
Abstract
We previously identified a low-frequency (1.1 %) coding variant (G45R; rs200573126) in the adiponectin gene (ADIPOQ) which was the basis for a multipoint microsatellite linkage signal (LOD = 8.2) for plasma adiponectin levels in Hispanic families. We have empirically evaluated the ability of data from targeted common variants, exome chip genotyping, and genome-wide association study data to detect linkage and association to adiponectin protein levels at this locus. Simple two-point linkage and association analyses were performed in 88 Hispanic families (1,150 individuals) using 10,958 SNPs on chromosome 3. Approaches were compared for their ability to map the functional variant, G45R, which was strongly linked (two-point LOD = 20.98) and powerfully associated (p value = 8.1 × 10−50). Over 450 SNPs within a broad 61 Mb interval around rs200573126 showed nominal evidence of linkage (LOD > 3) but only four other SNPs in this region were associated with p values < 1.0 × 10−4. When G45R was accounted for, the maximum LOD score across the interval dropped to 4.39 and the best p value was 1.1 × 10−5. Linked and/or associated variants ranged in frequency (0.0018–0.50) and type (coding, non-coding) and had little detectable linkage disequilibrium with rs200573126 (r 2 < 0.20). In addition, the two-point linkage approach empirically outperformed multipoint microsatellite and multipoint SNP analysis. In the absence of data for rs200573126, family-based linkage analysis using a moderately dense SNP dataset, including both common and low-frequency variants, resulted in stronger evidence for an adiponectin locus than association data alone. Thus, linkage analysis can be a useful tool to facilitate identification of high-impact genetic variants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Family-based linkage analysis has been highly successful in identifying genetic loci underlying Mendelian disorders. In contrast, linkage analysis of complex traits and diseases in the general population has resulted in little success. While many complex trait and disease variants have been identified through genome-wide association studies (GWAS), the majority of these loci have small effect sizes and cumulatively explain relatively little of the overall risk (Kiezun et al. 2012; Manolio et al. 2009). These observations have led investigators to assess new approaches and to reassess methods such as family-based linkage analysis.
Large-scale exome- and genome-wide sequencing have facilitated creation of extensive resources for analysis of common, primarily non-coding variants and more recently common, low-frequency, and rare coding variants through exome chip genotyping. This provides an ability to re-address the failures of family-based linkage approaches to identify complex trait loci. Family-based approaches remain a potentially powerful methodology for identification of complex trait loci. Patterns of segregation of uncommon or rare variants amplify power for detection compared to conventional population-wide association studies. With these new data resources, the question arises as to how linkage analysis will perform in this framework and especially what can be expected when translated to empirical studies.
In prior reports, we have described one of the few contemporary examples of family-based linkage (and association) with a complex trait (An et al. 2013; Bowden et al. 2010). In a microsatellite-based multipoint linkage analysis, a linkage peak for plasma adiponectin protein levels (LOD = 8.2) overlying the ADIPOQ locus on chromosome 3 was identified in Hispanic families in the Insulin Resistance Atherosclerosis Family Study (IRASFS) (Guo et al. 2006). Common, non-coding variations did not account for this linkage, but a combination of conventional and exome sequencing revealed a novel coding variant (G45R; rs200573126) that segregated with low adiponectin levels (average 80 % reduction) and was highly associated with plasma adiponectin (p = 5.03 × 10−40) (Bowden et al. 2010). This G45R variant was present at 1.1 % frequency in the sample, contributed significantly to the variance in adiponectin levels (20 %), and accounted for the previously observed linkage signal.
This low-frequency coding variant which was the source of linkage and association with a complex trait was identified using targeted methods. An agnostic search for novel variants contributing to complex traits would likely include a genome-wide approach searching for linkage (and association) to complex traits in families. Here we have evaluated the performance of a combined linkage and association analysis approach in a locus-wide re-analysis of the ADIPOQ region. This analysis provides insight into the empirical signature of a low-frequency, high-impact causal variant in a background of genotype data from GWAS and exome chip sources. The characteristics of this linkage are relevant especially to the scenario in which a novel trait-defining variant has not been directly genotyped.
Materials and methods
Samples
The samples used in this study were from the Hispanic cohort of the IRASFS (Henkin et al. 2003). Briefly, subjects were ascertained on the basis of large family size in San Luis Valley, Colorado, and San Antonio, Texas. The sample consisted of a maximum of 1,414 individuals from 88 families with available genotype data. Detailed relationship information about these samples is included in Supplemental Table 1. A subset of these individuals (n = 1,150) had plasma adiponectin levels measured by radioimmunoassay (RIA; Linco Research, St. Charles, MO, USA), as previously reported (Bowden et al. 2010; Guo et al. 2006). IRB approval was obtained at all clinical and analysis sites, and all participants provided informed consent.
Genotype data
Single-nucleotide polymorphism (SNP) genotype data from a variety of sources were collected. These included individually genotyped SNPs at the ADIPOQ locus (n = 33) (An et al. 2012, 2013) which were combined with data from 7,497 SNPs in the ADIPOQ region (chr3:161,560,463-197,838,262, hg19, 25 MB proximal and to the distal end of the chromosome) derived from Illumina OmniExpress (GWAS chip) genome-wide genotyping as part of the GUARDIAN Consortium (Goodarzi et al. 2013) and data from all chromosome 3 SNPs (n = 3,428) on the Illumina HumanExome Beadchip (Hellwege et al. 2014) for a total of 10,958 non-redundant, polymorphic SNPs. Quality control for each group of SNP data has previously been described in detail (An et al. 2012; Hellwege et al. 2014). All datasets underwent Mendelian error checking using PedCheck (O’Connell and Weeks 1998).
Statistical analyses
All SNPs were evaluated for both two-point family-based linkage and single SNP association using Sequential Oligogenic Linkage Analysis Routines (SOLAR) (Almasy and Blangero 1998). Both analyses used age, sex, body mass index (BMI), and study center as covariates. Association analysis additionally included three admixture variables as covariates. Admixture was estimated using ADMIXTURE (Alexander et al. 2009) assuming five ancestral populations from exome chip-wide SNP data pruned for linkage disequilibrium (LD) to produce admixture estimates for the greatest number of samples. HapMap samples from CEU, YRI and CHN-JPT were included in the analysis. Three variables were selected as representing the variation in these Hispanic samples as larger values for K began to isolate individual pedigrees. Plasma adiponectin levels were transformed (natural log) to best approximate the distributional assumptions of the test. To test the influence of kurtosis, empirical LOD scores were calculated for each series of analyses using the lodadj procedure in SOLAR as appropriate. This procedure simulates the data to generate a distribution of LOD scores under the null hypothesis of no linkage at a given number of repetitions. For the purposes of this study, 100 replicates were used to generate the empirical LOD adjustment. Additional linkage and association analyses were performed adjusting for G45R as a covariate in the respective models.
To evaluate the relative performance of multipoint versus two-point linkage analysis, the previously published (Guo et al. 2006) microsatellite multipoint linkage analysis encompassing 25 microsatellite markers from chromosome 3 was contrasted with the two-point linkage analyses. The analyses were computed with and without the G45R polymorphism to observe the impact of functional variant on the evidence for linkage. A SNP-based linkage analysis panel was created from the GWAS data for multipoint analysis to reflect a more contemporary approach to linkage analysis. This SNP panel used the Illumina Human Linkage Panel IVb genotyping panel as its backbone. Of the 409 SNPs present from the linkage panel on chromosome 3, 160 (39 %) of these were directly available from the GWAS. The remaining SNPs were selected using proxies for linkage panel SNPs (D′ = 1.00; r 2 > 0.98) while maintaining low linkage disequilibrium (D′ < 0.40; r 2 < 0.20) between other markers in the panel. Unanticipated linkage disequilibrium between markers required pruning these 409 markers to an uncorrelated subset of 156 SNPs for multipoint analysis. Multipoint identity by descent (IBD) matrices were calculated using Loki (Heath 1997; Heath et al. 1997) as specified previously (Guo et al. 2006) and multipoint linkage analysis was performed using SOLAR.
Results
Two-point linkage analysis
Two-point linkage analysis was chosen as the primary linkage tool to avoid signal inflation due to LD between closely linked markers. Linkage analysis was carried out with a total of 10,958 SNPs on chromosome 3 comprising common, low-frequency, and rare non-coding and coding variants in 1,414 DNAs from 88 families. The highest density of SNPs was in a 36-Mb interval surrounding the ADIPOQ locus. Family sizes ranged from 2 to 33 individuals. A breakdown of the demographic characteristics by SNP variant class is summarized in Table 1. Comprehensive sample characteristics have been described previously (Bowden et al. 2010). SNP minor allele frequencies (MAF) ranged from 0.0018 to 0.5. The previously documented causal variant G45R (rs200573126) was the most strongly linked variant with a two-point LOD = 20.98 (Table 2; Fig. 1; Supplemental Table 2). Additionally, many variants across chromosome 3 (n = 453) also showed evidence of linkage, with LOD scores ranging from 3 to 14.85 (Table 2; Fig. 1). These SNPs spanned 56 MB proximal and 5.2 MB distal to the ADIPOQ locus (n = 7,203 SNPs in the region) and included both common non-coding and lower frequency coding variants. In contrast, a very large number of SNPs (n = 8,910) showed little evidence of linkage (LOD <1.0), including 73 % of SNPs (n = 5,293) within the 61 MB region specified above. Minor allele frequencies for the nominally linked variants ranged from 0.0044 to 0.5. Adjusting for G45R as a covariate, the maximum observed two-point LOD score across the chromosome was reduced to 4.39 at rs10937349 (Fig. 2). This SNP rs10937349 is an intronic variant in the LPP gene (encoding LIM domain containing preferred translocation partner in lipoma), which is 1.65 Mb distal of ADIPOQ. This LPP SNP was not associated with adiponectin levels with or without adjustment for the G45R variant (p = 0.53 and 0.76, respectively) (Figs. 1 and 2). The G45R-adjusted analysis identified only four variants with LOD scores greater than 3 (Fig. 2) which were widely dispersed across the greater ADIPOQ region, some being greater than 15 Mb away from the ADIPOQ locus.

Adiponectin two-point linkage and association on chromosome 3

Adiponectin two-point linkage and association on chromosome 3, adjusted for the presence of G45R
Multipoint linkage analysis: microsatellite and SNP performance
We have also revisited multipoint approaches such as the previously observed microsatellite multipoint linkage (maximum LOD = 8.2; Table 3) (Guo et al. 2006) as a complementary analysis to the two-point linkage results summarized above. When not adjusting for G45R (LOD = 20.98), the largest two-point LOD score is observed with rs10937185 (LOD = 14.85). By comparison, adding G45R as a marker to the microsatellite multipoint increased the LOD score marginally from 8.2 to 8.4 (Table 3). Review of the microsatellite data revealed that one allele of a neighboring microsatellite marker TTTA040 (allele 160 bp) was in linkage disequilibrium with G45R (D′ = 0.96). Two other microsatellite markers were also in LD (D3S2427; D′ = 0.62 and D3S2398; D′ = 0.48) with G45R. Thus, how much this modest increase in the LOD score is due to increased linkage content versus bias due to linkage disequilibrium is unclear (Table 3). To evaluate the performance of SNPs in the region in a multipoint framework, GWAS genotypes were pruned to replicate the Illumina HumanLinkage IVb genotyping panel (N = 8,000 SNPs), and further pruned to eliminate linkage disequilibrium between SNPs. Multipoint linkage analysis results with these SNP markers on chromosome 3 (N = 156) yielded a LOD score of 6.4. When G45R was included, the LOD score rose to 10.9. Thus, comparison of the approaches yielded similar overall results, albeit with substantially different maximum LODs.
Association analysis
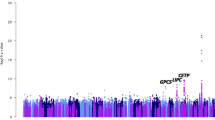
In parallel with the two-point linkage analysis, the SNP dataset was analyzed for association with adiponectin. The SNP association analysis across the greater ADIPOQ region confirmed the G45R association with adiponectin (p value = 8.1 × 10−50) but provided little evidence of association at other loci (Table 4). The lack of association of other SNPs across the region (Fig. 3) is consistent with the existence of only a single functional variant in the region and a lack of linkage disequilibrium between G45R and any other SNP (r 2 ≤ 0.20). This observation was reinforced when focusing on association results for SNPs with LOD scores over 8 (Table 2) where association p values ranged from nominal 2.5 × 10−4 to non-significant. Overall, a total of 866 variants demonstrated at least nominal evidence of association (p < 0.05), including only three with p values less than 1.0 × 10−5. In addition to G45R, among these highly associated SNPs were an intronic variant (rs12054151; p value = 1.0 × 10−7) in the ST6GAL1 gene over 200 kb distal of G45R and a coding variant (rs200567888; K207 N; p value = 4.3 × 10−6) in the EPHA3 gene which is located nearly 100 Mb distal to ADIPOQ. Results of association in the immediate region of 400 kb surrounding ADIPOQ are presented in Supplementary Fig. 1.

Linkage disequilibrium surrounding G45R (indicated by the red star) in a subset of 225 unrelated individuals from IRAS-FS. Color scheme represents pairwise r 2 values
Association analysis was also computed adjusting for the presence of G45R (Fig. 2; Table 3). It is of note that the associations above (rs12054151 and rs200567888) were reduced to p values of 0.88 and 1.0, respectively, upon adjustment for G45R. A total of 477 variants also showed evidence of association (p < 0.05), with the most strongly associated SNPs having p values of 1.1 × 10−5 and 4.1 × 10−5. These two variants, however, were located 49 MB distal from G45R and separated by 22 kb. Both variants are intergenic and were genotyped as part of the exome chip. Of the 477 associated variants, 240 of them were nominally associated (p < 0.05) in the original model (i.e., without adjustment for G45R), suggesting that adjusting for G45R identifies additional variants which were masked when the strongly associated causal variant was present.
Discussion
In this study, we explored the characteristics of a genetic linkage signal due to a low-frequency, high-impact mutation in the ADIPOQ gene. These characteristics are especially relevant to ongoing searches for high-impact variants contributing to the variance of complex traits. The targeted coding variant, ADIPOQ G45R, is a definitive example of a low-frequency, high-impact variant: it was present in 7 of 88 families in IRASFS with a 1.1 % frequency, yet accounts for a high multipoint LOD score (8.2) and contributes substantially to variance in adiponectin levels in this Hispanic sample (17 %) (Bowden et al. 2010) and is thus a high-impact variant. We examined the characteristics of linkage using a contemporary genetic dataset of Illumina Omni Express GWAS SNPs and exome chip data from chromosome 3. Due to the characteristics of this variant (low frequency and only observed in Hispanics), we sought to determine whether surrounding variants would have been sufficient to draw the attention of an investigator to this region, i.e., if we did not know of or have data for the functional variant, would we be able to detect its presence? The two-point linkage analysis across this chromosome implicated many SNPs aside from G45R, with the most strikingly linked being the intergenic SNP rs10937185 (LOD = 14.85; MAF = 0.15) which is nearly 2 Mb away from G45R (Fig. 1). Thus, linkage results without G45R would present a broad region effectively similar to linkage peaks observed in past multipoint linkage analyses of Mendelian traits. Therefore, evidence of linkage can be observed with both common and low-frequency surrogates for a low-frequency coding and functional variant in the absence of data for the functional variant. To emphasize, this result is obtained with a high-density contemporary dataset consisting primarily of GWAS and exome chip data. After adjustment for the functional G45R, the LOD scores dropped substantially with just one SNP with a LOD score greater than 3. For example, the LOD score for rs10937185 dropped to 1.89 (Fig. 2; Table 2). There was very little linkage evidence implicating additional variants nearby. In the opposite case, adjusting for rs10937185 as a covariate reduced the LOD score at G45R only slightly, from 20.98 to 17.17 (data not shown).
In parallel with the linkage analysis, we performed single SNP association analysis. The two-point linkage analysis results are an easily interpretable complement to the association analysis which readily facilitates alignment of linkage and association signals. The strongest association signal other than G45R was rs12054151 (p value = 1.0 × 10−7), located in the ST6GAL1 gene which is 213 kb from ADIPOQ and has a LOD score of 7.99 (Table 4). This variant nominally explained an estimated 3.1 % of the variance in adiponectin levels. In the context of gene discovery, this combination of linkage and association would be fairly promising; however, there was no residual association after adjusting for the presence of G45R (p value = 0.88; Table 4). Overall, there was limited evidence of association across chromosome 3. When viewed from the perspective of a GWAS, there were some notable SNPs, but none with compelling evidence of association that would draw the eye of an investigator and few had any evidence of support by nearby SNPs.
Examining the intersection of linkage and association identified 119 variants which had both p-values less than 0.05 and a LOD score greater than 3. Looking at the results more stringently (p < 0.001 and LOD >3) left just 20 variants (Supplemental Figs. 2 and 3), though the G45R mutation with association p value of 8.1 × 10−50 and LOD score of over 20 shows that the functional variant substantially outperforms surrogate markers. Conversely, when examining the G45R adjusted results, there were just four variants with a LOD score greater than 3 and only 10 with a p value less than 0.001. These two groups were, however, mutually exclusive: there were no nominally associated SNPs with a LOD score greater than 3.
This analysis of the characteristics of linkage and association across a region when an uncommon causal variant is known leads to several conclusions. Perhaps most obvious is that the evidence of two-point linkage for the functional G45R variant was substantial, yielding a LOD score of 20. Parenthetically, the adiponectin trait demonstrates high kurtosis (lack of normality after adjustment for covariates) in the analyses presented here. When accounting for kurtosis, the maximum LOD score with G45R is reduced (LOD ≈ 10) but overall inferences are the same (Supplemental Table 2). Large numbers of variants surrounding the causal variant over an extended region show substantial evidence of linkage (LODs of up to 14.85). This linkage evidence is driven by the G45R variant, however (Fig. 2), indicating the power to detect a strikingly causal variant even if it were not typed and analyzed. Thus, linkage in this scenario of a low-frequency, high-impact variant is perhaps surprisingly consistent with conventional linkage patterns even though the 1.1 % MAF variant appeared in only 7 of 88 pedigrees.
The previously observed striking microsatellite linkage may be due to the chance LD between G45R and a microsatellite allele (D′ = 0.96). In comparing multipoint with two-point methods using SNP data, this was not as strongly detected when using a SNP panel constructed from 409 common SNPs (pruned to n = 156) which apparently do a poorer job of capturing LD with the functional variant. It is unclear whether this is due to the primarily European-derived information used in developing the original linkage chip being different from that of our Hispanic samples, or if there is some other confounding aspect to the SNPs chosen. It is unclear if information content is the reason for differences in performance, but it seems implicit that a two-point analysis of a high-density dataset of GWAS and exome chip based SNPs would perform at least at equal with “traditional” approaches.
The ability of multiple markers in the simple two-point analysis to detect a linkage signal has important implications that suggest extension of this approach to other traits and other study samples. The two-point linkage approach has clear advantages especially for a seamless alignment with complementary association data (e.g., Fig. 1). First, this documents the power of linkage analysis to facilitate identification of high-impact genetic variants even in a sample of families which would be considered modest in size by current standards for genetic association studies. Importantly, this could encompass more sophisticated phenotypes rather than the generic phenotypes addressable in most GWAS studies (e.g., acute insulin response rather than type 2 diabetes). Second, and most importantly, significant evidence of linkage can be observed even with both common and low-frequency surrogates for a low-frequency coding and functional variant. Notably, in this example, the low-frequency causal variant had a much higher LOD score than non-causal neighboring SNPs, providing further support to its relevance. Consequently, linkage analysis has the potential for discovery of novel variants from genome-wide resources of common (GWAS) or coding (exome chip) data sets even when the functional variant was not directly genotyped such as a private variant in a single family.
Further implications are that common, non-coding variants identify the linkage signal. Thus, GWAS datasets that have been generated in most large ongoing studies are readily applicable for this approach. In addition, while this manuscript has focused on the scenario of low-frequency high-impact variant in our prior work (Hellwege et al. 2014), we showed that application of this approach is also relevant to common high-impact variant discovery. It is implicit that such variants (APOE and CETP) should be identified in GWAS, it is noteworthy that APOE was strongly linked to APOB and much more weakly to LDL (for example). Thus, the approach may be useful for common variants with more sophisticated phenotypes. Finally, it is increasingly clear that high-impact non-coding variants exist and are potential contributors to variance in traits. This linkage method should be directly applicable to discovery of such variants.
References
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664. doi:10.1101/gr.094052.109
Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211. doi:10.1086/301844
An SS, Hanley AJ, Ziegler JT, Brown WM, Haffner SM, Norris JM, Rotter JI, Guo X, Chen YD, Wagenknecht LE, Langefeld CD, Bowden DW, Palmer ND (2012) Association between ADIPOQ SNPs with plasma adiponectin and glucose homeostasis and adiposity phenotypes in the IRAS Family Study. Mol Genet Metab 107:721–728. doi:10.1016/j.ymgme.2012.10.003
An SS, Palmer ND, Hanley AJ, Ziegler JT, Brown WM, Haffner SM, Norris JM, Rotter JI, Guo X, Chen YD, Wagenknecht LE, Langefeld CD, Bowden DW (2013) Estimating the contributions of rare and common genetic variations and clinical measures to a model trait: adiponectin. Genet Epidemiol 37:13–24. doi:10.1002/gepi.21685
Bowden DW, An SS, Palmer ND, Brown WM, Norris JM, Haffner SM, Hawkins GA, Guo X, Rotter JI, Chen YD, Wagenknecht LE, Langefeld CD (2010) Molecular basis of a linkage peak: exome sequencing and family-based analysis identify a rare genetic variant in the ADIPOQ gene in the IRAS Family Study. Hum Mol Genet 19:4112–4120. doi:10.1093/hmg/ddq327
Goodarzi MO, Langefeld CD, Xiang AH, Chen YD, Guo X, Hanley AJ, Raffel LJ, Kandeel F, Buchanan TA, Norris JM, Fingerlin TE, Lorenzo C, Rewers MJ, Haffner SM, Bowden DW, Rich SS, Bergman RN, Rotter JI, Watanabe RM, Wagenknecht LE (2013) Insulin sensitivity and insulin clearance are heritable and have strong genetic correlation in Mexican Americans. Obesity. doi:10.1002/oby.20639
Guo X, Saad MF, Langefeld CD, Williams AH, Cui J, Taylor KD, Norris JM, Jinagouda S, Darwin CH, Mitchell BD, Bergman RN, Sutton B, Chen YD, Wagenknecht LE, Bowden DW, Rotter JI (2006) Genome-wide linkage of plasma adiponectin reveals a major locus on chromosome 3q distinct from the adiponectin structural gene: the IRAS family study. Diabetes 55:1723–1730. doi:10.2337/db05-0428
Heath SC (1997) Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am J Hum Genet 61:748–760. doi:10.1086/515506
Heath SC, Snow GL, Thompson EA, Tseng C, Wijsman EM (1997) MCMC segregation and linkage analysis. Genet Epidemiol 14:1011–1016. doi:10.1002/(sici)1098-2272(1997)14:6<1011:aid-gepi75>3.0.co;2-l
Hellwege JN, Palmer ND, Raffield LM, Ng MC, Hawkins GA, Long J, Lorenzo C, Norris JM, Ida Chen YD, Speliotes EK, Rotter JI, Langefeld CD, Wagenknecht LE, Bowden DW (2014) Genome-wide family-based linkage analysis of exome chip variants and cardiometabolic risk. Genet Epidemiol 38:345–352. doi:10.1002/gepi.21801
Henkin L, Bergman RN, Bowden DW, Ellsworth DL, Haffner SM, Langefeld CD, Mitchell BD, Norris JM, Rewers M, Saad MF, Stamm E, Wagenknecht LE, Rich SS (2003) Genetic epidemiology of insulin resistance and visceral adiposity. The IRAS Family study design and methods. Ann Epidemiol 13:211–217
Kiezun A, Garimella K, Do R, Stitziel NO, Neale BM, McLaren PJ, Gupta N, Sklar P, Sullivan PF, Moran JL, Hultman CM, Lichtenstein P, Magnusson P, Lehner T, Shugart YY, Price AL, de Bakker PI, Purcell SM, Sunyaev SR (2012) Exome sequencing and the genetic basis of complex traits. Nat Genet 44:623–630. doi:10.1038/ng.2303
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM (2009) Finding the missing heritability of complex diseases. Nature 461:747–753. doi:10.1038/nature08494
O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266. doi:10.1086/301904
Acknowledgments
This work was supported by the grant R01 HG007112 from the National Human Genome Research Institute (D.W.B. and C.D.L.). The GUARDIAN study which contributed the IRASFS GWAS genotypes to this project is supported by grant R01 DK085175 from the NIDDK (L.E.W.). The provision of GWAS genotyping data was supported in part by UL1TR000124 (CTSI), and DK063491 (DRC). A subset of the IRASFS exome chips were contributed with funds from the Department of Internal Medicine at the University of Michigan (E.K.S.). Computational support was provided by the Center for Public Health Genomics at Wake Forest School of Medicine.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hellwege, J.N., Palmer, N.D., Brown, M.W. et al. Empirical characteristics of family-based linkage to a complex trait: the ADIPOQ region and adiponectin levels. Hum Genet 134, 203–213 (2015). https://doi.org/10.1007/s00439-014-1511-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-014-1511-8