
Abstract
Plasma concentrations of Aβ40 and Aβ42 rise with age and are increased in people with mutations that cause early-onset Alzheimer’s disease (AD). Amyloid beta (Aβ) plasma levels were successfully used as an (endo)phenotype for gene discovery using a linkage approach in families with dominant forms of disease. Here, we searched for loci involved in Aβ plasma levels in a series of non-demented patients with hypertension in the Erasmus Rucphen Family study. Aβ40 and Aβ42 levels were determined in 125 subjects with severe hypertension. All patients were genotyped with a 6,000 single nucleotide polymorphisms (SNPs) illumina array designed for linkage analysis. We conducted linkage analysis of plasma Aβ levels. None of the linkage analyses yielded genome-wide significant logarithm of odds (LOD) score over 3.3, but there was suggestive evidence for linkage (LOD > 1.9) for two regions: 1q41 (LOD = 2.07) and 11q14.3 (LOD = 2.97), both for Aβ40. These regions were followed up with association analysis in the study subjects and in 320 subjects from a population-based cohort. For the Aβ40 region on chromosome 1, association of several SNPs was observed at the presenilin 2 gene (PSEN2) (p = 2.58 × 10−4 for rs6703170). On chromosome 11q14-21, we found some association (p = 3.1 × 10−3 for rs2514299). This linkage study of plasma concentrations of Aβ40 and Aβ42 yielded two suggestive regions, of which one points toward a known locus for familial AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Together with neurofibrillary tangles, amyloid beta (Aβ) plaques in the brain are a pathological hallmark of Alzheimer’s disease (AD) (Takashima 2009). The formation of the extracellular peptide Aβ results from the cleavage of the amyloid precursor protein (APP) by the enzymes β and γ secretase, as opposed to α and γ secretase (Murphy and LeVine 2010). Aβ is partially degraded within the brain and partly cleared from brain to plasma through the blood–brain barrier, a process in which several of the recently discovered Alzheimer risk genes are involved (Bettens et al. 2010). Excess production or poor clearance of amyloid beta may lead to neurotoxicity and formation of amyloid beta plaques. This is also true for truncated Aβ forms, Aβn-42 and Aβn40. The latter are more likely to be spliced into pathogenic forms. Aβn-42 accounts for 60 % of all Aβ species in pre-clinical AD stages (Sergeant et al. 2003). High plasma concentrations of Aβ are associated with an increased risk of AD (van Oijen et al. 2006) and there is revived interest in the use of this marker in clinical care, despite the fact that there may not be a causal relationship. Recent research has shown a decreased clearance of Aβ from the brain in those with mild cognitive dysfunction (Mawuenyega et al. 2010).
Aβ plasma levels were successfully used as an (endo)phenotype for gene discovery using a linkage approach in families with dominant forms of disease, leading to the discovery of presenilin 1 (PSEN1) (Ertekin-Taner et al. 2000). From a genetic perspective, it is of interest that the gene encoding the angiotensin converting enzyme (ACE) is not only consistently associated with AD, but also plays a role in the degradation of Aβ in the brain (Kehoe et al. 2009). Since hypertension in early life to middle age has now been well established as a risk factor for AD, and plasma Aβ levels are associated with hypertension (Lambert et al. 2011), we have conducted a genetic study aiming to identify genes involved in plasma Aβ levels in persons with hypertension.
Materials and methods
Study population
This study was embedded in the Erasmus Rucphen Family (ERF) study, a population-based study in a genetically isolated population. All approximately 3,000 participants in this study are living descendants of 22 couples who, at the end of the nineteenth century, had at least six children baptized in the community church. Extensive genealogy data are available from the year 1600 AD. Extensive data on the participants, including cardiovascular risk factors, family history, body composition, health habits, cognitive function, blood chemistry and genotyping, are available.
For this study, hypertensive subjects aged 55–75 years who did not have a history of stroke or dementia were selected from the study population. Hypertension was defined as a systolic blood pressure of ≥160 mmHg, a diastolic blood pressure of ≥100 mmHg and/or the use of antihypertensive medication. We chose to limit ourselves to subjects with this high threshold for hypertension to get the maximally informative subjects out of our cohort. Shah et al. (2012) provide a good insight into the interaction between amyloid beta and hypertension in the risk for AD and vascular brain changes. Of the 261 eligible individuals invited for this study, 135 agreed to participate. The participants and non-participants were similar with respect to cardiovascular risk factors, but the participants had a slightly higher duration of education (9 years in participants as opposed to 7.5 years in non-participants). All participants gave informed consent. The study was approved of by the Erasmus University Medical Center Medical Ethics Committee.
A replication sample was obtained from the Rotterdam Study (RS), a population-based cohort from the Rotterdam region (Hofman et al. 2007). There were 320 individuals available who met the same inclusion criteria as our discovery subjects (age 55–75 years, hypertensive and free from stroke and dementia) and had genotype information and amyloid beta measurements available.
Aβ measurements
Non-fasting blood samples were obtained in EDTA tubes and immediately cooled on ice. Plasma was extracted and stored at −80 °C. Plasma Aβ concentrations were measured with a fluorimetric bead-based immunoassay using xMAP® technology (Innogenetics®). Aβ40, Aβ42, and the truncated forms Aβn-42 and Aβn-40 were measured. From these measurements, we also calculated the ratios for Aβ40/Aβ42 and Aβ42/Aβn-42 (Hansson et al. 2010; Lambert et al. 2009).
Genotyping
Genotyping and pedigree data were available for 125 of the 129 subjects with full phenotype data. For all participants, genomic DNA was extracted from peripheral venous blood utilizing the salting out method (Miller et al. 1988). For genome-wide linkage analysis, genotyping was performed using the Illumina 6K linkage panel. Of the 6,000 single nucleotide polymorphisms (SNPs) on the array, 5,250 were used for analysis after quality control and excluding X-chromosomal SNPs. The genotyping was performed at the Centre National de Génotypage in France according to the manufacturer’s protocol.
For association analysis, we used SNPs from dense genotyping platforms that included Illumina 318K, Illumina 370K, Illumina 610K and Affymetrix 250K, which were merged as previously described (Demirkan et al. 2011). Genotyping on these platforms was performed according to the described protocols. Additionally, the array data were used to impute genotypes (using MACH version 1.0.16) (Li et al. 2009, 2010) to the HapMap rel. 22 CEU panel for meta-analysis. The genome-wide imputed dataset consisted of approximately 2.5 million SNPs.
For the replication cohort, we extracted genotype data for the regions of interest from the imputed genotype dataset. This dataset consists of approximately 2.5 million SNPs, genotyped on the Illumina HumanHap550-Duo BeadChip® and imputed with MACH.
Statistical analysis
Statistical analysis of quantitative traits was performed using PASW Statistics version 17.0 (SPSS Inc.) for Windows. The Aβ measurements were normally distributed, but kurtosis was increased for all distributions (from 1.98 to 3.29). Inverse-normal transformation of ranks was applied to all individual traits for linkage analysis, and ln transformation was used for the association analyses for the ratios as these were not normally distributed. The rank transformation was performed using the GenABEL package for R (Aulchenko et al. 2007). The family-based design of the ERF study makes it possible to evaluate the heritability of Aβ levels (Aβ40, Aβ42 and truncated forms Aβn40 and Aβn-42) using the pedigrees in the SOLAR program (Almasy and Blangero 1998).
Linkage
We performed linkage analysis for the different amyloid beta subtypes as defined above with age and sex as covariates, using the variance components method as implemented in Merlin (Abecasis et al. 2002). For computational reasons (software limitations), the study population was divided into 34 sub-pedigrees each consisting of two to seven subjects. A significant linkage result was defined as an LOD peak of ≥3.3, and a suggestive linkage result as an LOD peak of ≥1.9 (Lander and Kruglyak 1995). Additionally, we investigated all the remaining regions with an LOD peak ≥1.5. Linkage regions were defined as the region bounded by the maximum LOD score minus 1.
Fine mapping with association analysis
With the same traits and covariates, we performed association analysis with a polygenic model using GenABEL package for R (Aulchenko et al. 2007). The SNPs for the regions under the linkage peaks were taken from the dense panel of 700,000 SNPs. Association analysis was also performed for the SNPs of interest in the Rotterdam Study.
A meta-analysis of the results from the two studies for the regions under the peak was performed using the Metal software package for meta-analysis (Willer et al. 2010). In this meta-analysis, we used the 2.5 million imputed SNP dataset for ERF.
eQTL analyses
Information on the power of the identified SNP as expression quantitative trait loci (eQTL), i.e., the association of the SNPs found in our analyses to gene expression, was extracted from the mRNA by the SNP browser (MRBS) by Liang et al. (Dixon et al. 2007). (www.sph.umich.edu/csg/liang/asthma). We applied the genome-wide significance threshold for eQTLs defined by the authors of this database: an LOD of 6.076, corresponding to a p value of 1.2 × 10−7. We also extracted data on eQTL associations from the ScanDB database (www.scandb.org), which is based on the eQTL analyses by Zhang et al. (2008).
Results
Descriptives
Table 1 provides a description of the study population. Of the 135 subjects, 6 were excluded from analysis due to unsuccessful phenotype collection and 4 more because of insufficient genotype or pedigree data. Plasma Aβ40 was significantly correlated with BMI (p = 0.007).
We assessed the role of possible confounding factors by performing linear regression analyses on Aβ values with and without suspected confounders. We found no evidence for confounding from either any medication acting on the RAS system, for Aβ40 (p = 0.683) or Aβ42 (p = 0.481), or for diabetes (p values: in Aβ40, diabetes status = 0.681, HbA1C = 0.588; in Aβ42, diabetes = 0.067, HbA1C = 0.133).
We also assessed partial correlations between Aβ levels and cognitive functioning in a neuropsychological test battery including Stroop, Trailmaking test, Block Design, 15 word memory test, and word fluency. There was a nominally significant (p = 0.005, we ran 11 correlations for 2 Aβ measurements) partial correlation, corrected for age, sex and eduction, between Aβ42 levels and the Z score for word fluency, but not for any other individual or composite measure to Aβ42 or Aβ40.
Heritability
First, we estimated the heritabilities of Aβ levels (Aβ40, Aβ42 and truncated forms Aβn40 and Aβn-42) using the full ERF pedigree. We estimated a heritability of 0.23 (p = 0.19), 0.30 (p = 0.20), 0.12 (p = 0.31) and 0.55 (p = 0.07) for Aβ40, Aβ42, Aβn40 and Aβn-42, respectively. None of the heritabilities were significant. However, in a rank-transformed analysis to adjust for non-normality in pedigree fragments, the heritability estimates for Aβ40 (p = 0.04) and Aβ42 (p = 0.046) were significant.
Linkage analysis
None of the linkage analyses yielded genome-wide significant log odd (LOD) scores over 3.3, but there was suggestive linkage (LOD > 1.9) to Aβ40 in two regions: 1q41 (LOD = 2.07) and 11q14.3 (LOD = 2.97) (Table 2). The other individual traits had maximum LODs between 1.5 and 1.9: 2p22.2 (LOD = 1.86) for Aβn40, 15q13.3 (LOD = 1.63) and 15q26.1 (LOD = 1.7) for Aβ42, and 1q31.1 (LOD 1.6) and 17q25.1 (LOD = 1.61) for Aβn-42. For the ratios, we found no evidence for linkage. Online resource 1 shows the genome-wide linkage plots for all traits and zooms in on the regions with LOD > 1.9. Although the Aβ40 and Aβ42 levels are highly correlated (partial correlation = 0.433, p < 0.001), there is no overlap in linkage peaks between the two. In the chromosome 1 region for Aβ40, the maximum LOD for Aβ42 was 0.35. In the chromosome 11 region, there was no evidence for any linkage for Aβ42 (LOD = 0). Also vice versa, the chr15 regions linked to Aβ42 showed no evidence for linkage to Aβ40 (LOD max 0.05).
Association and eQTL analyses
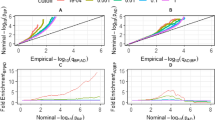
We further explored the suggestive regions on chromosome 1 and 11 using the dense SNP genotypes from the microarrays. There were 1,216 directly genotyped SNPs available for association analysis in the region identified on chromosome 1, and 562 SNPs for the region on chromosome 11. For the meta-analysis in the imputed SNP sets, there were 14986 SNPs for the region on chromosome 1, and 3981 SNPs for the region on chromosome 11. Figure 1a, b shows the local linkage and association plots for these regions. Results for the top hits in the directly genotyped dataset are provided in Online Resource 2.

a Local linkage and association plots for the Aβ40 region on chromosome 1. This figure shows the linkage and association results for the linkage region defined on chromosome 1. In A, a detail of the linkage plot is shown. B and C, respectively, show the association results under the peak from the imputed datasets for ERF and RS. D Gives the meta-analysis results for the two. A consistent signal can be seen in the PSEN2 region at 2.25 × 108 bp. b Local linkage and association plots for the Aβ40 region on chromosome 11. This figure gives the linkage and association results for the linkage region defined on chromosome 11. In A, a detail of the linkage plot is shown. B, C, respectively, show the association results under the peak from the imputed datasets for ERF and RS. D Gives the meta-analysis results for the two. There is no consistent signal across the two cohorts within the region on chromosome 11
For the Aβ40 region on chromosome 1, nominal association was observed for an SNP lying 82 kb upstream from the presenilin 2 gene (PSEN2) (rs6703170: p = 2.58 × 10−4) (Online Resource 2). In the meta-analysis of ERF and the Rotterdam Study, rs6697254 had the lowest p value, but the allele frequency was low (0.0057) making the finding unreliable. Rs12409752 had the lowest p value with a common risk allele (MAF 0.27, p = 1.1 × 10−4) which is an intergenic SNP between ITPKB and PSEN2. There was a large block of PSEN2 intronic SNPs spanning from 225130294 to 225149349 kb (NCBI build 36.3), which were associated with Aβ40. When considering eQTLs, several SNPs were associated (lowest p = 2.40 × 10−10 for rs2236914) with PSEN2 expression levels. The meta-analysis results and eQTL results are given in Online Resource 3.
When analyzing the chromosome 11 region in more detail, there were no strong association results. The SNPs showing some association in ERF (top hit rs2514299, p = 3.2 × 10−3) (Online Resource 2) did not replicate in the RS. The two SNPs most strongly associated with Aβ40 levels emerging from a meta-analysis of RS and ERF were rs947937 (p = 7.3 × 10−5) and rs947935 (p = 9.6 × 10−5) (Online Resource 3). eQTL analysis in this region showed no convincing results.
Discussion
In this family-based study, we found the highest heritability for Aβ42 and Aβn-42. This heritability is lower than that found in an extended family affected with late-onset Alzheimer’s disease (LOAD) (Ertekin-Taner et al. 2001). There was no genome-wide significant linkage of Aβ levels as none of the regions reached an LOD score of 3.3 or higher, although the region on chromosome 11 approached this genome-wide significance level at an LOD score of 2.97. In total, two regions showed suggestive linkage with an LOD of >1.9. Of these two regions, the chromosome 11 region showed the highest LOD score. In this region, the ERF and RS association analyses were not consistent. In the second region on chromosome 1 with an LOD score of 2.07, PSEN2 is the most remarkable gene, showing evidence both for association and an effect on expression levels of PSEN2.
On comparing our data with the literature, there are a number of remarkable findings. First, the PSEN2 gene is a known causative gene for some cases of familial AD (Van Broeckhoven 1995). PSEN2 was identified due to its homology to PSEN1 (Rogaev et al. 1995). Its penetrance is lower than that of PSEN1. In its turn, PSEN1 was identified in a study using Aβ plasma levels as an (endo)phenotype for gene discovery using a linkage approach in families with dominant forms of AD (St George-Hyslop et al. 1992). The presenilins are the proteases in the gamma secretase complex in the cell membrane responsible for the cleavage of APP into amyloid beta. Additionally, the presenilin 2 protein has been proven to downregulate cytokine-induced inflammatory responses in the brain, which can lead to neurodegeneration (Jayadev et al. 2010). On comparing the meta-analysis association results with the eQTL databases, several top SNPs showed significant association with PSEN2 expression levels, i.e., were eQTLs for this gene. This supports our theory that it is a variant in, or in the regulatory region of, PSEN2 causing this signal and pinpoints familial AD gene PSEN2 as playing a possible role in the multifactorial LOAD pathogenesis. Our study connects a common variant near PSEN2 to Aβ metabolism relatively early in life.
Second, it is interesting that we found a positive association of PSEN2 with Aβ40, while there was no evidence of any linkage or association with Aβ42 (best LOD for the region is 0.32). These findings support several recent reports that indicate that Aβ40 is a determinant at least as important as Aβ42, although whether its effect is protective or risk increasing is currently being debated (Kumar-Singh et al. 2006; van Oijen et al. 2006). Animal experiments have shown that all tested familial PSEN and APP mutations resulted in decreased Aβ40 production with an accumulation of APP C-terminal fragments, a sign of decreased PSEN activity, but only some mutations including the PSEN2 N141I (Volga German mutation) affected Aβ42 levels (Kumar-Singh et al. 2006). Our method does not allow us to pinpoint a specific mutation in the gene or its promoter regions, but it is possible that the variant underlying our signal selectively affects Aβ40 levels. We did not see evidence for association with the Aβ42/Aβ40 ratio.
The region on chromosome 11 is of particular interest for two reasons. First, the LOD score in this region approaches genome-wide significance. Second, this region has been associated with various neuropsychiatric disease, including autism (Anderson et al. 2009) and schizophrenia (Gurling et al. 2001; Petit et al. 1999) and it is close to the region 11q25 previously found associated with depressive disorder (Schol-Gelok et al. 2010), schizophrenia (Vorstman et al. 2006) and late-onset Alzheimer’s disease (Liu et al. 2007). Given the evidence of co-occurrence of depression and Alzheimer’s disease (Aznar and Knudsen 2011), this region is of particular interest from a clinical perspective. Within this linkage region, we cannot clearly identify a likely candidate gene based on the association or eQTL analyses.
There was no evidence of linkage with the APOE region on chromosome 19. APOE is the best-established risk gene for sporadic Alzheimer’s disease, plasma ApoE levels have been associated with amyloid beta burden in the brain, and ApoE is believed to play a role in the clearance of Aβ from the brain (Bettens et al. 2010; Thambisetty et al. 2010). A lack of power in the current study may be a possible explanation for this finding. Another explanation is that plasma Aβ42 and Aβ40 levels show no correlation to APOE. Only the Aβ42/Aβ40 ratio was correlated to APOE E4 carrier status in our sample (r 2 = −21; p = 0.023). Finally, compared to association, linkage analysis may not be as powerful an approach to identify a common susceptibility gene such as APOE. The strongest association signal within 1 Mb of the APOE gene was seen for rs1661197 (p = 0.001) with Aβ42. This SNP is located 312 kb away from the gene.
The main limitation of this linkage project is its limited sample size, due to the narrow inclusion criteria for this study and financial and logistics issues. Also, plasma amyloid beta is a rough estimate of the Aβ load in the brain and little is known about the variations and the changes of clearance from brain to plasma over time. There is no circadian variation in plasma Aβ levels (Lachno et al. 2009). However, our heritability studies show that Aβ42 and Aβ40 plasma levels are stable enough to yield significant evidence for familial clustering of increased Aβ levels at early age before the onset of dementia. Several studies have shown its value as an easily obtainable biomarker for risk of AD (Ertekin-Taner et al. 2008; van Oijen et al. 2006), although there are also negative studies (Hansson et al. 2010; Kester et al. 2010). It is the best method available for population-based research, as it is ethically not feasible to perform lumbar punctures on healthy volunteers at a large scale in a population-based setting.
Plasma amyloid beta 40 levels in healthy middle-aged subjects are associated with the locus containing the PSEN2 gene associated with early-onset Alzheimer’s disease, and with a locus on chromosome 11. Our findings support the involvement of these regions in the development of sporadic late-onset Alzheimer’s disease. Additionally, the identification of a known gene involved in the plasma Aβ levels in this hypothesis-free experiment can be considered a sign of robustness for this method in our inbred population. Lastly, our eQTL analysis underlines the new associations of interest in interpreting the results of an association analysis. Next-generation sequencing and expression analyses will hopefully allow us to investigate the linkage peaks identified in much more detail in the future.
References
Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101
Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211
Anderson BM, Schnetz-Boutaud NC, Bartlett J, Wotawa AM, Wright HH, Abramson RK, Cuccaro ML, Gilbert JR, Pericak-Vance MA, Haines JL (2009) Examination of association of genes in the serotonin system to autism. Neurogenetics 10:209–216
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM (2007) GenABEL: an R library for genome-wide association analysis. Bioinformatics 23:1294–1296
Aznar S, Knudsen GM (2011) Depression and Alzheimer’s disease: is stress the initiating factor in a common neuropathological cascade? J Alzheimers Dis 23:177–193
Bettens K, Sleegers K, Van Broeckhoven C (2010) Current status on Alzheimer disease molecular genetics: from past, to present, to future. Hum Mol Genet 19:R4–R11
Demirkan A, Penninx BW, Hek K, Wray NR, Amin N, Aulchenko YS, van Dyck R, de Geus EJ, Hofman A, Uitterlinden AG, Hottenga JJ, Nolen WA, Oostra BA, Sullivan PF, Willemsen G, Zitman FG, Tiemeier H, Janssens AC, Boomsma DI, van Duijn CM, Middeldorp CM (2011) Genetic risk profiles for depression and anxiety in adult and elderly cohorts. Mol Psychiatry 16:773–783
Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KC, Taylor J, Burnett E, Gut I, Farrall M, Lathrop GM, Abecasis GR, Cookson WO (2007) A genome-wide association study of global gene expression. Nat Genet 39:1202–1207
Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG (2000) Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science 290:2303–2304
Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Adamson J, Schaid DJ, Blangero J, Hutton M, Younkin SG (2001) Heritability of plasma amyloid beta in typical late-onset Alzheimer’s disease pedigrees. Genet Epidemiol 21:19–30
Ertekin-Taner N, Younkin LH, Yager DM, Parfitt F, Baker MC, Asthana S, Hutton ML, Younkin SG, Graff-Radford NR (2008) Plasma amyloid beta protein is elevated in late-onset Alzheimer disease families. Neurology 70:596–606
Gurling HM, Kalsi G, Brynjolfson J, Sigmundsson T, Sherrington R, Mankoo BS, Read T, Murphy P, Blaveri E, McQuillin A, Petursson H, Curtis D (2001) Genomewide genetic linkage analysis confirms the presence of susceptibility loci for schizophrenia, on chromosomes 1q32.2, 5q33.2, and 8p21-22 and provides support for linkage to schizophrenia, on chromosomes 11q23.3-24 and 20q12.1-11.23. Am J Hum Genet 68:661–673
Hansson O, Zetterberg H, Vanmechelen E, Vanderstichele H, Andreasson U, Londos E, Wallin A, Minthon L, Blennow K (2010) Evaluation of plasma Abeta(40) and Abeta(42) as predictors of conversion to Alzheimer’s disease in patients with mild cognitive impairment. Neurobiol Aging 31:357–367
Hofman A, Breteler MM, van Duijn CM, Krestin GP, Pols HA, Stricker BH, Tiemeier H, Uitterlinden AG, Vingerling JR, Witteman JC (2007) The Rotterdam Study: objectives and design update. Eur J Epidemiol 22:819–829
Jayadev S, Case A, Eastman AJ, Nguyen H, Pollak J, Wiley JC, Moller T, Morrison RS, Garden GA (2010) Presenilin 2 is the predominant gamma-secretase in microglia and modulates cytokine release. PLoS ONE 5:e15743
Kehoe PG, Miners S, Love S (2009) Angiotensins in Alzheimer’s disease—friend or foe? Trends Neurosci 32:619–628
Kester MI, Verwey NA, van Elk EJ, Scheltens P, Blankenstein MA (2010) Evaluation of plasma Abeta40 and Abeta42 as predictors of conversion to Alzheimer’s disease in patients with mild cognitive impairment. Neurobiol Aging 31:539–540
Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C (2006) Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat 27:686–695
Lachno DR, Vanderstichele H, De GG, Kostanjevecki V, De Meyer G, Siemers ER, Willey MB, Bourdage JS, Konrad RJ, Dean RA (2009) The influence of matrix type, diurnal rhythm and sample collection and processing on the measurement of plasma beta-amyloid isoforms using the INNO-BIA plasma Abeta forms multiplex assay. J Nutr Health Aging 13:220–225
Lambert JC, Schraen-Maschke S, Richard F, Fievet N, Rouaud O, Berr C, Dartigues JF, Tzourio C, Alperovitch A, Buee L, Amouyel P (2009) Association of plasma amyloid beta with risk of dementia: the prospective Three-City Study. Neurology 73:847–853
Lambert JC, Dallongeville J, Ellis KA, Schraen-Maschke S, Lui J, Laws S, Dumont J, Richard F, Cottel D, Berr C, Ames D, Masters CL, Rowe CC, Szoeke C, Tzourio C, Dartigues JF, Buee L, Martins R, Amouyel P (2011) Association of plasma Abeta peptides with blood pressure in the elderly. PLoS ONE 6:e18536
Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247
Li Y, Willer C, Sanna S, Abecasis G (2009) Genotype imputation. Annu Rev Genomics Hum Genet 10:387–406
Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR (2010) MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 34:816–834
Liu F, Arias-Vasquez A, Sleegers K, Aulchenko YS, Kayser M, Sanchez-Juan P, Feng BJ, Bertoli-Avella AM, van Swieten J, Axenovich TI, Heutink P, Van Broeckhoven C, Oostra BA, van Duijn CM (2007) A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am J Hum Genet 81:17–31
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330:1774
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Murphy MP, LeVine H III (2010) Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis 19:311–323
Petit J, Boisseau P, Taine L, Gauthier B, Arveiler B (1999) A YAC contig encompassing the 11q14.3 breakpoint of a translocation associated with schizophrenia, and including the tyrosinase gene. Mamm Genome 10:649–652
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T (1995) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376:775–778
Schol-Gelok S, Janssens AC, Tiemeier H, Liu F, Lopez-Leon S, Zorkoltseva IV, Axenovich TI, van Swieten JC, Uitterlinden AG, Hofman A, Aulchenko YS, Oostra BA, van Duijn CM (2010) A genome-wide screen for depression in two independent Dutch populations. Biol Psychiatry 68:187–196
Sergeant N, Bombois S, Ghestem A, Drobecq H, Kostanjevecki V, Missiaen C, Wattez A, David JP, Vanmechelen E, Sergheraert C, Delacourte A (2003) Truncated beta-amyloid peptide species in pre-clinical Alzheimer’s disease as new targets for the vaccination approach. J Neurochem 85:1581–1591
Shah NS, Vidal JS, Masaki K, Petrovitch H, Ross GW, Tilley C, DeMattos RB, Tracy RP, White LR, Launer LJ (2012) Midlife blood pressure, plasma beta-amyloid, and the risk for Alzheimer disease: the Honolulu Asia Aging Study. Hypertension 59:780–786
St George-Hyslop P, Haines J, Rogaev E, Mortilla M, Vaula G, Pericak-Vance M, Foncin JF, Montesi M, Bruni A, Sorbi S (1992) Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat Genet 2:330–334
Takashima A (2009) Amyloid-beta, tau, and dementia. J Alzheimers Dis 17:729–736
Thambisetty M, Tripaldi R, Riddoch-Contreras J, Hye A, An Y, Campbell J, Sojkova J, Kinsey A, Lynham S, Zhou Y, Ferrucci L, Wong DF, Lovestone S, Resnick SM (2010) Proteome-based plasma markers of brain amyloid-beta deposition in non-demented older individuals. J Alzheimers Dis 22(4):1099–1109
Van Broeckhoven C (1995) Presenilins and Alzheimer disease. Nat Genet 11:230–232
van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM (2006) Plasma Abeta(1–40) and Abeta(1–42) and the risk of dementia: a prospective case–cohort study. Lancet Neurol 5:655–660
Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L (2006) Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry 11:1, 18-1, 28
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191
Zhang W, Duan S, Kistner EO, Bleibel WK, Huang RS, Clark TA, Chen TX, Schweitzer AC, Blume JE, Cox NJ, Dolan ME (2008) Evaluation of genetic variation contributing to differences in gene expression between populations. Am J Hum Genet 82:631–640
Acknowledgments
This study was financially supported by the Netherlands Organization for Scientific Research (NWO), the Internationale Stichting Alzheimer Onderzoek (ISAO), the Hersenstichting Nederland (HSN) and the Centre for Medical Systems Biology (CMSB) in the framework of the Netherlands Genomics Initiative (NGI) and by the Russian Foundation for Basic Research (RFBR). We thank the participants from the Genetic Research in Isolated Populations, Erasmus Rucphen Family, who made this work possible. Also, we thank Petra Veraart for collecting all genealogical data.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
All research described in this paper was carried out in compliance with the laws and customs for scientific research in the Netherlands. The Erasmus University Medical Center Medical Ethics Committee approved both the Erasmus Rucphen Family Study and the Rotterdam Study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2012_1210_MOESM1_ESM.pdf
Online resource 1 Linkage plots Figure 1 shows the genome-wide linkage results for each of the individual Aβ measurements, plotting each SNP’s chromosome and position against its calculated LOD/HLOD. Figure 2 zooms in on the regions reaching suggestive linkage on chromosome 1 and 11 (PDF 779 kb)
439_2012_1210_MOESM2_ESM.doc
Online resource 2 Supplementary table. This table shows the association results under the linkage peak for the SNPs with association p < 0.001 to Aβ40 in the ERF directly genotyped set. Beside SNP details and the association p value to Aβ40, also the association p value to Aβ42 is given (DOC 90 kb)
439_2012_1210_MOESM3_ESM.xls
Online resource 3 Meta-analysis results and eQTL data. For both suggestive linkage regions, all SNPs with a meta-analysis association p value of < 0.001 are given. Beside SNP details and meta-analysis results, the file also contains information on eQTL associations for the SNPs from two different eQTL databases (XLS 126 kb)
Rights and permissions
About this article
Cite this article
Ibrahim-Verbaas, C.A., Zorkoltseva, I.V., Amin, N. et al. Linkage analysis for plasma amyloid beta levels in persons with hypertension implicates Aβ-40 levels to presenilin 2. Hum Genet 131, 1869–1876 (2012). https://doi.org/10.1007/s00439-012-1210-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-012-1210-2