
Abstract
Our recent genome-wide association study (GWAS) had discovered a new locus at 8p23 (rs2738048) associated with IgA nephropathy (IgAN) in Chinese Han patients, implicating the DEFA gene family within this locus as susceptibility genes. However, it is still unknown whether there are additional variations within these genes associated with the disease susceptibility. The aim of this study is to investigate the polymorphisms of DEFA genes in the susceptibility to IgAN and explore possible disease mechanisms. Sixteen tag single-nucleotide polymorphisms (tag SNPs) were selected for association study in 1,000 IgAN cases and 1,000 controls by using Sequenom MassArray system or TaqMan SNP genotyping assays. We found seven SNPs within DEFA genes that were significantly associated with IgAN, including rs2738048 discovered in our previous GWAS (p = 0.0007, OR = 0.77) and additional 6 SNPs (rs2615787, p = 0.0001, OR = 0.74; rs2738081, p = 0.0003, OR = 0.72; rs2738058, p = 0.0001, OR = 0.73; rs4288398, p = 0.0008, OR = 0.78; rs6984215, p = 0.002, OR = 0.63; rs12716641, p = 0.00002, OR = 0.71). Electrophoretic mobility shift assays and luciferase assays demonstrated that fragments containing rs2738048, rs2738081 and rs6984215 were transcription factor binding sites for CTF, SP1 and CdxA, respectively, and the allele status of rs2738048 and rs6984215 could significantly change the luciferase activity. These results suggest that polymorphisms within DEFA genes are involved in gene transcriptional regulation, and this may have some effect in mediating susceptibility to IgAN in southern Chinese.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
IgA nephropathy (IgAN) is one of the most common form of glomerulonephritis throughout the world and also the leading cause of kidney failure among Asian populations (D’Amico 1987; Tsukamoto et al. 2009). It is characterized by predominant IgA deposition in the glomerular mesangial area and the disease diagnosis requires a kidney biopsy (Donadio and Grande 2002; Barratt and Feehally 2005). The precise pathogenesis of IgAN is unclear, but genetic and environmental factors are thought to play important roles in this disease. The familial clustering of the disease, variation of the prevalence and clinical manifestations (Scolari 2003; Barratt and Feehally 2005; Izzi et al. 2006), all support a genetic component to IgAN.
Previous linkage study and candidate gene association study have identified several genetic factors predisposing to IgAN (Fennessy et al. 1996; Gharavi et al. 2000; Narita et al. 2002; Li et al. 2004; Bisceglia et al. 2006; Paterson et al. 2007; Cao et al. 2008; Lim et al. 2008; Yu et al. 2012). However, the results from candidate gene association studies have not been very reproducible and the genes underlying linkage signals were unknown. Recently, three genome-wide association studies (GWASs) have largely advanced our understanding of the disease from the genetic perspective (Feehally et al. 2010; Gharavi et al. 2011; Yu et al. 2012). In one study comprising 4,137 IgAN cases and 7,734 controls of Chinese Han ancestry that was performed by our group, we discovered a new locus at 8p23 (rs2738048) that implicated DEFA genes as one of the susceptibility genes. DEFA genes encode α-defensins, which are known to demonstrate important antimicrobial, antiviral, and immunomodulatory properties (Yamaguchi and Ouchi 2012). The DEFA region displays multiple copy number variants (CNVs), with a CNV extending from 5 to 14 copies per diploid genome across the DEFA1/A3 genes (Linzmeier and Ganz 2005). Recent studies have shown that higher copy number of DEFA1/A3 genes was associated with increased risk of Crohn’s disease (CD) and severe sepsis (Chen et al. 2010; Jespersgaard et al. 2011). However, the effects of the single-nucleotide polymorphisms (SNPs) in this region are less well-understood. In this study, we aim to investigate the association patterns of tag single-nucleotide polymorphisms (tagSNPs) across the DEFA gene cluster in Chinese Han patients with IgAN.
Materials and methods
Subjects
A total of 1,000 patients with renal biopsy-proven IgAN were recruited from our department. Patients with evidence of cirrhosis, Henoch–Schönlein purpura nephritis, hepatitis B-associated glomerulonephritis, HIV infection and systemic lupus erythematosus were excluded. All case samples were selected from previous GWAS samples, while 1,000 healthy subjects with no history of renal diseases as well as negative urine routine test were newly recruited as normal controls, which had not been included in previous GWAS. All subjects were of self-reported southern Chinese Han ancestry. The study was approved by the Ethics Committee of the First Affiliated Hospital, Sun Yat-sen University. All subjects gave a written informed consent for participation in this study. Genomic DNA was isolated from whole blood using a commercial DNA extraction kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and quantified by spectrophotometer.
SNP selection and genotyping
SNP genotype information was derived from the HapMap CHB and JPT populations (HapMap release 28, NCBI build 36) spanning 1 kb upstream and downstream of each DEFA gene. Haploview 4.2 was used for analyzing the HapMap data and selecting tag SNPs using the Tagger function, with the criteria of minor allele frequency (MAF) ≥0.05 in the HapMap population (both CHB and JPT) and pairwise linkage disequilibrium (LD) of r 2 < 0.8. A total of 16 tag SNPs were chosen for the DEFA locus.

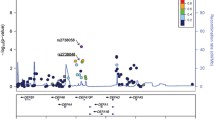
A linkage disequilibrium (LD) plot for tagSNPs of DEFA measured by r 2. A total of 992 controls were used for analysis. The gene structure of DEFA is shown with exons and relative exon size denoted by the width of the vertical bars. The 16 SNPs annotated to DEFA are represented as black dots and arrayed by position along chromosome 8, with tangential lines connecting each SNP to its haplotype block. Pairwise marker–marker LD (shown below the gene structure) was generated using Haploview v4.2 (Barrett et al. 2005). Regions of low-to-high LD, as measured by the r 2 statistic, are represented by light gray to black shading, respectively. The r 2 value is displayed as a percentage in the plot. One LD block has been identified (black solid line) and is involvement of 8 SNPs by Haploview v4.2. The four SNPs predicted to be the transcription factor binding sites (rs2615787, rs2738081, rs2738048 and rs6984215) are outlined in black rectangles
Thirteen SNPs were genotyped by using Sequenom Mass Array system (Sequenom iPLEXassay, San Diego, USA), while another 3 SNPs were genotyped by TaqMan SNP genotyping assays (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Allelic discrimination assays were performed by ABI Prism 7500 Sequence Detection System (Applied Biosystems).
SNP function prediction
SNP function was predicted using the bioinformatics tools Functional Single Nucleotide Polymorphism (F-SNP; http://compbio.cs.queensu.ca/F-SNP/), AliBaba 2.1 (TRANSFAC, conservation >80 %; http://www.gene-regulation.com) and SNP Function Prediction online (FuncPred; http://snpinfo.niehs.nih.gov/).
Nuclear protein extraction and electrophoretic mobility shift assay
For electrophoretic mobility shift assay (EMSA) analysis, oligonucleotides containing predicted transcription factor binding sites (TFBSs) surrounding rs2738081, rs2738081, rs4288398 and rs6984215 were designed and synthesized with biotin-labeled at 5′ end (supplementary Table 1). The open reading frame (ORF) sequence of four predicted transcription factors (HSTF, NCBI accession no. NM_004506.2; SP1, NCBI accession no. NM_138473.2; CTF, NCBI accession no. X12492.1; CdxA, NCBI accession no. NM_001804.2) was individually cloned into OmicsLink expression clone M90 vector (Genecopoeia Inc., Germantown, MD). Transcription factor expression vectors HSTF-M90, SP1-M90, CTF-M90, and CdxA-M90 were constructed and confirmed by DNA sequencing. Empty expression vector M90 (Genecopoeia Inc., Germantown, MD) was used as negative control.
HEK293T cells (ATCC) were cultured in Dulbecco’s modified Eagle medium (DMEM) (Gibco BRL, Life Technologies, New York, USA) supplemented with 10 % fetal bovine serum (Gibco BRL, Life Technologies, New York, USA), penicillin G (100 U/ml), and streptomycin (100 U/ml) (Gibco BRL, Life Technologies, New York, USA) at 37 °C in a humidified 5 % CO2 incubator. HEK293T cells were seeded into 6-well plates at a density of 3 × 105 cells per well 20 h before transfection. HEK293T cells plated in triplicate were transfected with expression vector of each respective transcription factor (HSTF-M90, SP1-M90, CTF-M90, CdxA-M90) and empty vector control M90, using Lipofectamine 2000 (Invitrogen, Carlsbad, USA) according to the manufacture’s protocol.
After 48 h of incubation, nuclear protein extracts were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Rockford, IL, USA). Binding reactions were performed using the Light Shift Chemiluminescent EMSA Kit (Thermo Scientific) according to the manufacturer’s protocol. Nuclear extract was incubated with biotin-labeled probe (50 fmol) for 20 min at room temperature. Unlabeled DNA probe or antibodies were added 20 min before the addition of the biotin-labeled DNA probe. The samples were separated on a 5 % polyacrylamide–TBE gel, and then transferred onto nylon membranes, UV cross-linked, and detected using X-ray film (Kodak). The image on the X-ray film was scanned at a resolution of 600 dpi. Each EMSA experiment was repeated three times. The anti-CTF, anti-SP1 and anti-CdxA antibodies were purchased from Abgent and Millipore.
Transient transfections and luciferase activity assays
Luciferase reporter plasmids were constructed as follows: about 100 bp SNP-containing DNA sequence was amplified from healthy human genomic DNA by PCR using a sense primer with an additional KpnI restriction site and an antisense primer with an additional BglII restriction site (Supplementary Table 2). The PCR fragments were purified and cleaved with KpnI and BglII, then cloned into pGL-3 promoter vector (Promega, Madison, USA). Identification of isolated clones was confirmed by DNA sequencing.
HEK293T cells were seeded into 12-well plates at a density of 2 × 105 cells per well 20 h before transfection. The pGL3 promoter plasmid (500 ng) and pRL-TK (25 ng) (Promega, Madison, USA) were co-transfected into HEK293T cells with either a transcription factor expression vector (CTF-M90, SP1-M90, HSTF-M90, CdxA-M90) (500 ng) or a negative-control empty vector M90 (500 ng) using Lipofectamine 2000 (Invitrogen, Carlsbad, USA). The plasmid pRL-TK was used as an internal control to normalize differences in transfection efficiency. After 24–48 h of incubation, cells were lysed and luciferase activities were measured using a dual-luciferase assay system (Promega, Madison, USA) on an Infinite F500 microplate reader (Tecan, Männdorf, Switzerland). For each plasmid construct, the experiment (three replicates per experiment) was independently repeated three times.
Statistical analyses
Hardy–Weinberg equilibrium (HWE) was tested by Chi-square test, and SNPs deviating from HWE in controls were excluded (p < 0.05). The associations between DEFA polymorphisms under additive, dominant and recessive genetic models with IgAN were tested in binary logistic regression model with R software (http://www.r-project.org/), where age and gender were included as covariates. An additive model assumes that the tested allele exerts an additive effect, so that carriers with 0, 1, 2 risk alleles would have zero, moderate and the strongest effect, respectively. A dominant model assumes that all carriers of the tested allele (both homozygote and heterozygote) would have an effect, while a recessive model assumes that only carriers with two copies of the tested allele (homozygote) would have an effect (Ma et al. 2010). In each model, the p value, odds ratio (OR), and 95 % confidence interval (95 % CI) were presented. The linkage disequilibrium patterns and statistics were obtained by Haploview v4.2 (Barrett et al. 2005). Bonferroni correction for 16 independent tests was used to correct for the number of SNPs studied, resulting in a p value threshold of 0.0031 for claiming statistical significance. Student’s t test was used to analyze the difference in luciferase activity between two alleles of each SNP by GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA, USA).
Results
Patient characteristics
Nineteen subjects (11 cases, 8 controls) with genotype call rate less than 90 % were excluded. Finally, 989 cases and 992 controls were included for further analysis. Sample information was shown in Table 1. All samples were collected from southern China.
Genetic association analysis
All the 16 tagSNPs were successfully genotyped by sequenom or TaqMan methods, and the genotype counts were consistent with the HWE (control group, p > 0.05). The physical locations of and inter-marker LD between these SNPs are schematically presented in Fig. 1. We tested for association under different inheritance models by using logistic regression adjusted for age and gender (Table 2). The minor alleles of the following six SNPs were more significantly associated with IgAN under additive models (rs2615787, p = 0.0001, OR = 0.74, 95 % CI: 0.64–0.86; rs2738081, p = 0.0003, OR = 0.72, 95 % CI: 0.60–0.86; rs2738058, p = 0.0001, OR = 0.73, 95 % CI: 0.63–0.85; rs2738048, p = 0.0007, OR = 0.77, 95 % CI: 0.66–0.89; rs4288398, p = 0.0008, OR = 0.78, 95 % CI: 0.68–0.90; rs12716641, p = 0.00002, OR = 0.71, 95 % CI: 0.61–0.83), while the minor allele of rs6984215 was more significantly associated under a recessive model (p = 0.002, OR = 0.63, 95 % CI: 0.47–0.85).
We also compared the allele frequencies of the seven associated SNPs among different HapMap populations. Whereas the minor allele of rs2738058 is more common in non-Asian than Asian populations, the minor alleles of rs4288398 and rs6984215 are similar between Asian and European ancestry populations but much lower in African populations (Supplementary Table 3).
Bioinformatic analyses predict TFBS in the region surrounding SNP
In order to analyze the molecular mechanisms underlying the associations between those seven SNPs and disease, the function of SNPs were predicted using three independent bioinformatics tools (SNPinfo, F-SNP and AliBaba 2.1) (Table 3). Only four SNPs (rs2615787, rs2738081, rs2738048, and rs6984215) were predicted to be functional and to alter the transcription factor binding sites (TFBSs) for the transcription factors heat shock factor (HSTF), specificity protein 1 (SP1), CCAAT box-binding transcription factor (CTF) and caudal-type homeobox protein (CdxA), respectively. These SNPs were predicted to either disrupt or create transcription factor sequence motifs and were considered as candidates for further gene transcriptional regulation analysis.
EMSA demonstrates DNA–nuclear proteins binding ability of SNPs
We conducted EMSA to investigate whether the DNA sequences containing SNPs (rs2615787, rs2738081, rs2738048, or rs6984215) could interact with nuclear proteins and alter DNA–protein interactions. As shown in Fig. 2a, nuclear proteins from HEK 293T cells transfected with CTF expression vector retarded the migration of probes for both the C allele (lane 2) and the T allele (lane 7). The band corresponding to shifted complex showed weaker intensity when the labeled oligonucleotide contained the C allele, whereas a higher intensity was achieved when the labeled oligonucleotide contained the T allele. The DNA–protein complex was supershifted by anti-CTF antibody (lanes 3 and 8) and could be attenuated by competition with unlabeled probe (lanes 4 and 9). There was no detectable band corresponding to shifted complex when oligonucleotide interacted with nuclear proteins from mock HEK 293T cells (lanes 5 and 10). The results indicated that the T allele of rs2738048 might bind to transcription factor CTF more effectively than the C allele. The same assay was conducted with oligonucleotides containing the major and minor alleles of rs2738081 and rs6984215 (Fig. 2b, c). Both oligonucleotides containing rs2738081 or rs6984215 could interact with SP1 or CdxA. However, there was no significant difference of altered DNA–protein complex intensity between the major and minor alleles. We could not detect any interaction of rs2615787 with any of the nuclear proteins (Supplementary Fig. 1).

EMSAs performed with biotin-labeled oligonucleotide and nuclear extracts prepared from HEK293T cells transfected with an expression plasmid-encoding CTF (C), SP1 (S), CdxA (A). Specific binding of nuclear protein to the respective oligonucleotide was tested by adding competing unlabeled oligonucleotide probe and protein-specific antibody. a The rs2938048 oligonucleotide formed complex with nuclear proteins which was supershifted by the addition of anti-CTF antibody. b The rs2928081 oligonucleotide formed DNA–protein complex which was attenuated by unlabeled oligonucleotide. The complex band was abolished by addition of anti-SP1 antibody. c The rs6984215 oligonucleotide formed DNA–protein complex which was attenuated by unlabeled oligonucleotide. The complex band was abolished by addition of anti-CdxA antibody. Lanes 1 and 6, no nuclear extracts; lanes 2 and 7, 10 μg nuclear extracts with 20 fmol biotin-label probe; lanes 3 and 8, complex was supershifted with 2 μg antibody; lanes 4 and 9, complex was included unlabeled competitor in 200-fold molar excess; and lanes 5 and 10, nuclear extracts from HEk293T cells without transfection (M). I free biotin-labeled probes. II specific DNA–protein complex bands. The supershifted complex is denoted by a single arrow
The effect of SNPs on promoter activity by luciferase assay
The effect of the SNP-containing DNA sequence on gene expression was assessed by the luciferase assay. The pGL3-promoter reporter vector containing the SNP fragment was co-transfected with transcriptional factor expression construct into HEK293T cells. Compared with the empty expression plasmid M90, transfection of allele-specific reporter vector of rs2738048 with CTF expression vector, or transfection of allele-specific reporter vector of rs2738081 with SP1 expression vector resulted in a significant decrease in luciferase activity by ~3–8 fold (Fig. 3a, b). These results were consistent with the EMSA results, implying that rs2738048 and rs2738081 were transcription factor binding sites for CTF and SP1 and functioned as negative regulation of gene expression. However, only the allele status of rs2738048 significantly changed the luciferase activity. The T allele of rs2738048 showed a more significant decrease in luciferase activity than C allele (by ~50 %, p < 0.05, Fig. 3a), which may be attributed to the more efficiently binding to CTF of the T allele than the C allele (Fig. 2a). Co-transfection of allele-specific SNP (rs6984215T/C) containing vector with CdxA expression vector or M90 empty vector showed similar luciferase activity, which suggested that binding of transcription factor CdxA to this fragment was not significantly associated with repressed transcriptional activity (Fig. 3c). Furthermore, the C allele at rs6984215 had an obvious effect in decreasing luciferase activity compared to the T allele (by ~4 fold), thereby suggested that the C allele may cause a reduction in transactivation probably independent of CdxA. We also found that transfection of allele-specific SNP (rs2615787A/C) containing vector with or without HSTF expression vector did not change the luciferase activity (p = 0.5851, p = 0.7511, Fig. 3d). As EMSA results showed that there was no detectable interaction of rs2615787 with nuclear proteins (Fig. 2d), these results demonstrated that rs2615787A/C was likely not a binding site of HSTF and had no effect on transcriptional regulation by HSTF.

Functional analysis of SNPs by luciferase assay. a SNP (rs2738048C/T)-containing pGL3 promoter vector was transfected with or without CTF-M90 expression vector into HEK293T cells. The T allele of rs2738048 showed a more significant decrease in luciferase activity than C allele. b SNP (rs2738081A/C)-containing pGL3 promoter vector was transfected with or without SP1-M90 expression vector. Both C allele and A allele of rs2738081 significantly decreased the luciferase activity through binding to SP1. c SNP (rs6984215T/C)-containing pGL3 promoter vector was transfected with or without CdxA-M90 expression vector. Both could reduce luciferase activity. d SNP (rs2615787C/A)-containing pGL3 promoter vector was transfected with or without HSTF-M90 expression vector, which did not change the luciferase activity. The pGL3-promoter vector containing the SV40 promoter and empty expression vector M90 were used as negative controls. Luciferase activity was analyzed 36 h after transfection. All transfections were repeated in duplicates and the results were expressed as the mean of three different experiments ± SD. p values were evaluated using two-tailed Student’s t tests
Discussion
Over the past few years, genome-wide association studies (GWAS) have successfully identified numerous genetic variants for complex human diseases (McCarthy et al. 2008; Frazer et al. 2009). Our previous GWAS revealed that the SNP rs2738048 in DEFA was associated with IgAN (Yu et al. 2012). In this study, we further investigated tagSNPs within DEFA region to confirm the role of DEFA polymorphisms in susceptibility to IgAN and explore possible disease mechanisms.
There are seven SNPs within DEFA confirmed to be significantly associated with IgAN, including one SNP discovered in our previous GWAS and additional six SNPs. Individuals with minor alleles of rs2615787, rs2738081, rs2738058, rs2738048, rs4288398, rs12716641 and rs6984215 had significantly lower risk of developing IgAN, mostly with an additive genetic effect, suggesting that the minor alleles of these SNPs in DEFA genes might be a protective genetic factor against the development of IgAN.
Most of the disease-associated variants discovered in GWAS are noncoding variants, and a long-range regulation by enhancers, repressors or insulators may account for the association between these noncoding variants and complex polygenic diseases (Noonan and McCallion 2010; Ward and Kellis 2012; Lee et al. 2013). In our study, the significant SNPs discovered in the DEFA region for IgAN were also within noncoding regions. Since the distance between the predicted functional SNP and gene transcription unit is more than 2 kb, we tested whether the polymorphic segment could influence promoter and luciferase activity by binding to transcription factor to exert a long-range regulatory interaction. Although rs2738048 and rs2738081 are upstream variants of pseudogenes DEFA9P (≈5 kb) and DEFA8P (≈6 kb), several lines of evidence have shown that pseudogenes may have regulatory roles in gene expression (Healy et al. 1996; Korneev et al. 1999). In our study, a significant reduction of promoter activity was found after co-expression with transcription factor CTF or SP1, indicating that SNPs rs2738048 and rs2738081 in the 5′ flanking region of DEFA9P and DEFA8P were likely to regulate gene expression through binding of CTF and SP1. Our results also supported the previous opinion that transcription factor binding at the SNP site resulting in altered gene expression may be a possible molecular mechanism of how these SNPs influence susceptibility to disease (Hata et al. 2007; Butter et al. 2012; Karczewski et al. 2013).
We also found that the allele status of rs2738048 could significantly change the luciferase activity, where the major allele (T) had ~50 % lower luciferase activity than the minor allele (C). Another SNP rs6984215 was 9 kb upstream of DEFA1B. The minor C allele at rs6984215 had stronger transcriptional regulation activity than major T allele and showed increased repressor activity. SNP rs6984215 is likely to negatively regulate gene transcription independent of transcription factor CdxA. Our luciferase assay and EMSA results indicated that the DNA segment containing rs2738048 or rs6984215 may reduce gene expression through long-range interactions. Further studies are warranted to confirm whether the allele status of rs2738048 and rs6984215 influence the expression of DEFAs at transcriptional level.
Biologically, α-defensins encoded by DEFA genes are primarily produced by neutrophils and Paneth cells (Lehrer 2004). Increased levels of α-defensins were reported in type 1 diabetic patients with nephropathy, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and Sjögren’s syndrome (SS), demonstrating the important roles of α-defensins in autoimmune diseases (Bokarewa et al. 2003; Peluso et al. 2007; Saraheimo et al. 2008; Sthoeger et al. 2009). Low concentration of human α-defensins 1–3 (1 μg/ml) increase the secretion of TNFα, IL-1β and IL-8 in human monocyte-derived dendritic cells (Rodríguez-García et al. 2009), which are essential factors for initiating a proinflammatory cascade when mesangial cells expose to IgA antibodies in IgAN (Barratt and Feehally 2005). Alpha-defensins 1–3 can also enhance phagocytosis and clearance of intracellular pathogens (Arnett and Seveau 2011), and participate in antibodies and extracellular matrix productions (Lillard et al. 1999; Brogden et al. 2003; Martin and Leibovich 2005). The formation and impaired clearance of immune complexes and an inflammatory response to their deposition along with mesangial proliferation are critical for IgAN pathogenesis (Sancho et al. 1983; Barratt and Feehally 2005; Wyatt and Julian 2013). These studies suggest that α-defensins may affect the process of IgAN. Our study confirmed that variants within DEFA were associated with IgAN and some of them had effects on gene transcriptional regulation.
The present work is the first study to investigate the association between DEFA polymorphism and IgAN. However, there are some limitations in this study. Firstly, the associations of DEFA with IgAN were only studied in Chinese Han population, and further analysis in different ethnic populations will be needed to validate the result. Secondly, as the DEFA region displays a high frequency of CNVs, the correlations of the known CNVs with the SNPs should be further analyzed. Thirdly, although our results suggested that some SNPs of DEFAs had effects on gene transcriptional regulation, the expression level of α-defensins in IgAN patients carrying different alleles should be examined to confirm this finding.
In brief, we have confirmed the previous GWAS finding in IgAN and found more variations within DEFA genes, some of which may regulate promoter activity and gene expression. These results suggested that transcriptional activity of DEFA genes may influence susceptibility to IgAN in southern Chinese.
References
Arnett E, Seveau S (2011) The multifaceted activities of mammalian defensins. Curr Pharm Des 17:4254–4269
Barratt J, Feehally J (2005) IgA nephropathy. J Am Soc Nephrol 16:2088–2097
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Bisceglia L, Cerullo G, Forabosco P, Torres DD, Scolari F, Di Perna M, Foramitti M, Amoroso A, Bertok S, Floege J, Mertens PR, Zerres K, Alexopoulos E, Kirmizis D, Ermelinda M, Zelante L, Schena FP (2006) Genetic heterogeneity in Italian families with IgA nephropathy: suggestive linkage for two novel IgA nephropathy loci. Am J Hum Genet 79:1130–1134
Bokarewa MI, Jin T, Tarkowski A (2003) Intraarticular release and accumulation of defensins and bactericidal/permeability-increasing protein in patients with rheumatoid arthritis. J Rheumatol 30:1719–1724
Brogden KA, Heidari M, Sacco RE, Palmquist D, Guthmiller JM, Johnson GK, Jia HP, Tack BF, McCray PB (2003) Defensin-induced adaptive immunity in mice and its potential in preventing periodontal disease. Oral Microbiol Immunol 18:95–99
Butter F, Davison L, Viturawong T, Scheibe M, Vermeulen M, Todd JA, Mann M (2012) Proteome-wide analysis of disease-associated SNPs that show allele-specific transcription factor binding. PLoS Genet 8:e1002982
Cao HX, Li M, Nie J, Wang W, Zhou SF, Yu XQ (2008) Human leukocyte antigen DRB1 alleles predict risk and disease progression of immunoglobulin A nephropathy in Han Chinese. Am J Nephrol 28:684–691
Chen Q, Hakimi M, Wu S, Jin Y, Cheng B, Wang H, Xie G, Ganz T, Linzmeier RM, Fang X (2010) Increased genomic copy number of DEFA1/DEFA3 is associated with susceptibility to severe sepsis in Chinese Han population. Anesthesiology 112:1428–1434
D’Amico G (1987) The commonest glomerulonephritis in the world: IgA nephropathy. Q J Med 64:709–727
Donadio JV, Grande JP (2002) IgA nephropathy. N Engl J Med 347:738–748
Feehally J, Farrall M, Boland A, Gale DP, Gut I, Heath S, Kumar A, Peden JF, Maxwell PH, Morris DL, Padmanabhan S, Vyse TJ, Zawadzka A, Rees AJ, Lathrop M, Ratcliffe PJ (2010) HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol 21:1791–1797
Fennessy M, Hitman GA, Moore RH, Metcalfe K, Medcraft J, Sinico RA, Mustonen JT, D’Amico G (1996) HLA-DQ gene polymorphism in primary IgA nephropathy in three European populations. Kidney Int 49:477–480
Frazer KA, Murray SS, Schork NJ, Topol EJ (2009) Human genetic variation and its contribution to complex traits. Nat Rev Genet 10:241–251
Gharavi AG, Yan Y, Scolari F, Schena FP, Frasca GM, Ghiggeri GM, Cooper K, Amoroso A, Viola BF, Battini G, Caridi G, Canova C, Farhi A, Subramanian V, Nelson-Williams C, Woodford S, Julian BA, Wyatt RJ, Lifton RP (2000) IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22–23. Nat Genet 26:354–357
Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, Sanna-Cherchi S, Men CJ, Julian BA, Wyatt RJ, Novak J, He JC, Wang H, Lv J, Zhu L, Wang W, Wang Z, Yasuno K, Gunel M, Mane S, Umlauf S, Tikhonova I, Beerman I, Savoldi S, Magistroni R, Ghiggeri GM, Bodria M, Lugani F, Ravani P, Ponticelli C, Allegri L, Boscutti G, Frasca G, Amore A, Peruzzi L, Coppo R, Izzi C, Viola BF, Prati E, Salvadori M, Mignani R, Gesualdo L, Bertinetto F, Mesiano P, Amoroso A, Scolari F, Chen N, Zhang H, Lifton RP (2011) Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet 43:321–327
Hata J, Matsuda K, Ninomiya T, Yonemoto K, Matsushita T, Ohnishi Y, Saito S, Kitazono T, Ibayashi S, Iida M, Kiyohara Y, Nakamura Y, Kubo M (2007) Functional SNP in a Sp1-binding site of AGTRL1 gene is associated with susceptibility to brain infarction. Hum Mol Genet 16:630–639
Healy MJ, Dumancic MM, Cao A, Oakeshott JG (1996) Localization of sequences regulating ancestral and acquired sites of esterase6 activity in Drosophila melanogaster. Mol Biol Evol 13:784–797
Izzi C, Sanna-Cherchi S, Prati E, Belleri R, Remedio A, Tardanico R, Foramitti M, Guerini S, Viola BF, Movilli E, Beerman I, Lifton R, Leone L, Gharavi A, Scolari F (2006) Familial aggregation of primary glomerulonephritis in an Italian population isolate: Valtrompia study. Kidney Int 69:1033–1040
Jespersgaard C, Fode P, Dybdahl M, Vind I, Nielsen OH, Csillag C, Munkholm P, Vainer B, Riis L, Elkjaer M, Pedersen N, Knudsen E, Andersen PS (2011) Alpha-defensin DEFA1A3 gene copy number elevation in Danish Crohn’s disease patients. Dig Dis Sci 56:3517–3524
Karczewski KJ, Dudley JT, Kukurba KR, Chen R, Butte AJ, Montgomery SB, Snyder M (2013) Systematic functional regulatory assessment of disease-associated variants. Proc Natl Acad Sci USA 110:9607–9612
Korneev SA, Park JH, O’Shea M (1999) Neuronal expression of neural nitric oxide synthase (nNOS) protein is suppressed by an antisense RNA transcribed from an NOS pseudogene. J Neurosci 19:7711–7720
Lee YJ, Horie Y, Wallace GR, Choi YS, Park JA, Choi JY, Song R, Kang YM, Kang SW, Baek HJ, Kitaichi N, Meguro A, Mizuki N, Namba K, Ishida S, Kim J, Niemczyk E, Lee EY, Song YW, Ohno S, Lee EB (2013) Genome-wide association study identifies GIMAP as a novel susceptibility locus for Behcet’s disease. Ann Rheum Dis 72:1510–1516
Lehrer RI (2004) Primate defensins. Nat Rev Microbiol 2:727–738
Li YJ, Du Y, Li CX, Guo H, Leung JC, Lam MF, Yang N, Huang F, Chen Y, Fang JQ, Maxwell PH, Lai KN, Wang Y (2004) Family-based association study showing that immunoglobulin A nephropathy is associated with the polymorphisms 2093C and 2180T in the 3′ untranslated region of the Megsin gene. J Am Soc Nephrol 15:1739–1743
Lillard JW Jr, Boyaka PN, Chertov O, Oppenheim JJ, McGhee JR (1999) Mechanisms for induction of acquired host immunity by neutrophil peptide defensins. Proc Natl Acad Sci USA 96:651–656
Lim CS, Kim SM, Oh YK, Joo KW, Kim YS, Han JS, Kim S (2008) Megsin 2093T-2180C haplotype at the 3′ untranslated region is associated with poor renal survival in Korean IgA nephropathy patients. Clin Nephrol 70:101–109
Linzmeier RM, Ganz T (2005) Human defensin gene copy number polymorphisms: comprehensive analysis of independent variation in alpha- and beta-defensin regions at 8p22–p23. Genomics 86:423–430
Ma RC, Tam CH, Wang Y, Luk AO, Hu C, Yang X, Lam V, Chan AW, Ho JS, Chow CC, Tong PC, Jia W, Ng MC, So WY, Chan JC (2010) Genetic variants of the protein kinase C-beta 1 gene and development of end-stage renal disease in patients with type 2 diabetes. JAMA 304:881–889
Martin P, Leibovich SJ (2005) Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol 15:599–607
McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN (2008) Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9:356–369
Narita I, Saito N, Goto S, Jin S, Omori K, Sakatsume M, Gejyo F (2002) Role of uteroglobin G38A polymorphism in the progression of IgA nephropathy in Japanese patients. Kidney Int 61:1853–1858
Noonan JP, McCallion AS (2010) Genomics of long-range regulatory elements. Annu Rev Genomics Hum Genet 11:1–23
Paterson AD, Liu XQ, Wang K, Magistroni R, Song X, Kappel J, Klassen J, Cattran D, St George-Hyslop P, Pei Y (2007) Genome-wide linkage scan of a large family with IgA nephropathy localizes a novel susceptibility locus to chromosome 2q36. J Am Soc Nephrol 18:2408–2415
Peluso G, De Santis M, Inzitari R, Fanali C, Cabras T, Messana I, Castagnola M, Ferraccioli GF (2007) Proteomic study of salivary peptides and proteins in patients with Sjögren’s syndrome before and after pilocarpine treatment. Arthritis Rheum 56:2216–2222
Rodríguez-García M, Oliva H, Climent N, Escribese MM, García F, Moran TM, Gatell JM, Gallart T (2009) Impact of alpha-defensins1-3 on the maturation and differentiation of human monocyte-derived DCs. Concentration-dependent opposite dual effects. Clin Immunol 131:374–384
Sancho J, Egido J, Rivera F, Hernando L (1983) Immune complexes in IgA nephropathy: presence of antibodies against diet antigens and delayed clearance of specific polymeric IgA immune complexes. Clin Exp Immunol 54:194–202
Saraheimo M, Forsblom C, Pettersson-Fernholm K, Flyvbjerg A, Groop PH, Frystyk J, FinnDiane Study Group (2008) Increased levels of alpha-defensin (-1, -2 and -3) in type 1 diabetic patients with nephropathy. Nephrol Dial Transplant 23:914–918
Scolari F (2003) Inherited forms of IgA nephropathy. J Nephrol 16:317–320
Sthoeger ZM, Bezalel S, Chapnik N, Asher I, Froy O (2009) High alpha-defensin levels in patients with systemic lupus erythematosus. Immunology 127:116–122
Tsukamoto Y, Wang H, Becker G, Chen HC, Han DS, Harris D, Imai E, Jha V, Li PK, Lee EJ, Matsuo S, Tomino Y, Tungsanga K, Yamagata K, Hishida A (2009) Report of the Asian Forum of Chronic Kidney Disease Initiative (AFCKDI) 2007. “Current status and perspective of CKD in Asia”: diversity and specificity among Asian countries. Clin Exp Nephrol 13:249–256
Ward LD, Kellis M (2012) Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol 30:1095–1106
Wyatt RJ, Julian BA (2013) IgA Nephropathy. N Engl J Med 368:2402–2414
Yamaguchi Y, Ouchi Y (2012) Antimicrobial peptide defensin: identification of novel isoforms and the characterization of their physiological roles and their significance in the pathogenesis of diseases. Proc Jpn Acad Ser B Phys Biol Sci 88:152–166
Yu XQ, Li M, Zhang H, Low HQ, Wei X, Wang JQ, Sun LD, Sim KS, Li Y, Foo JN, Wang W, Li ZJ, Yin XY, Tang XQ, Fan L, Chen J, Li RS, Wan JX, Liu ZS, Lou TQ, Zhu L, Huang XJ, Zhang XJ, Liu ZH, Liu JJ (2012) A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet 44:178–182
Acknowledgments
We are grateful to all the subjects and healthy volunteers who participated in this work. We thank Jia-Nee Foo for reviewing the manuscript and providing constructive suggestions. This work was funded by Key Program of National Natural Science Foundation of China (81130012), National Natural Science Foundation of China for Distinguished Young Scholars (30925019), Young Scientists Fund of National Natural Science Foundation of China (81200489), Natural Science Foundation of Guangdong Province, China (S2012040006453) and Young Scholars Fund for the Doctoral Program of Higher Education of China (20120171120087).
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Additional information
R. Xu and S. Feng contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xu, R., Feng, S., Li, Z. et al. Polymorphism of DEFA in Chinese Han population with IgA nephropathy. Hum Genet 133, 1299–1309 (2014). https://doi.org/10.1007/s00439-014-1464-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-014-1464-y