
Abstract
Hemophilia B (HB) is an X-linked disorder caused by defects of F9 encoded coagulation factor IX, which is an ideal model for gene therapy. Most existing HB gene therapies are based on viral mediated gene supplementation, which could increase immunoreaction. In this study, CRISPR/Cas9 system was used for gene correction in an F9 mutant HB mouse model in both adult mice (in vivo) and in germline cells (ex vivo). In vivo, naked Cas9-sgRNA plasmid and donor DNA were delivered to HB mice livers to recover the mutation via hydrodynamic tail vein (HTV) injection. 62.5% of the HTV-treated mice showed a detectable gene correction (>1%) in the F9 alleles of hepatocytes, which was sufficient to remit the coagulation deficiency. Ex vivo, three different forms of Cas9 were microinjected into germline cells of HB mice to investigate their efficiency and safety in gene correction. Cas9 protein showed higher gene recovery rates, less embryo toxicity, and lower mosaic repair percentage, making it more suitable for germline gene therapy. Our study strongly supports that CRISPR/Cas9-mediated genome editing is feasible in gene therapy of genetic disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hemophilia B (HB) is an X-linked inherited bleeding disorder caused by deficiency or dysfunction of coagulation factor IX (FIX). According to FIX plasma procoagulant levels (FIX:C), HB is classified as mild (5–40% of normal level), moderate (1–5% of normal level), or severe (less than 1% of normal level) (White et al. 2001). Approximately 60% of HB cases are severe sufferers, who may have life-threatening spontaneous bleeding episodes. The first line of treatment for HB is factor replacement therapy by concentrates or recombinant FIX. However, the half-life of FIX is approximately 24 h; therefore, the patients require lifelong blood transfusions. As HB is a monogenic disease, patients will restore their hemostasis as long as functional FIX increased to 1% or above (Wang et al. 1999). Thus, gene therapy may be a better approach to cure HB. Up to now, the main strategy for HB gene therapy is gene supplementation treatment, which genetically supplies normal F9 cDNA to increase the FIX level in plasma. Traditional strategies use viral mediated transduction to introduce F9 cDNA into the liver, muscle, or fibroblast cells of HB animals and patients (Chen et al. 2003; Jiang et al. 2006; Kay et al. 1993; Lu et al. 1993; Manno et al. 2003, 2006; Nathwani et al. 2014), and recent studies employed the Sleeping Beauty transposon or gene editing system (zinc finger nucleases, ZFN) to randomly or site-specifically insert F9 cDNA into the genome (Anguela et al. 2013; Li et al. 2011; Sharma et al. 2015; Yant et al. 2000). All these treatments have successfully remitted the coagulation deficiency of patients, and the site-specific insertion to Alb of the F9 promoter performed better in long-term FIX expression for more than 8 weeks (Sharma et al. 2015). However, the safety of viral vectors is still under consideration, and random integration caused by AAV or PB is also a risk to genome stability.
Gene correction is an ideal strategy of gene therapy for genetic disorders. By presenting a donor DNA template, the mutant DNA site can be corrected fundamentally by homology-directed repair (HDR). As the spontaneous HDR rate is as low as 1 × 10−6 (Iiizumi et al. 2008), a gene-editing system is especially needed for site-specific DNA cleavage to increase this rate. CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats and CRISPR associated) is a kind of gene editing system derived from the bacterial immune mechanism (Jinek et al. 2012). The endonuclease Cas9 can specifically bind to and cleave its target DNA with the guidance of sgRNA (single guide RNA). Due to its obvious advantages of high gene-editing efficiency, high operability and low cost, this technology is widely used in DNA mutation homologous repair in mouse embryos (Wu et al. 2013; Zhou et al. 2014), cell lines (Cong et al. 2013; Jinek et al. 2013) and induced pluripotent stem cells (iPS) from patients (Dever et al. 2016; Schwank et al. 2013; Xu et al. 2015). The CRISPR/Cas9 system has shown broad prospects for application to gene correction therapy. A few pioneer studies have also succeeded in in situ gene recovery in the livers of adult mice at an HDR rate of 0.4–0.5% (Guan et al. 2016; Yin et al. 2014), but more evidence is needed to confirm its feasibility and to try to improve the DNA correction rate. In addition, different forms of Cas9, including DNA vectors, mRNA and protein, have been used in these studies aiming at precise Cas9 expression (Cho et al. 2014; Kim et al. 2014b; Sung et al. 2014; Wang et al. 2013), controlling off-target rates, etc. Therefore, it is also necessary to compare their efficiency and explore their usage in gene correction therapy.
Here, we generated a severe HB mouse model to imitate human disease and showed two CRISPR/Cas9-mediated gene correction therapy approaches in vivo in adult mice and ex vivo in germline cells. In vivo, Cas9 expression vectors delivered by hydrodynamic tail vein (HTV) injection led to a remission in hemostasis, at an ~1% repair rate on cDNA-based detection. In germline cells, microinjection of Cas9 protein was safer for gene therapy and had higher repair efficiency. Our study provides strong evidence that CRISPR/Cas9 is feasible in gene correction therapy.
Materials and methods
Animal model and study approval
Wild-type C57BL/6 mice were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. All mice were housed in a pathogen-free facility at 25 ± 1 °C on a 12-h light/dark cycle with free access to food and tap water. All animal operations conformed to the regulations drafted by the Association for Assessment and Accreditation of Laboratory Animal Care in Shanghai, and the study protocol was reviewed and approved by the Institutional Animal Care and Use Committee, Fudan University, China.
Generation of the hemophilia B mouse model and embryo microinjection
To generate the hemophilia B mouse model, 4-week-old female wild-type C57BL/6J mice were superovulated with pregnant mare serum gonadotropin (PMSG) and human chorionic gonadotropin (HCG) 48 h before mating to male mice. Embryos were collected from the oviducts of female mice and injected with Cas9 mRNA (50 ng/μL) and sgRNA (10 ng/μL) into the pronuclei at the one-cell stage. The injected embryos were cultured in KSOM at 37 °C, 5% CO2 for 2 days and then transplanted into pseudopregnant mice for breeding. Two weeks after birth, 0.5 cm tail tips were obtained from the newborn mice for genomic DNA extracting and sequencing to identify the gene mutation mice. Homologous HB mice were selected for following study.
Plasmid construction and hydrodynamics-based DNA transfection
The pSK-Cas9-sgRNA-expressing plasmid was subcloned from plasmids pSP6-2sNLS-spCas9 and p-T7-gRNA, gifts from Prof. Jihua Yao (Fudan University); promoters were modified for eukaryotic expression. Three sgRNAs targeting different sites around the mutation were chosen for gene editing, and the protospacer sequences (synthesized by Genery Co., Ltd.) for sgRNA targeting were inserted into the pSK-Cas9-sgRNA expression plasmid through BbsI ligation. The HDR donor plasmid was constructed by inserting the 1.4 kb F9 homologous amplicon into a pSK vector and adding corresponding sgRNA binding sequences at both ends of the homologous arm for self-linearization in the liver. Synonymous mutations were introduced into the donor by overlap PCR.
Plasmids were diluted into 8% (V/m) saline for hydrodynamics-based DNA transfection into HB mice (Liu et al. 1999). All plasmid solution was injected through the tail vein within 5–7 s for maximum liver absorption. For the Cas9 treatment group, 30 μg of pSK-Cas9-sgRNA expression plasmid and 30 μg of corresponding donor plasmid were co-injected. For the control group, 30 μg donor plasmid was injected alone.
Cas9 preparation and embryo microinjection
To compare the treatment effect of different Cas9 forms in germline gene therapy, Cas9 mRNA and protein were expressed and purified. Cas9 mRNA and the 98-nt HR3 sgRNA were transcribed in vitro using a MEGAshortscript™ T7 Transcription Kit (Thermo Fisher) according to the manufacturer’s instructions. Wild-type SpCas9 protein and high-fidelity SpCas9 (SpCas9-HF) were gifts from Dr. Huaxing Zhu (Novoprotein Co., Ltd). The 212 nt single-strand DNA donor was obtained through the DNA separation procedure of M-280 Streptavidin beads (Thermo Fisher). In Brief, double-strand DNA donor were PCR amplified with a biotin-labeled forward primer and a non-labeled reverse primer, and then incubated with streptavidin beads. The non-labeled DNA strand was separated from the other stand and the beads by 0.1 N NaOH elution, and purified by DNA purification beads (Thermo Fisher). The sequences of PCR primers for donor amplification were ssF: 5′-biotin-GGAGACAGGCTTCCAT TCTTC-3′ and ssR: 5′-TTCACCCCAGCTAATAATGC-3′.
The embryos for germline gene therapy were acquired from HB mice in a similar manner to wild-type mice. For Cas9 mRNA treatment, 10 ng/μL of Cas9 mRNA was co-injected with 15 ng/μL sgRNA and 10 ng/μL ssDonor. For protein treatment, 200 nM (~30 ng/μL) of Cas9-wt or Cas9-HF protein was preincubated with 400 nM (~14 ng/μL) F9 HR3 sgRNA in the injection buffer (0.25 mM EDTA, 10 mM Tris at pH 7.4) at 37 °C for 10 min and then injected with 10 ng/μL ssDonor. All components were microinjected into pronuclei of one-cell embryos. The injected embryos were cultured in KSOM at 37 °C, 5% CO2 for 3 days. A portion of the embryos were placed in 1 × PCR buffer (Takara) with 0.5 μg proteinase K and heated to 65 °C for 30 min, 95 °C for 10 min to release their genomic DNA. Then, the DNA was PCR-amplified by Taq HS (Takara) with primers F9LF, F9LR for Sanger sequencing to measure the gene correction rate. Other embryos were transplanted into pseudopregnant mice for breeding and further detection. The sequences of the primers are listed in Supplementary Table 1.
cDNA-based unlabeled probe hybridization detection
For cDNA-based gene detection and real-time PCR, total RNA was extracted from a fraction of mouse liver tissue (100 mg) homogenized in Trizol (Invitrogen). cDNAs were synthesized using HiScript Q RT SuperMix (Vazyme) from 0.5 μg RNA.
Asymmetric PCR with F9SF/F9SR was utilized to amplify these cDNAs for unlabeled probe detection (van der Stoep et al. 2009) using HRM Master Mix (Roche) on a Roche LightCycle96. The final concentrations of the forward and reverse primers were 0.5 and 0.05 μmol/L, respectively. The cycling conditions were 95 °C for 10 min followed by 50 three-step cycles of 95 °C for 15 s, 60 °C for 20 s, and 70 °C for 15 s. Unlabeled probe was added into the PCR system (final concentration 0.5 μmol/L) for hybridization; fluorescence values were recorded every 0.02 °C. The normalized melting peaks were used for high-resolution melting analysis. The sequences of the primers and unlabeled probe are listed in Supplementary Table 1.
Cas9-derived RNA-guided engineered nucleases (RGEN) mediated in vitro gene enrichment
The cDNAs from HTV-treated mouse liver tissues were PCR-amplified with the primer pair F9 LF/F9 LR and purified by ethanol precipitation assay. Purified amplicons (200 ng) were first incubated with Cas9 protein (2 pmol) and HR3 sgRNA (2 pmol) in reaction buffer (Jinek et al. 2012) (20 mmol/L HEPES, 150 mmol/L KCl, 0.1 mmol/L EDTA, 10 mmol/L MgCl2, 0.5 mmol/L DTT) for 1 h DNA cleavage, and then amplified in the second round of amplification with a pair of inner primers F9SF/F9SR for Sanger sequencing. As the HR3 sgRNA specifically binds to the incorrect F9 allele with an 8-bp deletion, the corrected gene can be enriched by this cleavage.
Off-target analysis
Off-target prediction was carried out using the BLAST/BLAT tool of Ensembl (http://asia.ensembl.org/index.html) and a CRISPR design website (http://crispr.mit.edu/). High-resolution melting analysis was used for off-target detection. The primers for PCR amplification are listed in Supplementary Table 1 as indicated.
In suit RNA hybridization and real-time PCR
Mouse liver tissues were fixed in 4% paraformaldehyde, dehydrated, embedded in OCT, and cryosectioned at 10 μm thickness. The sections were then incubated with acetic acid blocking agent (Beijing Leagene Biotechnology Co., Ltd.) to inactivate the activity of basal alkaline phosphatase. A digoxin-labeled DNA probe was used for in situ RNA hybridization to these sections at 55 °C overnight. After washes and blocking, the hybridized probe was finally developed by anti-dig/AP staining (Roche).
The expression of F9 was quantitated by real-time PCR with cDNAs from these liver tissues using SYBR Green Real-time PCR Master Mix (Toyobo) performed on an ABI 7900 (Life Technology). Mouse beta-actin (mActb) was used as the endogenous control to obtain normalized values. The sequences of the primers are listed in Supplementary Table 1.
FIX concentration, FIX plasma procoagulant level test
Mouse plasma was obtained via tail-clip bleeding and anticoagulated with 1/10 (V/V) sodium citrate immediately. Blood samples were spun at 1000 rpm for 20 min to separate the upper layer (plasma). FIX activity tests were conducted using the ACL Top 700 automatic blood coagulation analyzer (Werfen); the FIX concentration in plasma was detected using a mouse coagulation factor IX ELISA kit (Shanghai Meilian Biotechnology).
Statistics
The statistical significance of comparisons between the two groups was analyzed with Student’s t test. Differences are considered statistically significant when P values are less than 0.05.
Results
Generation of F9 mutant mouse strain
We utilized the CRISPR/Cas9 system to modify the 8th exon of the mouse F9 gene as previously described (Qihan et al. 2015), and obtained an 8-bp deletion C57BL/6 mouse strain, F9 KO. The deletion, which is located at a region highly conserved with human F9, forms a premature stop codon, leading to the absence of a key residue (Ser421) for FIX activation (Fig. 1a, b). Both mRNA and protein levels of F9 in KO mice (n = 8) were significantly decreased compared to wild-type (WT) controls (Fig. 1c, d). Consistently, the procoagulant levels (FIX:C) of KO mice fell to 1% or less of the normal level, and the mice did not show obvious blood clotting in the 5-min tail-clip test. All these evidences indicated that the F9 KO mouse is a good model of severe HB.

Generation of F9 mutant mouse strain. a Schematic diagram of sectional F9 locus, showing target DNA sites designed for CRISPR/Cas9-mediated gene knockout (black underline) and the mutant site of the KO mouse strain (dash lines). Corresponding amino acids are shown in the upper row; the asterisk (*) indicates the stop codon; e7, exon 7. b Amino acid alignment of a partial sequence of FIX in human and mouse. An asterisk (*) indicates residues that are fully conserved, a plus sign (+) indicates residues that are strongly conserved. c Expression of F9 mRNA in the hepatic tissue of WT and KO mouse strains. d Concentration of FIX protein in the plasma of WT and KO mouse strains. Data in c and d were acquired from 8 mice per group, and are shown as the mean ± the s.d. Two-tailed unpaired Student’s t tests were used to determine the P value. **P < 0.01
Gene correction of adult HB mice through hydrodynamics-based DNA transfection of Cas9 components and self-linearized HR donor
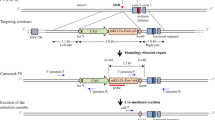
To correct the mutant F9 gene in adult HB mice and restore their hemostasis, the CRISPR/Cas9 system was employed to induce genome editing and homologous recombination. As the liver is the only tissue expressing FIX, we used hydrodynamic-based naked DNA transfection (Liu et al. 1999), a non-immunogenicity strategy, to inject Cas9-sgRNA-expressing plasmids and donor plasmids to hepatocytes. Three sgRNAs (HR1/2/3) targeting different sites around the mutation were chosen for gene editing, which were all pretested and found effective in DNA cleavage (Table 1; Supplementary Fig. 1). sgRNA binding sequences were added to both ends of the homologous arm for donor plasmid linearization (Fig. 2a), and synonymous mutations were induced into the donor to protect it from Cas9/sgRNA cleavage (Table 1).

In vivo gene correction of adult HB mice through hydrodynamics-based DNA transfection of Cas9 components. a Schematic diagram of plasmids used for in vivo gene correction. The pSK-Cas9-sgRNA plasmid containing Cas9 and sgRNA expression cassettes. The HDR donor plasmid contains a 1.4-kb homologous arm and two sgRNA-binding sequences (RBS) for self-linearization. L-arm/R-arm, left and right homologous arm, 800 and 600 bp, respectively. b Schematic diagram of unlabeled DNA probe hybridization assay to defective (KO) and corrected (WT) F9 DNA. Cyan line indicates the probe, blue bars represent the hydrogen bonds of paired bases, gray box indicates the absent 8 bp of KO DNA. c, d High-resolution melting peak of unlabeled probe hybridization assay performed on hepatic cDNA obtained from each HTV-treated group. c cDNA from HR3 sgRNA treated mice. Pink line represents result of a mixture of KO and WT DNA. d cDNA from the control group with only the donor plasmid injected. e Sanger sequencing results of cDNA amplicon from a representative Cas9-treated sample (RGEN-enriched before sequencing); arrows indicate the synonymous nucleotides provided by the donor template, box shows the corrected absent fragment. f Expression of F9 mRNA in hepatic tissue of mice treated as indicated. UNTR, untreated F9 KO mice. g, i FIX concentration and activity in the plasma of mice treated as indicated. h mRNA in situ hybridization of liver biopsies from mice treated as indicated. j Representative results of a tail-clip assay. Datasets in f, g and i are shown as the mean ± the s.d. Two-tailed unpaired Student’s t tests were used to determine the P value. *P < 0.05; ***P < 0.001
The effect of the therapy was first evaluated by cDNA-based gene detection 6 weeks after HTV treatment. An unlabeled probe with the detection sensitivity of 1% was designed to distinguish the mutant and corrected F9 alleles by melting temperature (Fig. 2b, Supplementary Fig. 2a). By unlabeled probe hybridization detection, 62.5% (15 out of 24) of the Cas9-treated mice had a detectable rate of gene correction (Fig. 2c, Supplementary Fig. 2b–d), which means that more than 1% of hepatocytes were repaired in these mice. The correction percentage was especially high in the Cas9-HR3 sgRNA-treated group, with six out of eight mice carrying repaired alleles, while no corrected gene was detected in the control group mice, which were only injected with donor plasmids (Fig. 2d). The synonymous mutations intentionally introduced into the homologous arms were also discovered in RGEN-enriched (Kim et al. 2014a) cDNA of Cas9-treated mice. It further confirmed the occurrence of homology-directed gene repair (Fig. 2e). In addition, the expression of F9 was increased in both liver and plasma (Fig. 2f, g). The FIX concentration rose to an average of 3.39 times the concentration in control mice (Fig. 2g). However, we did not find any enrichment region of gene correction in liver biopsies (Fig. 2h).
The treatment also led to a remission in coagulation deficiency. The plasma procoagulant levels of FIX in Cas9 HTV-treated groups increased to 4.21, 9.38 and 5.4% (Fig. 2i, j), which indicates that they converted to the moderate or mild type of HB, reflecting a good therapeutic effect. Representative tail-clip results were shown in Fig. 2j. No significant differences in blood aspartate transaminase (AST) or alanine transaminase (ALT) levels were observed 6 weeks after Cas9-treatment.
Cas9 protein is a better tool for HB mice germline gene therapy
Hydrodynamics-based Cas9 DNA transfection can be used for gene correction therapy in adult mice, which is concurrent with previous reports (Guan et al. 2016; Yin et al. 2014). However, the corrected gene cannot be inherited to the next generation, so it is also necessary to explore and optimize a more effective approach for CRISPR/Cas9-mediated germline gene therapy. To compare the gene recovery efficacy and safety of different forms of Cas9, we microinjected a normalized dose of Cas9 mRNA, wild-type SpCas9 protein (Cas9-wt), or high-fidelity SpCas9 protein (Cas9-HF) (Kleinstiver et al. 2016) into the male pronucleus of one-cell-stage embryos of F9 KO mice. HR3 sgRNA and a 212 nt single-strand DNA donor were co-injected. After 2 days of in vitro culture, more embryos developed to blastulas in protein injection groups (Table 2), and the birth rate was also 1.56 times higher than that of the mRNA injection group after transplantation into surrogate mothers. This suggests that Cas9 protein-mediated treatment is less deleterious to the embryos.
Meanwhile, protein injection also induced a higher gene correction efficiency. Sanger sequencing performed on in vitro cultured embryos demonstrated that 52.6% of the mutant F9 DNA was corrected in the Cas9-wt injection group, 1.62-fold higher than the mRNA injection group (Table 2). In addition, 70% of embryos in the Cas9-wt injection groups were homozygously or heterozygously repaired, which means they may generate gametes without the defective gene. While the mosaic rate of mRNA injection was as high as 71.4%, it is also unfavorable in germline gene therapy (Fig. 3; Table 2, Supplementary Table 2). Five potential off-target sites were detected by high-resolution melting analysis, but no off-targets were found in any treatment group (Supplementary Fig. 3a, b). Cas9-HF also performed better in embryo survival and gene correction than mRNA injection, but the DNA correction efficiency is slightly lower than that of Cas9-wt. The treatment effect was finally evaluated in the newborn mice. The mice in both groups carrying the corrected F9 gene showed good hemostasis in the tail-clip test and a higher FIX concentration than F9 KO mice (Supplementary Fig. 3c), which indicates that Cas9-mediated germline gene therapy is effective.

Representative sequencing results of germline gene-corrected embryos. Representative sequencing results show different DNA recovery rates in microinjected embryos. The sgRNA binding sequence is underlined. Arrows highlight the synonymous nucleotides provided by the donor template, box shows the corrected absent fragment
Discussion
Of all F9 mutations, more than 56% were found in the serine protease (SP) domain, the catalytic domain of FIX (Rallapalli et al. 2013). Thus, in this study, we introduced a mutation into the homologous SP domain in mice and generated a mouse strain presenting a severe HB phenotype. The absence of the key catalytic residue Ser421 in this mouse is similar to a common mutation in human hemophilia, Ser411Gly; therefore, it is a good model to imitate human disease.
In the gene therapy of adult mice, Cas9-sgRNA expression vectors and self-linearizable double-strand donor DNA were HTV injected into mouse livers for in situ gene correction. Most mice showed a >1% gene recovery according to cDNA-based detection, which supports the previous evidence of Cas9-mediated HB gene correction to another F9 mutant site (Guan et al. 2016). We have also made two improvements: first, to the best of our knowledge, we used the lowest dose of DNA of Cas9 components for in vivo therapy, while a high concentration of Cas9 and donor DNA may reduce the possibility of off-targets and random DNA insertion (Cho et al. 2014). Second, the self-linearizable donor we used may have induced a higher rate of homology repair than circular donors. However, as the F9 mRNA level decreased significantly in KO mice due to nonsense-mediated mRNA decay, the corrected F9 mRNA would be more stable than the mutant one, so the actual recovery rate in the genome may be slightly lower than we detected in cDNA. We did not estimate the actual HDR rate by next-generation sequencing, because the delayed degradation of the dsDNA donor (Andrianaivo et al. 2004; Lin et al. 2014; Liu et al. 1999) may mix with genomic DNA and affect the accuracy of sequencing tested on interrupted DNA fragments. We have indeed detected a higher correction rate on DNA-based HRMA analysis after 6 weeks of HTV treatment (data not shown), but sequencing on longer fragments revealed it to be a false positive. Meanwhile, HDR with an ssDNA donor may be suitable for correction efficiency estimation. Considering previous gene correction results by ssDNA donor together, a gene correction rate of 0.5% in hepatocytes may restore hemostasis in HB mice.
Naked DNA hydrodynamic tail vein injection was initially used as a replacement of the directly portal vein injection to animal models. By rapidly injecting a large volume of DNA solution to mice, naked DNA can be intercepted by hepatocytes under the high hydrostatic pressure (Yang et al. 2001), but the high-pressure may be harmful or even lethal to large animals, so HTV was thought to be inoperable in human. Nowadays, modifications have been made to make it possible to be applied to larger animals: Khorsandi et al. put a balloon catheter into pig’s liver by invasive surgery to introduce hydrostatic pressure and transfer DNA. This method only caused a brief pressure increase in the liver which was not harmful to heart. Moreover, modified HTV has been demonstrated to be safe in a phase I clinical trial (Khorsandi et al. 2008). Although it does not behave as well as traditional HTV in gene transfer efficiency, the low host immunogenicity makes it applicable for repetitive treatments, which shows the prospect to be used as gene transfer tool in human gene therapy.
Target selection is an important issue for CRISPR/Cas9-mediated gene therapy. In this study, three sgRNAs (HR1, HR2 and HR3) were used for Cas9 HTV-treated therapy. Among them, HR2 sgRNA nears the F9 8 bp-deletion mutation, while HR1 showed a higher cleave ability. As the gene correction rate in the Cas9-HR2 sgRNA treatment group is much higher than the Cas9-HR1 sgRNA group, it indicated that the distance between the cleavage site and the mutant site plays a more important role in gene homologous recombination than sgRNA cleavage efficiency, but target distance no further than 70 bp may also be effective. Thus, the target selection of sgRNAs in gene therapy may not be limited to the exact mutation site but have alternative options around the mutation.
It is also a problem that the relatively low gene correction rate may limit the broader usage of CRISPR/Cas9 in other heritable diseases that need more gene recovery. Trying to increase DNA HDR rate by other Cas9 delivery methods and repopulating the repaired cells may be two practicable strategies, while germinal alteration of the mutant gene is also a good choice.
Germline gene therapy has been controversial for a long time; besides ethical considerations, the safety of germline gene therapy is one of the most important issues (Anderson 1985; Lebo and Golbus 1991). There is an active demand for an approach that can repair mutant genes but induce less toxicity and fewer unwanted changes at the same time. In this study, we used the longest ssDNA donor template as far as we know and have achieved a higher gene correction rate than previous studies (Kim et al. 2014b; Lin et al. 2014; Sung et al. 2014). In addition, we discovered that microinjection of Cas9-wt protein performed better in gene correction, with higher HDR rates, less embryotoxicity, and lower mosaic rates, indicating that it is a better method for germline gene therapy. The higher mosaic rate induced by Cas9 mRNA is due to its delayed expression (Sung et al. 2014) at 8–12 h after microinjection into cells, when the zygotes have started to undergo mitosis. The divided cells may receive different doses of Cas9 components, leading to different gene-editing effects, which is not good for germline gene therapy. The longer expression duration of Cas9 mRNA will also lead to an accumulation of the protein in the zygotes and significantly increase the risk of off-target gene editing (Sung et al. 2014). However, we have not detected any off-target gene editing in either of the groups in our study, so the advantage of Cas9 protein, especially Cas9-HF protein, is not obvious. Deep sequencing could be better for off-target detection. To the best of our knowledge, it is the first evidence to compare the safety of different Cas9 forms (especially Cas9 HF) in germline gene therapy.
In conclusion, we provide evidence that CRISPR/Cas9 is a versatile tool for gene correction of genetic diseases both in vivo and ex vivo in germline gene editing, supporting its broader use in gene therapy and precision medicine.
References
Anderson WF (1985) Human gene therapy: scientific and ethical considerations. Recomb DNA Tech Bull 8:55–63
Andrianaivo F, Lecocq M, Wattiaux-De Coninck S, Wattiaux R, Jadot M (2004) Hydrodynamics-based transfection of the liver: entrance into hepatocytes of DNA that causes expression takes place very early after injection. J Gene Med 6:877–883. doi:10.1002/jgm.574
Anguela XM, Sharma R, Doyon Y, Miller JC, Li H, Haurigot V et al (2013) Robust ZFN-mediated genome editing in adult hemophilic mice. Blood 122:3283–3287. doi:10.1182/blood-2013-04-497354
Chen L, Chen H, Lu H, Wu X, Lu D, Qiu X, Xue J (2003) Muscle injection of rAAV/mFIX to secrete clotting factor IX corrects the hemorrhagic tendencies in hemophilia B mice. Sci China C Life Sci 46:422–430. doi:10.1007/BF03192585
Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS (2014) Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24:132–141. doi:10.1101/gr.162339.113
Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi:10.1126/science.1231143
Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE et al (2016) CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 539:384–389. doi:10.1038/nature20134
Guan Y, Ma Y, Li Q, Sun Z, Ma L, Wu L et al (2016) CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol Med 8:477–488. doi:10.15252/emmm.201506039
Iiizumi S, Kurosawa A, So S, Ishii Y, Chikaraishi Y, Ishii A et al (2008) Impact of non-homologous end-joining deficiency on random and targeted DNA integration: implications for gene targeting. Nucleic Acids Res 36:6333–6342. doi:10.1093/Nar/Gkn649
Jiang H, Pierce GF, Ozelo MC, de Paula EV, Vargas JA, Smith P et al (2006) Evidence of multiyear factor IX expression by AAV-mediated gene transfer to skeletal muscle in an individual with severe hemophilia B. Mol Ther 14:452–455. doi:10.1016/j.ymthe.2006.05.004
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi:10.1126/science.1225829
Jinek M, East A, Cheng A, Lin S, Ma EB, Doudna J (2013) RNA-programmed genome editing in human cells. Elife. doi:10.7554/eLife.00471
Kay MA, Rothenberg S, Landen CN, Bellinger DA, Leland F, Toman C et al (1993) In vivo gene therapy of hemophilia B: sustained partial correction in factor IX-deficient dogs. Science 262:117–119
Khorsandi SE, Bachellier P, Weber JC, Greget M, Jaeck D, Zacharoulis D et al (2008) Minimally invasive and selective hydrodynamic gene therapy of liver segments in the pig and human. Cancer Gene Ther 15:225–230. doi:10.1038/sj.cgt.7701119
Kim JM, Kim D, Kim S, Kim JS (2014a) Genotyping with CRISPR-Cas-derived RNA-guided endonucleases. Nat Commun 5:3157. doi:10.1038/ncomms4157
Kim S, Kim D, Cho SW, Kim J, Kim JS (2014b) Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012–1019. doi:10.1101/gr.171322.113
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK (2016) High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529:490–495. doi:10.1038/nature16526
Lebo RV, Golbus MS (1991) Scientific and ethical considerations in human gene therapy. Bailliere’s Clin Obstet Gynaecol 5:697–713
Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS et al (2011) In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475:217–221. doi:10.1038/nature10177
Lin S, Staahl B, Alla RK, Doudna JA (2014) Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. doi:10.7554/Elife.04766
Liu F, Song Y, Liu D (1999) Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther 6:1258–1266. doi:10.1038/sj.gt.3300947
Lu DR, Zhou JM, Zheng B, Qiu XF, Xue JL, Wang JM et al (1993) Stage I clinical trial of gene therapy for hemophilia B. Sci China B 36:1342–1351
Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR et al (2003) AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 101:2963–2972. doi:10.1182/blood-2002-10-3296
Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ et al (2006) Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 12:342–347. doi:10.1038/nm1358
Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J et al (2014) Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371:1994–2004. doi:10.1056/NEJMoa1407309
Qihan W, Cong H, Ruilin S, Huaxing Z, Hongyan C, Jian F, Daru L (2015) A quick and efficient method to generate hemophilia B mouse models by the CRISPR/Cas system. Yi chuan 37:1143–1148. doi:10.16288/j.yczz.15-117
Rallapalli PM, Kemball-Cook G, Tuddenham EG, Gomez K, Perkins SJ (2013) An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B. J Thromb Haemost 11:1329–1340. doi:10.1111/jth.12276
Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T et al (2013) Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13:653–658. doi:10.1016/j.stem.2013.11.002
Sharma R, Anguela XM, Doyon Y, Wechsler T, DeKelver RC, Sproul S et al (2015) In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 126:1777–1784. doi:10.1182/blood-2014-12-615492
Sung YH, Kim JM, Kim HT, Lee J, Jeon J, Jin Y et al (2014) Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res 24:125–131. doi:10.1101/gr.163394.113
van der Stoep N, van Paridon CD, Janssens T, Krenkova P, Stambergova A, Macek M et al (2009) Diagnostic guidelines for high-resolution melting curve (HRM) analysis: an interlaboratory validation of BRCA1 mutation scanning using the 96-well LightScanner. Hum Mutat 30:899–909. doi:10.1002/humu.21004
Wang L, Takabe K, Bidlingmaier SM, Ill CR, Verma IM (1999) Sustained correction of bleeding disorder in hemophilia B mice by gene therapy. Proc Natl Acad Sci USA 96:3906–3910
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918. doi:10.1016/j.cell.2013.04.025
White GC 2nd, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J et al (2001) Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost 85:560
Wu YX, Liang D, Wang YH, Bai MZ, Tang W, Bao SM et al (2013) Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13:659–662. doi:10.1016/j.stem.2013.10.016
Xu P, Tong Y, Liu XZ, Wang TT, Cheng L, Wang BY et al (2015) Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C>T) mutation in beta-thalassemia-derived iPSCs. Sci Rep 5:12065. doi:10.1038/srep12065
Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z, Kay MA (2000) Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet 25:35–41. doi:10.1038/75568
Yang J, Chen S, Huang L, Michalopoulos GK, Liu Y (2001) Sustained expression of naked plasmid DNA encoding hepatocyte growth factor in mice promotes liver and overall body growth. Hepatology 33:848–859. doi:10.1053/jhep.2001.23438
Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M et al (2014) Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 32:551–553. doi:10.1038/Nbt.2884
Zhou J, Shen B, Zhang W, Wang J, Yang J, Chen L et al (2014) One-step generation of different immunodeficient mice with multiple gene modifications by CRISPR/Cas9 mediated genome engineering. Int J Biochem Cell Biol 46:49–55. doi:10.1016/j.biocel.2013.10.010
Acknowledgements
We are grateful to Prof. Jihua Yao for her kindness gift of Cas9 expressing plasmid, to Dr. Huaxing Zhu and Mr. Yuanping Liao (Novoprotein Co., Ltd.) for their assistance in Cas9 protein expression and purification, to Dr. Xiao Song and Dr. Shingming Wang for their help in language polishing, to Dr. Kun Chen for providing technical assistance. This work was supported by the grants from the Natural Science Foundation of China (31571371).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
We declare that no competing interests exist.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Huai, C., Jia, C., Sun, R. et al. CRISPR/Cas9-mediated somatic and germline gene correction to restore hemostasis in hemophilia B mice. Hum Genet 136, 875–883 (2017). https://doi.org/10.1007/s00439-017-1801-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-017-1801-z