
Abstract
In this study, we have performed autozygosity mapping on a large consanguineous Pakistani family segregating with intellectual disability. We identified two large regions of homozygosity-by-descent (HBD) on 16q12.2–q21 and 16q24.1–q24.3. Whole exome sequencing (WES) was performed on an affected individual from the family, but initially, no obvious mutation was detected. However, three genes within the HBD regions that were not fully captured during the WES were Sanger sequenced and we identified a five base pair deletion (actually six base pairs deleted plus one base pair inserted) in exon 7 of the gene FBXO31. The variant segregated completely in the family, in recessive fashion giving a LOD score of 3.95. This variant leads to a frameshift and a premature stop codon and truncation of the FBXO31 protein, p.(Cys283Asnfs*81). Quantification of mRNA and protein expression suggests that nonsense-mediated mRNA decay also contributes to the loss of FBXO31 protein in affected individuals. FBXO31 functions as a centrosomal E3 ubiquitin ligase, in association with SKP1 and Cullin-1, involved in ubiquitination of proteins targeted for degradation. The FBXO31/SKP1/Cullin1 complex is important for neuronal morphogenesis and axonal identity. FBXO31 also plays a role in dendrite growth and neuronal migration in developing cerebellar cortex. Our finding adds further evidence of the involvement of disruption of the protein ubiquitination pathway in intellectual disability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intellectual disability (ID) is a neurodevelopmental disorder that affects ~1 % of children worldwide (Maulik et al. 2011). The symptoms associated with ID are impaired capacity of learning and processing new or complex information, culminating in decreased adaptive behavior and cognitive functioning (Ellison et al. 2013; Leonard and Wen 2002). Causes of ID may include environmental factors, such as teratogens, infection, and neurological trauma; however, ID also has a strong genetic etiology, with a diverse range of genetic defects known, including chromosomal aberrations, sub-microscopic copy number variations (CNVs), and DNA sequence mutations within genes, including genes located on the autosomes, X chromosome and the mitochondrial genome. While research into chromosomal aberrations, CNVs, autosomal dominant and X-linked forms of ID have continued apace for some years, research into autosomal recessive forms of ID (ARID) has been largely overlooked, and only in recent years concerted efforts have been made to identify ARID genes (reviewed in Musante and Ropers 2014). Recent estimates suggest that 13–24 % of ID cases in Europe are likely to be due to AR causes, and may be the most common cause of ID in populations where consanguinity is prevalent (Musante and Ropers 2014). ID can be divided into two groups: non-syndromic ID is characterized as the only clinical feature in patients, while syndromic ID occurs in combination with one or more additional clinical features (reviewed in Kaufman et al. 2010). Forty genes for non-syndromic ARID have been reported to date (reviewed in Musante and Ropers 2014).
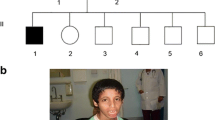
In this study, we report the investigation of a consanguineous family (ASMR72) from Bahawalpur District, Punjab Province, Pakistan, segregating with non-syndromic ID in a pattern consistent with autosomal recessive inheritance (Fig. 1). The family has six affected individuals (five living) in the youngest generation, across two branches, and presents an excellent opportunity for disease gene identification. There are two affected sisters from one branch and three brothers (one of whom presents with some dysmorphic facial features) from another branch, in whom two homozygosity-by-descent (HBD) regions on chromosome 16q12.2–q21 and 16q24.1–q24.3 are shared, as determined by homozygosity mapping (Fig. 1). These regions were identified by genome-wide homozygosity mapping of DNA samples from five of those patients using 250 K single nucleotide polymorphism (SNP) array and next generation sequencing (NGS). SNP arrays and NGS, together, are a powerful tool for discovering genes associated with Mendelian disorders.

a Pedigree of family ASMR72. Affected individuals are indicated by dark-shaded symbols. b Left HomozygosityMapper output, showing complete HBD only on chromosome 16 (indicated by red arrow); right dCHIP output showing chromosome 16 homozygous regions (blue; heterozygous stretches in yellow), with shared HBD regions indicated by red parenthesis for 16q12.2–q21 (rs11076059–rs818406; chr16:54,850,716-65,477,189; hg19), red arrow for 16q24.1–q24.3 (rs1562110–rs4785775; chr16:86,759,160-90,163,275). c Ideogram of chromosome 16 indicating region of HBD, and the FBXO31 gene, indicating the location of c.847_852delinsA in exon 7. d Electropherograms showing the forward strand homozygous wild type sequence and the homozygous c.847_852delinsA sequence
Materials and methods
Family recruitment
In the present study, a family from Bahawalpur District, Punjab Province, Pakistan, was recruited through COMSATS Institute of Information Technology, Islamabad, after informed written consent was given (for affected individuals and children 16 or under parental consent was given). Institutional research ethics board consent was given for the study through COMSATS. The family has five living affected individuals in the same generation, all of them from consanguineous relationships. There are two affected sisters (a further affected sister was deceased) from one loop and three brothers from another (Fig. 1). Blood was drawn for genetic studies, and genomic DNA was extracted by standard methods. Neurological and medical assessment in the Pakistani family was performed on all affected members by a team of experienced clinicians who come from the same culture and speak the local language of the family. In addition, a psychiatrist trained and experienced in psychiatry of intellectual disability (M.A.), also familiar with the local culture, assessed the family using Vineland Adaptive Behavior Scales, Second Edition (Sparrow et al. 2005) as framework to gather information. The information was used to estimate the level of intellectual disability for all the affected members of the family. Photographs of family members were assessed by an experienced clinical geneticist (J.S.) for dysmorphic features.
Microarray and whole exome sequencing
DNA from all five affected and one unaffected (IV:1) individuals was genotyped using Affymetrix Genome-Wide Human 250 K SNP Array (Affymetrix; Santa Clara, CA, USA). Microarrays were run as a service by the London Regional Genomics Centre (http://www.lrgc.ca). The microarray data analysis was performed using dCHIP software (http://biosun1.harvard.edu/complab/dchip; Lin et al. 2004) and HomozygosityMapper (http://www.homozygositymapper.org; Seelow et al. 2009).
We performed whole exome sequencing (WES) on DNA from individual IV-8 using the SOLiD 4 (Life Technologies) platform, with coding exons and flanking introns captured using the SureSelect Human All Exon 50 Mb kit (Agilent Technologies, Santa Clara, CA, USA), according to the manufacturers’ recommended protocols. The raw reads were aligned using SHRiMP2 (David et al. 2011). Next, duplicated reads were removed with the MarkDuplicate subroutine of PICARD (http://picard.sourceforge.net). To improve the base resolution consensus, a micro re-alignment was performed on each read with the SRMA program (Homer and Nelson 2010). Genome Analysis Tool Kit Package (McKenna et al. 2010) was used for Q score recalibration. The tools in this package recalibrated base quality scores of sequencing-by-synthesis reads in an aligned BAM file. The BAM files thus produced were used as input for the DNASTAR software. Seqman NGen ver 3.0.4 was used to assemble BAM-based reads against the human reference genome, build hg19. The detailed reports were exported for further analyses and filtering for homozygous coding or splice variants that are not known SNPs (dbSNP131), and for non-synonymous changes predicted to be damaging. BAM files were imported into the Integrated Genomics Viewer (http://www.broadinstitute.org/igv/), and the HBD regions checked for adequate coverage of exons. Sanger sequencing was used for validation of variants and segregation analysis within the family. Also, exons within the two 16q HBD regions that were not successfully captured and covered by the WES (with ≥reads) were Sanger sequenced. Primers were designed using Primer3 (http://www.broadinstitute.org/genome_software/other/primer3.html; see Supplementary Materials for list of primers), and Sanger sequencing was run under service by The Centre for Applied Genomics (TCAG; http://www.tcag.ca).
Microsatellite genotyping and linkage analysis
Microsatellite markers flanking and spanning the HBD region on 16q23–q24 (D16S3091, D16S402, D16S520, D16S2621 and D16S305) were genotyped for DNAs from all available family members. Genotyping was performed at TCAG. Linkage analysis was performed using SimWalk2 (version 2.91) (Sobel and Lange 1996; Sobel et al. 2001, 2002) under a recessive model of inheritance, initially for these microsatellite markers, and after the identification of the FBXO31 c.847_852delinsA variant we also included that in the linkage analysis.
EBV transformation and culture of lymphoblasts
Patient and controls blood samples collected in acid-citrate-dextrose BD Vacutainer® blood tubes (Becton, Dickinson and Company) were diluted (1:1) with Roswell Park Memorial Institute medium, and white blood cells separated using ACCUSPIN tubes (Sigma), and transformed with Epstein–Barr virus.
RNA extraction, cDNA synthesis and sequencing
RNA was extracted from lymphoblast cells using the Trizol method. cDNA was synthesized through reverse transcription of 1 μg of RNA using Superscript III™ Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) and random hexamers according to the manufacturer’s guidelines. Correct splicing for FBXO31 from lymphoblast mRNA from affected family members was confirmed by RT-PCR followed by Sanger sequencing using the primers 5′-GAGATCACAGGCGTGTCTTG and 5′-GCTGAGCATCACAATCTCCA (from exons 2 and 7 respectively).
Quantitative RT-PCR
Primers were designed to amplify the coding DNA sequence (CDS) of the FBXO31 gene (Fig. 2a), using the primer express software (Applied Biosystems Inc., Foster city, CA, USA) and their details are given in Supplementary Table 1. CDS primers for β-Actin and HPRT were used as an internal reference for all the runs. PCR was performed in quadruplicates, and on a 384-well single plate to avoid inter-plate variability. Universal SYBR Green® PCR conditions were used for real-time PCR with the ViiA7 system (Life Technologies). Each plate included distilled water, RT-minus and RNA-minus negative controls. The Ct for all reactions was calculated automatically by the Applied Biosystems ViiA7 TM software. Gene expression analysis was calculated using the comparative Ct method (Livak and Schmittgen 2001).

a Relative quantification of RT-PCR results show decreased expression of FBXO31 in patients (IV:8 and IV:10) compared to unaffected individual (IV:6). Comparison by pairwise student t test showed significant difference in expression: **p < 10−5 for all three amplicons used. Location of amplicons on FBXO31 is also shown. b Ideogrammatic representation of the FBXO31 protein, indicating the location of known domains, including the FBox domain, DUF3506 (a domain of unknown function), a serine phosphorylation site at residue 278 (predicted by SMART: http://smart.embl.de/). Putative cyclin interacting domains are indicated by pink bars (predicted by Mini Motif Miner 3.0: http://mnm.engr.uconn.edu/MNM/SMSSearchServlet). The location of the frameshifting mutation at residue 283 is indicated as well as the position of the resulting premature termination codon (PTC), with red arrows. The positions of the epitopes for the N-terminal and C-terminal antibodies are indicated by blue horizontal bars. c Western blots using affected and unaffected family member and control lymphoblast cells for FBXO31. The Bethyl Labs FBXO31 antibody binds to an epitope in the C-terminal region (residues 489–539), which lies within the truncated region of FBXO31. The 63 kDa band is likely cross reacting with another protein. Little or no expression of upper (66 kDa) band can be seen in the affected individuals (IV:4, IV:3, IV:10 and IV:8). The upper band is present in the unaffected individual (IV:6) and controls (C1, C2, C3 and C4). The housekeeping protein β-actin is indicated as a loading control. Western blot for an N-terminal antibody (Abcam) is also shown for the same individuals. The epitope for this antibody lies upstream of the truncated region. A 40 and a 66 kDa band were seen, which most likely correspond to alternative isoforms of FBXO31. Reduced intensity of the 66 kDa bands is noted for the affected individuals. d After normalizing against β-actin, quantification of the mean intensity of the FBXO31 40 kDa bands using the N-terminal antibody shows a significant (~1.4-fold) decrease in FBXO31 protein expression in the affected individuals compared to the unaffected family members and control individuals (unpaired student t test: *p < 0.05)
Protein extraction
Protein was also extracted from lymphoblasts cells for comparative quantitation. After centrifugation and pelleting in a 50-ml falcon tube (500 g, 5 min), the cell pellet was resuspended in ice-cold phosphate buffered saline (PBS) followed by additional centrifugation and lysed using lysis buffer (250 mM NaCl, 50 mM Tris–Cl pH 8.0, 5 mM EDTA, 0.5 % Nonidet P-40) containing protease inhibitors (Roche mini, EDTA-free) 1 mM PMSF. After transferring cells to 1.5 ml tubes, incubating on ice (30 min) and vortexing, the lysates were centrifuged (10,000g, 10 min), and the supernatant collected. The protein concentrations for patient and control lymphoblasts were measured using the bicinchoninic acid method (Smith et al. 1985).
Western blot
Protein samples were separated by SDS page (4–20 % Mini-PROTEAN TGX polyacrylamide gel, Bio-Rad Laboratories, Hercules, CA, USA) and electrically transferred to a nitrocellulose membrane (BioTrace NT nitrocellulose membrane, PALL Life Sciences, Ann Arbor, MI, USA). The membrane was blocked for 1 h using 5 % skimmed milk in TBS-Tween, incubated overnight with the primary antibody, and then incubated with secondary antibody horseradish peroxidase-conjugated donkey anti-rabbit IgG (1:1,000, GE Healthcare, Little Chalfont, Buckinghamshire, UK). We used two primary antibodies: the first antibody [rabbit anti-FBXO31 (1:1,000), Bethyl Laboratories, Montgomery, TX, USA] binds to an epitope between the residues 489 and 539 of human FBOX31, C-terminal to the mutation in the affected individuals, and the second antibody (rabbit anti-FBXO31 (1:1,000), Abcam, Cambridge, UK) binds to the N-terminal region; epitope residues are 1–367 (Supplemental Material-protein sequence). The CT antibody data sheet indicates an ~66 kDa band is expected, which corresponds roughly to the full length 539-amino acid protein encoded by mRNA accession #NM_024735.4. The data sheet for the AbCam NT antibody indicates an expected 40 kDa band. This would correspond to a 369-amino acid alternate isoform of FBXO31 encoded by mRNA NM_001282683.1. However, we also see the 66 kDa band, as expected. In our hands, both antibodies appear to cross react with an ~63 kDa band of unknown origin. Both isoforms would be affected similarly by the frameshifting mutation. The immunoblot was developed by enhanced chemiluminescence western blot detection system (GE Healthcare). To verify if the amount of proteins for all samples were equal, the anti-FBXO31 was stripped from the membrane. The blot was incubated in stripping solution [100 mM β-mercaptoethanol, 2 % (w/v) SDS, 62.5 mM Tris–HCl, pH 6.7] with agitation for 30 min at 50 °C, washed five times in 1× TBS-T, followed by the regular immunoblotting protocol for loading control β-actin (1:10,000; Abcam, Cambridge, UK) followed by secondary antibody horseradish peroxidase-conjugated donkey anti-rabbit IgG (1:1,000, GE Healthcare, Little Chalfont, Buckinghamshire, UK). Quantification of the mean intensity of the FBXO31 40 kDa bands using the N-terminal antibody, using Image J software (http://rsb.info.nih.gov/ij/) and normalized with β-actin, processed using Student t test by Graphpad Prism (http://www.graphpad.com).
Immunocytochemistry
Primary hippocampal neuronal cells were obtained from E15 DBA mouse embryos using standard procedures (Seibenhener and Wooten 2012). For immunofluorescent staining of neurons, the cells were washed twice in PBS, and fixed in 4 % paraformaldehyde (Electron Microscopy Sciences), then washed twice with PBS. Primary antibodies were diluted in PBS with 1 % BSA and incubated for 1 h, followed by three washes with PBS. Anti-secondary antibodies were diluted in PBS with 1 % BSA and incubated for 1 h followed by three washes with PBS. Finally, coverslips were mounted with DAKO Fluorescent Mounting Medium, sealed and then stored at 4 °C until imaging. We used two different antibodies targeting FBXO31: a C-terminal antibody from Bethyl Laboratories (Montgomery, TX, USA), and a C-terminal antibody from Novus Biologicals (Oakville, Canada). The secondary antibody used was goat polyclonal antibody to rabbit (TR; Abcam). All antibodies were used at 1:200 dilution.
Microscopy
The immunostained hippocampal neurons were imaged using Olympus IX83 confocal microscope. Images were documented using Fluoview software version 4.0 (Olympus). Fluorescent images and the DIC images were acquired sequentially with their specific settings.
Results
Photographs of all living affected family members were assessed for dysmorphic features by an experienced clinical geneticist. They showed common recognizable coarse facial features including broad nasal bridge, fleshy nares, and thick prominent lips. Other clinical features include: patient IV:3 has a round face shape with prominent supraorbital ridge and deep-set eyes, a bulbous nasal tip and fleshy nares. Patient IV:4 has coarse facial features, with a low anterior hairline, short forehead, prominent eyebrows with medial flaring and synophrys, a broad nasal bridge, bulbous nasal tip and fleshy nares. Patient IV:8—a lateral image reveals long, well formed, normally set ears, flat facial profile and a short, upturned nose. Later, he developed coarser facial features, including a square-shaped face with a tall, dimpled forehead, prominent, arched eyebrows with synophrys, upslanting palpebral fissures with apparent hypertelorism and retrognathia. The ears continue to be large, well formed and normally set. This individual’s facial features appear more prominent and dysmorphic relative to his affected siblings and cousins. Patient IV:9 has somewhat coarse facial features, with a short forehead with prominent supraorbital ridge, prominent eyebrows with synophrys, long, slightly downslanting palpebral fissures. As he grew older, many of these features including large, well formed and set ears can be observed. Patient IV:10 shows a short forehead, prominent supraorbital ridge, arched thick eyebrows, deep-set eyes, and small chin with mild retrognathia. Ears are large, well formed and normally set. Patient IV:10 can be seen to have a widow’s peak, short forehead, thick, prominent eyebrows with synophrys and deep-set eyes with apparent hypertelorism. Ears are somewhat large, but normally formed and set.
All the patients had mild to moderate degree of ID. There was no history of seizures and neurological examination did not reveal any abnormality. Two patients had magnetic resonance imaging scans of brains and no abnormal findings were reported.
Following genotype analysis, two regions of autozygosity were found to be shared by all affected members, but not unaffected individual IV:1: a 10.6 Mb region, 16q12.2–q21 (rs11076059–rs818406; chr16:54,850,716-65,477,189; hg19), and 3.4 Mb on 16q24.1–q24.3 (rs1562110–rs4785775; chr16:86,759,160-90,163,275). The first region contains the Bardet–Biedl syndrome 2 gene, BBS2 (MIM 209900), as well as GPR56 (polymicrogyria; MIM 606854), and CDH8, in which deletions have previously been reported in individuals with ID and/or autism (Pagnamenta et al. 2011). The second region includes the gene ANKRD11, which has been linked to the dominant ID syndrome, KBG syndrome (MIM 148050). These four genes were excluded by Sanger sequencing prior to WES. After WES analysis, no likely mutations were identified within these regions, so all missing exons within the two HBD regions on 16q were Sanger sequenced. This included exons of the genes FBXO31, MAP1LC3B, and ZCCHC14. We identified a homozygous frameshifting variant within exon 7 of the gene encoding F-box protein 31, FBXO31, NM_024735.3 (FBXO31_v001): c.847_852delinsA; p.(Cys283Asnfs*81), on 16q24.2. This variant is effectively a 5 bp deletion, although it involves a 6 bp deletion (delCAGGCA) and 1 bp insertion (insA), which causes a frameshift mutation starting at p.Cys283Asn and a premature stop codon at position 362 resulting in a 177-amino acid truncation of FBXO31 protein. Sanger sequence analysis showed complete segregation with ARID within the Pakistani family, and the variant allele was not present in 300 Pakistani control individuals. This variant was not in any SNP databases, or present in 1,000 genomes (http://browser.1000genomes.org/index.html; accessed Jan 2014) or NHLBI EVS (http://evs.gs.washington.edu/EVS/; accessed Jan 2014) datasets. This variant occurs at the 5th nucleotide after the acceptor splice site. In silico analysis of splice sites (using the Berkeley Drosophila Genome Project software: http://www.fruitfly.org/index.html) suggests a decreased probability of splice acceptor usage from 0.76 (wild type) to 0.46 (mutant). This could suggest alternative splice site usage as another possible mechanism for disruption of FBXO31. However, sequence analysis of lymphoblast mRNA shows no effect on splicing.
Linkage analysis was performed using microsatellite genotyping from across the 16q24 HBD region. Using Simwalk2, a maximum location score (directly comparable to multipoint LOD score) of 2.008 was calculated for marker D16S2621, which is just ~500 Kb distal to FBXO31. After we incorporated the FBXO31 variant in the analysis, the maximum location score which increased to 3.948 was calculated (Fig. 3).

Linkage analysis using SIMWALK2. Position in Haldane cM is plotted on the x-axis, and Location Score (assuming no heterogeneity; α = 1.0) on the y-axis. The significance threshold for linkage of 3.0 is indicated by the red dashed line. Maximum Location Score of 3.948 was achieved for the FBXO31 variant and microsatellite marker D16S2621
Quantification of lymphoblast mRNA from the affected individuals shows a ~threefold decrease in expression levels compared to the unaffected individual (student’s t test: Amplicon 1, IV:8: p = 1.48 × 10−7, IV:10: p = 1.40 × 10−7; Amplicon 2, IV:8: p = 2.18 × 10−8, IV:10: p = 8.73 × 10−8; Amplicon 3, IV:8: p = 3.49 × 10−7, IV:10: p = 2.73 × 10−6; Fig. 2a). These results suggest that nonsense-mediated decay may cause degradation of the FBXO31 mRNA carrying the 5 bp deletion/premature termination codon. Western blot analysis appeared to confirm this, showing reduced levels of FBXO31 protein in the affected individuals (student’s t test: p = 0. 034; Fig. 2b). Additionally, subcellular localization of FBXO31 was performed by immunocytochemistry and showed the protein located in the cell body and axons in primary hippocampal neurons from wild-type E15 mouse embryos (Fig. 4).

Cultured hippocampal neurons were fixed using PFA, followed by immunostaining with FBX031 antibodies. Representative images are shown for a FBXO31 antibody (Bethyl Laboratories) and b differential image contrast (DIC) image; c FBXO31 antibody (Novus Biologicals) and d corresponding DIC image. Scale bar 50 μm
The truncating mutation identified in FBXO31 may perturb protein function by disrupting the FBXO31/Par6c or FBXO31/SCF complexes which for the first time shows an important link between ID and the gene FBXO31.
Discussion
In this study, we have shown a frameshifting, truncating mutation in the gene FBXO31 associated with a form of autosomal recessive ID in family ASMR72. The affected individuals appear to have a spectrum of mild to moderate dysmorphic features, but since photographic images of unaffected relatives were not available, the milder traits cannot be viewed in a familial context, and these individuals may be considered non-syndromic. On the other hand, some facial features seen in several affected family members suggest that there may be common dysmorphisms associated with mutation of FBXO31. The 5 bp deletion and premature stop codon in the mutant FBXO31 appear to correspond to diminished mRNA and protein levels. Moreover, the depletion of FBXO31 protein would likely affect the formation of axons and dendrites during neurogenesis.
The FBXO31 gene comprises 9 exons, and encodes a 539-amino acid protein containing a 47-amino acid F-box binding domain which binds proteins S phase kinase-associated protein 1 (Skp1), Ring-box 1 (Rbx1) and Cullin-1 to form a Skp/Cullin/F-box (SCF) ubiquitin E3 ligase complex (Kumar et al. 2005). A shorter, 8-exon transcript encodes a 431-amino acid protein which does not include the F-box motif. The E3 ligase complex is an important regulator of the ubiquitin proteasome system (UPS) and performs the last stage of the ubiquitination reaction along with E1 and E2 ligases (reviewed in Deshaies and Joazeiro 2009). The variable F-box component determines the substrate specificity of this complex. Protein ubiquitination can lead to different outcomes, depending on which lysine residue of the interacting substrate protein ubiquitin binds, can result in complete proteasomal degradation or can modify protein function (reviewed in Deshaies and Joazeiro 2009). It has been noted that the UPS system is a vital component for neuronal development and morphogenesis (reviewed in Yi and Ehlers, 2007).
The role of FBXO31 has been well characterized as a cell-cycle regulator and as a tumor suppressor gene in cancer research (Kumar et al. 2005; Huang et al. 2010; Kanie et al. 2012); however, reports have shown a variety of roles in brain development, especially in axonal and dendritic growth. Recently, functional studies of neuronal FBXO31-SCF have been implicated in controlling the initiation of axonal growth by an interaction with the polarity protein Par6c through a PDZ binding domain (Vadhvani et al. 2013). The functional mechanism which initiates axonal growth is regulated by FBXO31-SCF ligase, which ubiquinates Par6c resulting in complete proteosomal degradation. However, FBXO31-SCF has also been implicated in dendritic growth independently of Parc6, suggesting that FBXO31-SCF controls dendrite growth via a substrate other than Parc6 (Vadhvani et al. 2013). Our immunocytochemistry (ICC) analysis in mouse hippocampal neurons shows FBXO31 located in axons and in soma, but not at the centrosomes as reported by Vadhvani et al. (2013). Although our study used the same anti-FBXO31 antibodies, however, it is not clear at what stage of development and how old the primary culture was in the Vadhvani et al. study, and thus these differences are not directly comparable.
The importance of FBXO31 as a centrosomal protein and a regulator in neuronal morphogenesis, where it controls the positioning and migration of axons as well as dendritic growth, gives an insight into the function of this gene in the neuronal development. Additionally, the interaction between FBXO31-SCF/Par6c which regulates axon initiation, and FBXO31-SCF which controls dendritic growth establishes a controlled signaling network. Hence, information gathered by the mutation to the FBXO31 gene could shed light as to how the signaling complex is disrupted and how this affects brain development, leading to ID.
Other genes involved in the protein ubiquitination pathway have previously been implicated in intellectual disability, including the ubiquitin ligase genes: HUWE1 (Froyen et al. 2008), MID1 (encoding a member of the tripartite motif family of E3 ubiquitin ligases; Quaderi et al. 1997), MID2 (Geetha et al. 2014), Cereblon (CRBN; Higgins et al. 2008), which encodes a substrate receptor for the Cullin/RING E3 ligase complex (Xu et al. 2013), UBE3A (Angelman syndrome; Kishino et al. 1997; Matsuura et al. 1997), UBE2A (Nascimento et al. 2006), UBE3B (blepharophimosis-ptosis-intellectual disability syndrome; Basel-Vanagaite et al. 2012), UBR1 (Johanson–Blizzard syndrome; Zenker et al. 2005), and CUL4B (Tarpey et al. 2007). TRIM50 in the Williams–Beuren syndrome region encodes a member of the tripartite motif family of E3 ubiquitin ligases (Micale et al. 2008). Thus, the identification of FBXO31 as a gene for ID adds to the growing importance of this pathway in normal neurodevelopment and cognition. Genes encoding Parc6 and other substrates of the FBXO31-SCF complex would also be potential candidate genes for ID.
References
Basel-Vanagaite L, Dallapiccola B, Ramirez-Solis R, Segref A, Thiele H, Edwards A, Arends MJ, Miró X, White JK, Désir J, Abramowicz M, Dentici ML, Lepri F, Hofmann K, Har-Zahav A, Ryder E, Karp NA, Estabel J, Gerdin AK, Podrini C, Ingham NJ, Altmüller J, Nürnberg G, Frommolt P, Abdelhak S, Pasmanik-Chor M, Konen O, Kelley RI, Shohat M, Nürnberg P, Flint J, Steel KP, Hoppe T, Kubisch C, Adams DJ, Borck G (2012) Deficiency for the ubiquitin ligase UBE3B in a blepharophimosis-ptosis-intellectual-disability syndrome. Am J Hum Genet 91:998–1010. doi:10.1016/j.ajhg.2012.10.011
David M, Dzamba M, Lister D, Ilie L, Brudno M (2011) SHRiMP2: sensitive yet practical short read mapping. Bioinformatics 27:1011–2101. doi:10.1093/bioinformatics/btr046
Deshaies RJ, Joazeiro CA (2009) RING domain E3 ubiquitin ligases. Annu Rev Biochem 78:399–434. doi:10.1146/annurev.biochem.78.101807.093809
Ellison JW, Rosenfeld JA, Shaffer LG (2013) Genetic basis of intellectual disability. Annu Rev Med 64:441–450. doi:10.1146/annurev-med-042711-140053
Froyen G, Corbett M, Vandewalle J, Jarvela I, Lawrence O, Meldrum C, Bauters M, Govaerts K, Vandeleur L, Van Esch H, Chelly J, Sanlaville D, van Bokhoven H, Ropers HH, Laumonnier F, Ranieri E, Schwartz CE, Abidi F, Tarpey PS, Futreal PA, Whibley A, Raymond FL, Stratton MR, Fryns JP, Scott R, Peippo M, Sipponen M, Partington M, Mowat D, Field M, Hackett A, Marynen P, Turner G, Gécz J (2008) Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am J Hum Genet 82:432–443. doi:10.1016/j.ajhg.2007.11.002
Geetha TS, Michealraj KA, Kabra M, Kaur G, Juyal RC, Thelma BK (2014) Targeted deep resequencing identifies MID2 mutation for X-linked intellectual disability with varied disease severity in a large kindred from India. Hum Mutat 35:41–44. doi:10.1002/humu.22453
Higgins JJ, Hao J, Kosofsky BE, Rajadhyaksha AM (2008) Dysregulation of large-conductance Ca(2+)-activated K(+) channel expression in nonsyndromal mental retardation due to a cereblon p. R419X mutation. Neurogenetics 9:219–223. doi:10.1007/s10048-008-0128-2
Homer N, Nelson SF (2010) Improved variant discovery through local re-alignment of short-read next-generation sequencing data using SRMA. Genome Biol 11:R99. doi:10.1186/gb-2010-11-10-r99
Huang HL, Zheng WL, Zhao R, Zhang B, Ma WL (2010) FBXO31 is down-regulated and may function as a tumor suppressor in hepatocellular carcinoma. Oncol Rep 24:715–720
Kanie T, Onoyama I, Matsumoto A, Yamada M, Nakatsumi H, Tateishi Y, Yamamura S, Tsunematsu R, Matsumoto M, Nakayama KI (2012) Genetic evaluation of the role of F-box proteins in cyclin D1 degradation. Mol Cell Biol 32:590–605. doi:10.1128/MCB.06570-11
Kaufman L, Ayub M, Vincent JB (2010) The genetic basis of non-syndromic intellectual disability: a review. J Neurodev Disord 2:182–209
Kishino T, Lalande M, Wagstaff J (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15:70–73
Kumar R, Neilsen PM, Crawford J, McKirdy R, Lee J, Powell JA, Saif Z, Martin JM, Lombaerts M, Cornelisse CJ, Cleton-Jansen AM, Callen DF (2005) FBXO31 is the chromosome 16q24.3 senescence gene, a candidate breast tumor suppressor, and a component of an SCF complex. Cancer Res 65:11304–11313
Leonard H, Wen X (2002) The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev 8:117–134
Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C (2004) dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics 20:1233–1240
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408
Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL (1997) De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 15:74–77
Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S (2011) Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil 32:419–436. doi:10.1016/j.ridd.2010.12.018
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi:10.1101/gr.107524.110
Micale L, Fusco C, Augello B, Napolitano LM, Dermitzakis ET, Meroni G, Merla G, Reymond A (2008) Williams–Beuren syndrome TRIM50 encodes an E3 ubiquitin ligase. Eur J Hum Genet 16:1038–1049. doi:10.1038/ejhg.2008.68
Musante L, Ropers HH (2014) Genetics of recessive cognitive disorders. Trends Genet 30:32–39. doi:10.1016/j.tig.2013.09.008
Nascimento RM, Otto PA, de Brouwer AP, Vianna-Morgante AM (2006) UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X-linked mental retardation syndrome. Am J Hum Genet 79:549–555
Pagnamenta AT, Khan H, Walker S, Gerrelli D, Wing K, Bonaglia MC, Giorda R, Berney T, Mani E, Molteni M, Pinto D, Le Couteur A, Hallmayer J, Sutcliffe JS, Szatmari P, Paterson AD, Scherer SW, Vieland VJ, Monaco AP (2011) Rare familial 16q21 microdeletions under a linkage peak implicate cadherin 8 (CDH8) in susceptibility to autism and learning disability. J Med Genet 48:48–54. doi:10.1136/jmg.2010.079426
Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, Muenke M, Ropers HH, Ballabio A (1997) Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet 17:285–291
Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. (2009) HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res 37(Web Server issue):W593-9. doi:10.1093/nar/gkp369
Seibenhener ML, Wooten MW (2012) Isolation and culture of hippocampal neurons from prenatal mice. J Vis Exp (65). doi:10.3791/3634
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85. doi:10.1016/0003-2697(85)90442-7
Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker sharing statistics. Am J Hum Genet 58:1323–1337
Sobel E, Sengul H, Weeks DE (2001) Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum Hered 52:121–131
Sobel E, Papp JC, Lange K (2002) Detection and integration of genotyping errors in statistical genetics. Am J Hum Genet 70:496–508
Sparrow SS, Cicchetti DV, Balla DA (2005) Vineland-II adaptive behavior scales: survey forms manual. AGS Publishing, Circle Pines
Tarpey PS, Raymond FL, O’Meara S, Edkins S, Teague J, Butler A, Dicks E, Stevens C, Tofts C, Avis T, Barthorpe S, Buck G, Cole J, Gray K, Halliday K, Harrison R, Hills K, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Varian J, West S, Widaa S, Mallya U, Moon J, Luo Y, Holder S, Smithson SF, Hurst JA, Clayton-Smith J, Kerr B, Boyle J, Shaw M, Vandeleur L, Rodriguez J, Slaugh R, Easton DF, Wooster R, Bobrow M, Srivastava AK, Stevenson RE, Schwartz CE, Turner G, Gecz J, Futreal PA, Stratton MR, Partington M (2007) Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet 80:345–352
Vadhvani M, Schwedhelm-Domeyer N, Mukherjee C, Stegmuller J (2013) The centrosomal E3 ubiquitin ligase FBXO31-SCF regulated neuronal morphogenesis and migration. PLoS One 8:e57530. doi:10.1371/journal.pone.0057530
Xu G, Jiang X, Jaffrey SR (2013) A mental retardation-linked nonsense mutation in cereblon is rescued by proteasome inhibition. J Biol Chem 288:29573–29585. doi:10.1074/jbc.M113.472092
Yi JJ, Ehlers MD (2007) Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev 59:14–39
Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, Beier M, Hülskamp G, Guzman C, Rehder H, Beemer FA, Hamel B, Vanlieferinghen P, Gershoni-Baruch R, Vieira MW, Dumic M, Auslender R, Gil-da-Silva-Lopes VL, Steinlicht S, Rauh M, Shalev SA, Thiel C, Ekici AB, Winterpacht A, Kwon YT, Varshavsky A, Reis A (2005) Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson–Blizzard syndrome). Nat Genet 37:1345–1350
Acknowledgments
We wish to thank the family members for their willing participation and cooperation with this study. This research was supported by a grant from the Canadian Institutes of Health Research (#MOP-102758), and by the Pakistan Higher Education Commission (HEC). We declare that the authors have no competing interests for this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
A. Mir and K. Sritharan contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Mir, A., Sritharan, K., Mittal, K. et al. Truncation of the E3 ubiquitin ligase component FBXO31 causes non-syndromic autosomal recessive intellectual disability in a Pakistani family. Hum Genet 133, 975–984 (2014). https://doi.org/10.1007/s00439-014-1438-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-014-1438-0