
Abstract
The Hennekam lymphangiectasia–lymphedema syndrome is a genetically heterogeneous disorder. It can be caused by mutations in CCBE1 which are found in approximately 25 % of cases. We used homozygosity mapping and whole-exome sequencing in the original HS family with multiple affected individuals in whom no CCBE1 mutation had been detected, and identified a homozygous mutation in the FAT4 gene. Subsequent targeted mutation analysis of FAT4 in a cohort of 24 CCBE1 mutation-negative Hennekam syndrome patients identified homozygous or compound heterozygous mutations in four additional families. Mutations in FAT4 have been previously associated with Van Maldergem syndrome. Detailed clinical comparison between van Maldergem syndrome and Hennekam syndrome patients shows that there is a substantial overlap in phenotype, especially in facial appearance. We conclude that Hennekam syndrome can be caused by mutations in FAT4 and be allelic to Van Maldergem syndrome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Defects in lymphatic structures resulting in congenital lymphedema and lymphangiectasia are the main features of the Hennekam lymphangiectasia–lymphedema syndrome (HS) (OMIM#235510). Other characteristics include unusual facial morphology, variable intellectual disabilities and, at a low frequency, malformations (van Balkom et al. 2002). HS has been reported in 45 individuals (Alders et al. 2013). Mutations in CCBE1, an extracellular matrix protein essential for the development of the lymphatic vasculature, have been found responsible for the syndrome (Alders et al. 2009). However, mutations in this gene are detected only in a subset of patients (~25 %), indicating that the syndrome is genetically heterogeneous (Alders et al. 2013).
Here, we describe the identification of mutations in FAT4 as a second cause for HS. Mutations in this gene have been previously reported in patients with Van Maldergem syndrome (VMS) (Van Maldergem et al. 1992; Cappello et al. 2013). VMS is characterized by intellectual disability, periventricular heterotopia, an unusual face, camptodactyly and syndactyly, small kidneys, osteoporosis and tracheal anomalies sometimes necessitating tracheostomy, and not known to be associated with lymphedema (Mansour et al. 2012). We compare clinical features of both HS and VMS and show that each of the two entities shows specific manifestations but otherwise also a substantial overlap in phenotype exists.
Results
Identification of FAT4 mutations in HS patients
Homozygosity mapping in family F1 (Fig. 1), originally described by Hennekam et al. (1989) and of Dutch descend, identified a single homozygous region on chromosome 4q28, containing FAT4, in all affected individuals (Supplementary table S1). Whole-exome sequencing and filtering for homozygous variants that were shared in all three affected, absent in 20 other whole exomes that served as normal controls, and with a minor allele frequency of <1 % yielded only one variant: a homozygous nucleotide substitution at position c.7123G>A in FAT4 predicted to result in the amino acid substitution p.(Glu2375Arg). Sanger sequencing of FAT4 in 24 additional HS patients identified biallelic mutations in four other families: a homozygous duplication of 6 nt (c.7041_7046dup) resulting in a two-aminoacid duplication p.2348_2349dupGlyThr in proband F2-1 and his affected niece (Al-Gazali et al. 2003); two homozygous missense variants, c.1423T>C (p.Phe475Leu) and c.1456G>C (Glu486Gln) in F3-1; compound heterozygous mutations c.1195delC (p.Leu399Serfs*19) and c.12851C>T (p.Ser4284Phe) in F5-1; and a homozygous splice site mutation c.7200-2A>C in F4-1 (Table 1; Fig. 1; supplementary Fig. S1). According to, at that time only existing refseq for FAT,4 NM_015284.3, the latter mutation would be annotated as c.7200-8A>C. However, mRNA analysis in multiple tissues (Supplementary Fig. S3) demonstrated that splicing actually occurs 6 nt upstream of the annotated splice site of transcript NM_015284.3, placing the mutation in patient F4-1 at the invariant nucleotides of the exon 8 acceptor splice site (c.7200-2A>C) (new refseq NM_001291303.1). This mutation is predicted to result in the usage of the originally annotated (in NM_015284.3) splice site 6 nt downstream, resulting in the deletion of two aminoacids p.SerTyr2400_2401del. Unfortunately, no mRNA from this patient is available to confirm this predicted effect. All parents studied were heterozygous and all identified mutations were at evolutionary conserved aminoacid positions (Supplementary Fig. S2) and absent in control populations (dbSNP, 1000 genomes, NHBLI ESP, GoNL), except for c.12851C>T (p.Ser4284Phe), which has been found in 1/1302 alleles in the NHBLI ESP cohort. One of the mutations in FAT4 reported as causal for VMS (together with a truncating mutation on the other allele) is identical to the mutation homozygously identified in family F1, p.(Glu2375Lys) (Cappello et al. 2013) (Fig. 2).

Pedigrees of the families with homozygous FAT4 mutations. Families F1, F2 and F4 have been previously described (F1 in Hennekam et al. (1989), F2 in Al-Gazali et al. (2003) and F4 in Erkan et al. (1998). The FAT4 mutation status of genotyped individuals is indicated (+/+ for homozygotes, +/wt for heterozygotes and wt/wt for mutation-negative individuals). Two-point parametric linkage analysis in the families F1, F2, F3 and F4 yielded a maximum additive LOD score of 3.6

FAT4 domain structure and location of FAT4 mutations in HS (above) and VMS (below). The annotation of the mutations is based on the transcript that uses the alternative upstream splice site of exon 8 (see Supplementary Fig. S3), which means that after position p.2400, the annotation is different from refseq NM_ 015284.3. We show only the annotation for all mutations according to refseq NM_001291303.1
VMS can also be caused by mutations in the gene encoding for the ligand for FAT4, DCHS1. Therefore, we have sequenced the DCHS1 gene in 14 HS patients in whom no molecular abnormalities had been found in CCBE1 or FAT4, but no mutations were detected.
Clinical comparison HS and VMS
A comparison between the phenotype in HS and VMS shows a considerable overlap between the two entities (Table 2). Facial resemblance is remarkable (Fig. 3a, b), and in both entities cognitive impairment, decreased height, microcephaly and distal limb anomalies (camptodactyly and syndactyly) are common. Most VMS patients with a FAT4 mutation have a severe intellectual disability although milder intellectual disability does occur. In HS, the cognitive impairment is usually mild to moderate and in some individuals cognition is even normal (Alders et al. 2013). A major sign in HS is the marked lymphatic vessel dysplasia which shows edema especially evident at the distal limbs but regularly is generalized (Alders et al. 2013). Lymphedema in HS caused by CCBE1 mutations always is present from birth on, but in HS caused by FAT4 mutations the lymphedema can also start later in life during childhood, as is the case in patient F2-1 (Al-Gazali et al. 2003). Only in a single VMS individual pitting edema of one limb has been reported (Mansour et al. 2012). In VMS a major sign is periventricular heterotopia. This has been reported once in a HS patient (not available for further molecular studies) (Alders et al. 2013). Since there is usually no clinical reason to perform brain MRI studies in HS individuals, further MRI data are lacking. Other manifestation characteristics for VMS such as tracheal anomalies, small kidneys, and osteoporosis are very uncommon or absent in HS.

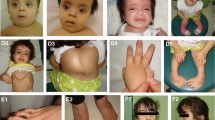
Clinical features in individuals with FAT4 mutations. a Individual F2-1 (front and side), F1-1, and F4-1 (total body) with Hennekam syndrome. Note flat face, hypertelorism, epicanthi, narrow palpebral fissures, and small ears. b Two-year-old boy with Van Maldergem syndrome and FAT4 mutation for comparison, note strong resemblances to a
Discussion
We report here that mutations in FAT4 are a second cause of Hennekam syndrome. Mutations in FAT4 were identified in 5/24 families (~20 %), a rate comparable to CCBE1 mutations in this patient group. As mutations in these two genes explain <50 % of HS cases, further heterogeneity is likely to be present. The mechanism behind FAT4 mutations causing defects in lymphangiogenesis needs further studies. Whereas CCBE1 has been shown to be critically involved in lymphangiogenesis in zebrafish and mice (Hogan et al. 2009; Bos et al. 2011), mice deficient for fat4 do not have signs of lymphedema (Saburi et al. 2008; Mao et al. 2011). It should be noted however that FAT4 knockout mice die perinatally and defects in the lymphatic vasculature can easily escape attention unless specifically sought for, and may only become more evident later in development. Generation of non-lethal mouse models, possibly by the use of hypomorphic alleles or conditional knockout of fat4 expression, may offer the possibility to study the effect of fat4 mutation on the lymphatic system in mice. Mouse fat4 knockout models do not resemble HS and do not resemble VMS either. The renal hypoplasia present in those mice is in concordance with the small kidneys found in VMS, but no other phenotypic features of VMS are present, including periventricular heterotopia as which is a major characteristic of this entity. Only using in utero intraventricular electroporation of shRNAs, the involvement of FAT4 in neuronal proliferation and differentiation could be demonstrated (Cappello et al. 2013).
FAT4 is the mammalian orthologue of the Drosophila fat gene (ft), and known to act on two pathways, the Planar Cell Polarity (PCP) pathway and the Hippo signaling pathway. The involvement in the PCP pathway seems conserved in mice since Fat4 −/− mice display several planar cell polarity phenotypes such as curved body axis and curly tails, disturbed orientation of hair cells in the cochlea, broadened spinal cord and loss of cell polarity in the renal tubular epithelium (Saburi et al. 2008). A role of FAT4 in the Hippo pathway in mice is less clear. Livers of FAT4 mutant mice did not show any phenotypic abnormality whereas knock out of other genes in the Hippo pathway causes hepatomegaly and bile duct hamartomas. In addition, the Hippo interacting domain in Drosophila is not conserved in mammals (Bossuyt et al. 2014). In the mouse brain the increased proliferation of neuronal progenitor cells could be rescued or mimicked by repression or upregulation of Yap, a downstream effector of the Hippo pathway (Cappello et al. 2013). The exact role of FAT4 in human and the pathways involved in the phenotypic expression of mutation in VMS and HS remains to be determined. Recently, it was reported that the core PCP proteins Celsr1 and Vangl2 are critically involved in the morphogenetic process of intraluminal valve formation in lymphatic vessels (Tatin et al. 2013). At present there are not detailed reports on valve morphology in HS patients.
The identification of mutations in FAT4 in both HS and VMS implicates FAT4 to be involved in both neuronal and lymphatic development in human. Such link is not novel and it has been hypothesized that similar mechanisms and pathways are involved for migration and differentiation of both neuronal and lymphendothelial progenitor cells. Several genes crucial for lymphangiogenesis have also been shown to play a role in brain development. For instance, PROX1 is involved in differentiation of neuronal progenitor cells (Kaltezoti et al. 2010), VEGFC and its receptor FLT4 cause proliferation of the neuronal progenitor cells (Hou et al. 2011; Le Bras et al. 2006) and ANGPT2 is involved in the radial migration of neuronal cells towards the cortical plate (Marteau et al. 2011).
Furthermore, we report here that HS and VMS can be allelic disorders, although both can also be caused by mutations in another gene. Detailed comparison of the phenotypes of HS and VMS shows a substantial overlap on one hand and features specific for each syndrome on the other hand. Especially the facial appearance is remarkably alike, and also cognitive impairment, decreased height, microcephaly, small ears and distal limb anomalies are common in both syndromes (Fig. 3). Within the HS patient group no major phenotypic differences are noticed between patients with a CCBE1 mutation and those with a FAT4 mutation. Similarly, within the VMS group phenotypes cannot distinguish between FAT4 mutation-positive patients and patients carrying mutations in the second gene causing VMS, DCHS1, the ligand of FAT4.
In HS patients no mutations were found in DCHS1. The number of patients tested (n = 14) is too low to definitely exclude DCHS1 as a HS candidate gene, but there may be differences in FAT4 and DCHS1 function that explain the differences in phenotype in VMS and HS. The periventricular heterotopia seen in VMS is partially penetrant and more consistently present in patients with DCHS1 mutations than in patients with a mutated FAT4 gene (Cappello et al. 2013). Also, type or localization of FAT4 mutations may play a role in explaining phenotypic variability between the two entities. The mutation p.(Glu2375Arg) was found homozygously in three affected individuals in family F1, all presenting with lymphedema and lymphangiectasias, and in combination with a truncating mutation in a patient with VMS. Differences in phenotypic expression may be due to different effects of a mutation present either in a homozygous or compound heterozygous state. Alternatively, other (epi-)genetic factors may be of influence on the phenotypic outcome of a mutation, for instance the influence on the phenotype in cystic fibrosis by the Cystic Fibrosis Modifier (Zielenski et al. 1999) and Dynactin 4 (Emond et al. 2012). Detailed phenotype–genotype comparisons (Hennekam and Biesecker 2012) may shed more light on this matter.
Methods
Homozygosity mapping
Homozgosity mapping in family F1 (F1-1, F1-2 and F1-3) was performed using the Affymetrix® Genome-Wide Human SNP Array 6.0 array. Results were analyzed using Nexus Copy Number™ software version 7 (BioDiscovery). Homozygous regions of more than 5 Mb were called (SNPRank Segmentation).
Whole-exome sequencing
Whole-exome sequencing was conducted using the SeqCap EZ Human Exome Library v3.0 (Roche NimbleGen) and a 5500 SOLiD™ instrument (Life Technologies). Samples were prepared using standard SolID 75 × 35 paired-end sequencing protocols. Alignment of sequence reads to human reference genome (hg19) was done using Lifescope 2.5.1, and variants were called using the GATK2.5 software package.
Whole-exome sequencing in three affected children in family F1 generated 195,673,578, 293,569,596 and 153,863,822 sequence reads, respectively. Coverage of targeted regions at 2×, 10× and 20× read depth (after removal of duplicate reads) was 93, 86 and 79 % for individual F1-1; 94, 89 and 84 % for individual F1-2; and 92, 85 and 75 % for individual F1-3.
Prioritization of variants identified with WES was done using the Cartagenia BENCHlab NGS software v3.0.4 (Cartagenia NV). Public databases used for determining the frequency of the identified variants in the general population were 1,000 genomes (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521/ALL.wgs.phase1_release_v3.20101123.snps_indels_sv.sites.vcf.gz), dbSNP137 (http://www.ncbi.nlm.nih.gov/projects/SNP) the ESP6500 dataset (http://evs.gs.washington.edu/EVS/), and the GoNL database (498 Dutch individuals, http://www.nlgenome.nl/).
Sanger sequencing of FAT4 and DCHS1
Primers were designed to amplify all coding exons of FAT4 and DCHS1 using the Primer3 software (http://frodo.wi.mit.edu/). Amplification was performed with M13-tagged primers using HOT FIREPol™ DNA polymerase (Solys Biodyne) and a touchdown PCR program. PCR fragments were sequenced using the Bigdye kit v1.1(Applied Biosystems). Reactions were run on an ABI3700 or ABI3730XL genetic analyzer (Applied Biosystems) and sequences were analyzed using Sequence Pilot (JSI Medical systems) or CodonCode aligner (CodonCode Corporation). Variants were further characterized using Alamut version 2.3 (Interactive Biosoftware, Rouen, France).
Statistics
Two-point parametric Linkage analysis was performed by EasyLinkage Plus v5.08 package (http://sourceforge.net/projects/easylinkage/) using the Superlink v1.6 program. Pedigree was analyzed as a recessive model and LOD scores were obtained at 0.05 recombination increment steps until a recombination value of 0.45. Penetrance of disease and disease allele frequency were given 99 % and 0.001, respectively.
References
Alders M, Hogan BM, Gjini E, Salehi F, Al-Gazali L, Hennekam EA, Holmberg EE, Mannens MM, Mulder MF, Offerhaus GJ, Prescott TE, Schroor EJ, Verheij JB, Witte M, Zwijnenburg PJ, Vikkula M, Schulte-Merker S, Hennekam RC (2009) Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Nat Genet 41:1272–1274
Alders M, Mendola A, Adès L, Al Gazali L, Bellini C, Dallapiccola B, Edery P, Frank U, Hornshuh F, Huisman SA, Jagadeesh S, Kayserili H, Keng WT, Lev D, Prada CE, Sampson JR, Schmidtke J, Shashi V, van Bever Y, Van der Aa N, Verhagen JM, Verheij JB, Vikkula M, Hennekam RC (2013) Evaluation of clinical manifestations in patients with severe lymphedema with and without CCBE1 mutations. Mol Syndromol 4:107–113
Al-Gazali LI, Hertecant J, Ahmed R, Khan NA, Padmanabhan R (2003) Further delineation of Hennekam syndrome. Clin Dysmorphol 12:227–232
Bos FL, Caunt M, Peterson-Maduro J, Planas-Paz L, Kowalski J, Karpanen T, van Impel A, Tong R, Ernst JA, Korving J, van Es JH, Lammert E, Duckers HJ, Schulte-Merker S (2011) CCBE1 is essential for Mammalian lymphatic vascular development and enhances the lymphangiogenic effect of Vascular Endothelial Growth Factor-C in vivo. Circ Res 109:486–491
Bossuyt W, Chen CL, Chen Q, Sudol M, McNeill H, Pan D, Kopp A, Halder G (2014) An evolutionary shift in the regulation of the Hippo pathway between mice and flies. Oncogene 33:1218–1228
Cappello S, Gray MJ, Badouel C, Lange S, Einsiedler M, Srour M, Chitayat D, Hamdan FF, Jenkins ZA, Morgan T, Preitner N, Uster T, Thomas J, Shannon P, Morrison V, Di Donato N, Van Maldergem L, Neuhann T, Newbury-Ecob R, Swinkells M, Terhal P, Wilson LC, Zwijnenburg PJ, Sutherland-Smith AJ, Black MA, Markie D, Michaud JL, Simpson MA, Mansour S, McNeill H, Götz M, Robertson SP (2013) Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat Genet 45:1300–1308
Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, Knowles MR, Wright FA, Rieder MJ, Tabor HK, Nickerson DA, Barnes KC, National Heart, Lung, and Blood Institute GO Exome Sequencing Project, Lung GO, Gibson RL, Bamshad MJ (2012) Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet 44:886–889
Erkan T, Kutlu T, Cullu F, Celik M, Demir T, Tüysüz B, Tümay GT (1998) Syndrome de Hennekam Arch Pediatr 5:1344–1346
Hennekam RC, Biesecker LG (2012) Next-generation sequencing demands next-generation phenotyping. Hum Mutat 33:884–886
Hennekam RC, Geerdink RA, Hamel BC, Hennekam FA, Kraus P, Rammeloo JA, Tillemans AA (1989) Autosomal recessive intestinal lymphangiectasia and lymphedema, with facial anomalies and mental retardation. Am J Med Genet 34:593–600
Hogan BM, Bos FL, Bussmann J, Witte M, Chi NC, Duckers HJ, Schulte-Merker S (2009) Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat Genet 41:396–398
Hou Y, Choi JS, Shin YJ, Cha JH, Choi JY, Chun MH, Lee MY (2011) Expression of vascular endothelial growth factor receptor-3 mRNA in the developing rat cerebellum. Cell Mol Neurobiol 31:7–16
Kaltezioti V, Kouroupi G, Oikonomaki M, Mantouvalou E, Stergiopoulos A, Charonis A, Rohrer H, Matsas R, Politis PK (2010) Prox1 regulates the notch1-mediated inhibition of neurogenesis. PLoS Biol 8(12):e1000565
Le Bras B, Barallobre MJ, Homman-Ludiye J, Ny A, Wyns S, Tammela T, Haiko P, Karkkainen MJ, Yuan L, Muriel MP, Chatzopoulou E, Breant C, Zalc B, Carmeliet P, Alitalo K, Eichmann A, Thomas JL (2006) VEGF-C is a trophic factor for neural progenitors in the vertebrate embryonic brain. Nat Neurosci 9:340–348
Mansour S, Swinkels M, Terhal PA, Wilson LC, Rich P, Van Maldergem L, Zwijnenburg PJ, Hall CM, Robertson SP, Newbury-Ecob R (2012) Van Maldergem syndrome: further characterisation and evidence for neuronal migration abnormalities and autosomal recessive inheritance. Eur J Hum Genet 20:1024–1031
Mao Y, Mulvaney J, Zakaria S, Yu T, Morgan KM, Allen S, Basson MA, Francis-West P, Irvine KD (2011) Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development 138:947–957
Marteau L, Pacary E, Valable S, Bernaudin M, Guillemot F, Petit E (2011) Angiopoietin-2 regulates cortical neurogenesis in the developing telencephalon. Cereb Cortex 21:1695–1702
Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, McNeill H (2008) Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat Genet 40:1010–1015
Tatin F, Taddei A, Weston A, Fuchs E, Devenport D, Tissir F, Makinen T (2013) Planar cell polarity protein Celsr1 regulates endothelial adherens junctions and directed cell rearrangements during valve morphogenesis. Dev Cell 26:31–44
Van Balkom ID, Alders M, Allanson J, Bellini C, Frank U, De Jong G, Kolbe I, Lacombe D, Rockson S, Rowe P, Wijburg F, Hennekam RC (2002) Lymphedema-lymphangiectasia-mental retardation (Hennekam) syndrome: a review. Am J Med Genet 112:412–421
Van Maldergem L, Wetzburger C, Verloes A, Fourneau C, Gillerot Y (1992) Mental retardation with blepharo-naso-facial abnormalities and hand malformations: a new syndrome? Clin Genet 41:22–24
Zielenski J, Corey M, Rozmahel R, Markiewicz D, Aznarez I, Casals T, Larriba S, Mercier B, Cutting GR, Krebsova A, Macek M Jr, Langfelder-Schwind E, Marshall BC, DeCelie-Germana J, Claustres M, Palacio A, Bal J, Nowakowska A, Ferec C, Estivill X, Durie P, Tsui LC (1999) Detection of a cystic fibrosis modifier locus for meconium ileus on human chromosome 19q13. Nat Genet 22:128–129
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Alders, M., Al-Gazali, L., Cordeiro, I. et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to Van Maldergem syndrome. Hum Genet 133, 1161–1167 (2014). https://doi.org/10.1007/s00439-014-1456-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-014-1456-y