian Institute of Health and Welfare, including the National Death Index and the Australian Cancer Database. MSKCC: The work at Sloan Kettering in New York was supported by the Robert and Kate Niehaus Center for Inherited Cancer Genomics and the Romeo Milio Foundation. Moffitt: This work was supported by the National Institutes of Health (R01 CA189184, P30 CA076292); Florida Department of Health Bankhead-Coley Grant (09BN-13); and the University of South Florida Oehler Foundation. Moffitt contributions were supported in part by the Total Cancer Care Initiative; Collaborative Data Services Core; and Tissue Core at the H. Lee Moffitt Cancer Center & Research Institute, a National Cancer Institute-designated Comprehensive Cancer Center (P30 CA076292). NQplus: The NQplus study is sponsored by a ZonMW investment grant (98-10030); by PREVIEW, the project PREVention of diabetes through lifestyle intervention and population studies in Europe and around the World (PREVIEW) project which received funding from the European Union Seventh Framework Programme (FP7/2007-2013) under grant no. 312057; by funds from TI Food and Nutrition (cardiovascular health theme), a public–private partnership on pre-competitive research in food and nutrition; and by FOODBALL, the Food Biomarker Alliance, a project from JPI Healthy Diet for a Healthy Life. OFCCR: As subset of ARCTIC, OFCCR is supported by a GL2 grant from the Ontario Research Fund; the Canadian Institutes of Health Research; and the Cancer Risk Evaluation Program grant from the Canadian Cancer Society Research Institute. This work is supported by the Ontario Institute for Cancer Research, through generous support from the Ontario Ministry of Research and Innovation (Senior Investigator Awards to T.J.H. and B.W.Z.) PLCO: This work is supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and the Division of Cancer Prevention, National Cancer Institute DHHS. Additionally, a subset of control samples were genotyped as part of the Cancer Genetic Markers of Susceptibility (CGEMS) Prostate Cancer GWAS (Yeager, M et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 2007 May;39[5]:645–9), CGEMS pancreatic cancer scan [PanScan] (Amundadottir, L et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009 Sep;41[9]:986 – 90, and Petersen, GM et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010 Mar;42[3]:224–8), and the Lung Cancer and Smoking study (Landi MT, et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am J Hum Genet. 2009 Nov;85[5]:679–91). The prostate and PanScan study datasets were accessed with appropriate approval through the dbGaP online resource [http://cgems.cancer.gov/data/] accession numbers phs000207.v1.p1 and phs000206.v3.p2, respectively, and the lung datasets were accessed from the dbGaP website (http://www.ncbi.nlm.nih.gov/gap) through accession number phs000093.v2.p2. Funding for the Lung Cancer and Smoking study was provided by National Institutes of Health, Genes, Environment and Health Initiative (Z01 CP 010200, NIH U01 HG004446, and NIH GEI U01 HG 004438). For the lung study, the GENEVA Coordinating Center provided assistance with genotype cleaning and general study coordination, and the Johns Hopkins University Center for Inherited Disease Research conducted genotyping. SEARCH: Cancer Research UK (C490/A16561). The Spanish study was supported by Instituto de Salud Carlos III, co-funded by FEDER funds—a way to build Europe—(PI14-613, PI09-1286); Catalan Government DURSI (2014SGR647); and Junta de Castilla y León (LE22A10-2). Spain: Catalan Government DURSI (2014SGR647); and Instituto de Salud Carlos III, co-funded by FEDER funds―a way to build Europe (PI14-00613). The Swedish Low-risk Colorectal Cancer Study: The study was supported by the Swedish research council (K2015-55X-22674-01-4, K2008-55X-20157-03-3, K2006-72X-20157-01-2); and the Stockholm County Council (ALF project). VITAL: This work is supported by the National Institutes of Health (K05 CA154337). WHI: The WHI program is supported by the National Heart, Lung, and Blood Institute; National Institutes of Health; United States Department of Health and Human Services through contracts (HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, HHSN271201100004C) The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health. Additional funds were provided by the National Cancer Institute, National Human Genome Research Institute, National Human Lung and Blood Institute, National Institute of Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke. Donors were enrolled at Biospecimen Source Sites supported by the National Cancer Institute and SAIC-Frederick, Inc. (SAIC-F) subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171), and Science Care, Inc. (X10S172). The Laboratory, Data Analysis, and Coordinating Center was supported by a contract (HHSN268201000029C to The Broad Institute, Inc). Biorepository operations were funded through an SAIC-F subcontract to Van Andel Institute (10ST1035). Additional data repository and project management were provided by SAIC-F (HHSN261200800001E). The Brain Bank was supported by a supplement to University of Miami (DA006227, DA033684, N01MH000028). Statistical Methods development grants were made to the University of Geneva (MH090941 & MH101814), the University of Chicago (MH090951, MH090937, MH101820, MH101825), the University of North Carolina - Chapel Hill (MH090936, MH101819), Harvard University (MH090948), Stanford University (MH101782), Washington University St Louis (MH101810), and the University of Pennsylvania (MH101822). The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 10/19/2016.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bien, S.A., Su, YR., Conti, D.V. et al. Genetic variant predictors of gene expression provide new insight into risk of colorectal cancer. Hum Genet 138, 307–326 (2019). https://doi.org/10.1007/s00439-019-01989-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-019-01989-8