
Abstract
Kabuki syndrome (KS) is one of the classical, clinically well-known multiple anomalies/mental retardation syndromes, mainly characterized by a very distinctive facial appearance in combination with additional clinical signs such as developmental delay, short stature, persistent fingerpads, and urogenital tract anomalies. In our study, we sequenced all 54 coding exons of the recently identified MLL2 gene in 34 patients with Kabuki syndrome. We identified 18 distinct mutations in 19 patients, 11 of 12 tested de novo. Mutations were located all over the gene and included three nonsense mutations, two splice-site mutations, six small deletions or insertions, and seven missense mutations. We compared frequencies of clinical symptoms in MLL2 mutation carriers versus non-carriers. MLL2 mutation carriers significantly more often presented with short stature and renal anomalies (p = 0.026 and 0.031, respectively), and in addition, MLL2 carriers obviously showed more frequently a typical facial gestalt (17/19) compared with non-carriers (9/15), although this result was not statistically significant (p = 0.1). Mutation-negative patients were subsequently tested for mutations in ten functional candidate genes (e.g. MLL, ASC2, ASH2L, and WDR5), but no convincing causative mutations could be found. Our results indicate that MLL2 is the major gene for Kabuki syndrome with a wide spectrum of de novo mutations and strongly suggest further genetic heterogeneity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epigenetic control of developmental processes is a fascinating mechanism by which spatial and temporal expression of distinct genes and pathways are regulated (Gibney and Nolan 2010). Alterations of epigenetic mechanisms have been mainly associated with the pathogenesis of cancer, but most recently also with the occurrence of congenital malformation syndromes (Fan et al. 2009; Eggermann et al. 2010; Choufani et al. 2010). Recent advances in massive parallel sequencing as well as adopted conceptual strategies, e.g., for analyzing thousands of variants previously identified by whole-exome sequencing have had a great impact on studying Mendelian disorders and have dramatically speeded up gene identification in medical genetics (Ng et al. 2010a; Gilissen et al. 2010; Hoischen et al. 2010; Kalay et al. 2011). An up-to-date example is the identification of de novo dominant mutations in the MLL2 gene (MIM 602113) using a whole-exome sequencing strategy in patients with Kabuki syndrome (KS) (Ng et al. 2010b).
Kabuki syndrome (MIM 147920) is a rare condition which comprises a typical facial appearance, postnatal short stature, organ malformations, and a varying degree of mental retardation. It was first described independently by two Japanese groups in 1981 (Niikawa et al. 1981; Kuroki et al. 1981). The name of the syndrome refers to the distinctive facial appearance which resembles the make-up of actors in Japanese Kabuki theater. The facial characteristics in Kabuki syndrome include long palpebral fissures with lateral eversion of the lower eyelid, long eyelashes, arched eyebrows with lateral thinning, large, prominent ears with dysplastic helices, and a depressed nasal tip. Additional findings include persistent fetal finger pads, recurrent middle ear infections in infancy, cardiac and renal abnormalities.
MLL2 is located on chromosome 12q13, and its 54 exons encode a protein of 5,537 amino acids. MLL2 belongs to the SET1 family of histone methyltransferases involved in epigenetic regulation of development. It acts in a multiprotein complex, and various binding partners within this complex have already been identified (Issaeva et al. 2007). The complex can bind to regulatory sites of genes and induces H3K4 trimethylation, and it is well established that methylation of the histone H3 on lysine K4 is associated with increased transcriptional activation of target genes (Chang et al. 2010). We now confirm that de novo dominant MLL2 mutations are the major cause of Kabuki syndrome, provide evidence for further genetic heterogeneity, and show that mutations in MLL2-binding partners are likely not the cause of MLL2-negative Kabuki syndrome.
Materials and methods
Patients
34 patients with the clinical diagnosis of Kabuki syndrome, in accordance with the suggested guidelines for clinical diagnosis (Adam and Hudgins 2005), were included in the study. Clinical features of some of the patients have already been published [Utine et al. 2008 (K1816 = patient 1, K1759 = patient 2, K1764 = patient 5, K1761 = patient 6, K1767 = patient 7, K1770 = patient 8, K1755 = patient 9, K1758 = patient 10, K1765 = patient 11); Armstrong et al. 2005 (K1718)]. All affected individuals or their legal representatives gave written informed consent to the study. The study was performed in accordance with the Declaration of Helsinki protocols. We collected peripheral blood samples from the affected children and parents, after informed consent was obtained, according to the protocols approved by the participating institutions. All the research procedures that followed were in accordance with the ethical standards of the responsible national and institutional committees on human subject research. Written consents for publication of the photographs presented in Fig. 2 were given. DNA from participating family members was extracted from peripheral blood lymphocytes by standard extraction procedures.
Mutation screening of MLL2 and relevant candidate genes
The following databases were used to obtain gene information: National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/), Ensembl Genome Server (http://www.ensembl.org/), UCSC Genome Bioinformatics (http://www.genome.ucsc.edu/) and 1000 genomes (http://browser.1000genomes.org/). For mutation screening we chose MLL2, its paralog MLL, and nine additional genes, i.e., SMAD1, CXXC1, ASC2, ASH2L, RBBP5, WDR5, DPY30, PAXIP1, and DKK1 that are involved in the ALR complex, MLL2 regulation and interaction. Primers were designed to amplify exons and adjacent splice sites according to the reference sequences (MLL2 reference sequence NM_003482), using ExonPrimer (http://www.genome.ucsc.edu/, http://ihg.helmholtz-muenchen.de/ihg/ExonPrimer.html). Primer sequences and PCR conditions are available on request. PCR products were sequenced by BigDye Terminator method on an ABI 3730 sequencer. We re-sequenced all identified mutations in independent experiments. The MLL2 mutation p.S543L in patient K1771 was screened in 100 healthy Turkish control individuals by PCR/restriction digestion using BsrD1. In addition, we tested 103 healthy Turkish control individuals for the presence of the putative MLL mutation p.L950F by PCR/restriction digestion using the enzyme MlyI and/or direct sequencing. Paternity testing was performed in 12 father–mother–patient trios using highly informative microsatellite markers D3S642, D3S1038, D3S3611, D10S1269, and D10S1760. Analysis of marker alleles confirmed paternity in all trios analyzed and, thereby, demonstrated a de novo origin of 11 MLL2 mutations.
In general, we analyzed the non-synonymous variants found in the screen using the servers PolyPhen (http://coot.embl.de/PolyPhen/) and ConSeq (http://conseq.tau.ac.il/), and tested for possible splice site alterations (http://www.fruitfly.org/seq_tools/splice.html, http://www.cbs.dtu.dk/services/NetGene2/, http://www.itb.cnr.it/sun/webgene/, and http://www.umd.be/HSF/). The protein sequences were analyzed with the server Pfam (http://pfam.sanger.ac.uk/search?tab=searchSequenceBlock) for protein domains. Mutation nomenclature was checked using the Mutalyzer software (http://www.mutalyzer.nl/2.0/). Statistical analysis of clinical findings of mutation carriers versus non-carriers was performed using SPSS version 19.0.
Results
MLL2 mutation screening
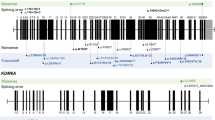
We sequenced all 54 coding exons of the MLL2 gene by Sanger sequencing in 34 patients who have been clinically diagnosed with Kabuki syndrome according to the suggested clinical diagnostic criteria (Adam and Hudgins 2005). In total, we identified heterozygous MLL2 mutations in 19 patients (56% overall detection rate), and 18 of these mutations were distinct (Table 1, Supplementary Figure 1). Only the missense mutation c.15461G>A (p.R5154Q) was found as a recurrent de novo mutation in two patients. Mutations included three nonsense mutations (p.G1320X, p.Q2811X, p.R2915X), two splice-site mutations (c.954+1G>T, c.13531−1G>T), six small deletions or insertions leading to a frameshift and premature stop codons (c.1483_1486delTCTC, c.1512_1513delTC, c.5268dupG, c.6595delT, c.7289dupT, c.9460delC), and seven missense variants (p.S543L, p.P647Q, p.V1192M, p.H1453R, p.A1718V, p.R5154Q, p.S5498F). Parents were available for testing in 12 families, and we found evidence for de novo origin of the mutations in 11 of these families (Fig. 1). No parental DNA was available for the missense variants p.P647Q, p.A1718V, and p.S543L. None of the three alterations is listed as SNP in databases or the 1000 Genome Project. In addition, p.P647Q and p.A1718V are conserved (ConSeq score: 5 of 9) and predicted as possibly damaging on PolyPhen. As the p.S543L was also present in the mother of patient K1771 (with no obvious clinical phenotype), we tested 100 ethnically matched Turkish controls and were able to show that p.S543L does not represent a common polymorphism. The p.S543L is conserved (ConSeq score: 5 of 9) but not located within a defined domain of MLL2, and, therefore, a PolyPhen prediction could not be established. The causative nature of this mutation remains unclear.

Distribution of MLL2 mutations. Schematic view of the MLL2 protein domains, all coding exons, and localization of identified MLL2 mutations. The mutations detected in the present study are shown on top and de novo mutations are marked with an asterisk. Mutations reported by Ng et al. and Paulussen et al. are indicated below. Except p.Y2199IfsX65, all mutations found in this study are novel
Analysis of the distribution of all identified mutations did not show any exon or domain preference within MLL2 (Fig. 1).
Additional candidate gene screening
We continued our molecular analysis in 15 MLL2-negative patients with Kabuki syndrome. Ten highly relevant candidate genes were chosen based on (1) a direct functional interaction of their gene products with MLL2 within the multiprotein complex (SMAD1, CXXC1, ASC2, ASH2L, RBBP5, WDR5, DPY30, PAXIP1), (2) predicted homology and function of the encoded protein (MLL), or (3) the description as a target gene (DKK1). We directly sequenced 36 exons of MLL (NM_005933, MIM 159555), 7 exons of SMAD1 (NM_005900, MIM 601595), 15 exons of CXXC1 (NM_001101654, MIM 609150), 15 exons of ASC2 (NM_014071, MIM 605299), 16 exons of ASH2L (NM_004674, MIM 604782), 14 exons of RBBP5 (NM_005057, MIM 600697), 14 exons of WDR5 (NM_017588, MIM 609012), 5 exons of DPY30 (NM_032574, MIM 612032), 21 exons of PAXIP1 (NM_007349, MIM 608254), and 4 exons of DKK1 (NM_012242, MIM 605189).
In the Turkish patient K1772 we found the heterozygous alteration c.2848C>T in exon 3 of MLL. This alteration is predicted to substitute a leucine at position 950 by phenylalanine (p.L950F). No further putative mutations were identified in any of the other candidate genes tested. We did not detect the MLL c.2848C>T alteration in 103 Turkish control individuals, and found that the variation was inherited from the healthy mother. No second putative mutation could be identified on the father’s allele, but we cannot exclude larger deletions and a possible recessive mode of inheritance within this family. No RNA was available from the patient for further analysis of MLL. The variation of unclear significance is conserved (ConSeq score: 5 of 9), but is not located within a known protein domain of MLL and the PolyPhen program predicted the p.L950F alteration to be “benign” (score 0.9). An accurate prediction concerning its causative nature can hardly be done.
Discussion
Clinical diagnosis of Kabuki syndrome is mainly based on the combination of unique facial features and the presence of developmental delay. While most patients show mild to moderate mental retardation (Niikawa et al. 1988), normal intelligence as well as severely impaired cognitive function has been reported (Matsumoto and Niikawa 2003; Ho and Eaves 1997). In our study, 95% of patients carrying a mutation in MLL2 showed some degree of developmental delay, although no formal IQ tests were performed in this study to quantify cognitive capacity. Among our patients with MLL2 mutation 89% were classified as having a typical facial gestalt in the initial physical evaluation (Table 1; Fig. 2), whereas in the patient group without MLL2 mutation only 60% met these criteria (Table 2). Interestingly, two MLL2 positive patients did not show the typical facial gestalt. Patient K1725, who carries a de novo splice-site mutation in MLL2, only presented with long palpebral fissures and high arched eyebrows, but did not have eversion of lateral lower eyelids, long eye-lashes, and prominent dysplastic ears leading to a not recognizable facial appearance of KS (consent for publication of the photograph was not given). Patient K1771 did not have the typical facial gestalt mainly because of lack of high arched eyebrows (again, consent for publication of the photograph was not given). In addition, it is important to mention that not all MLL2-positive patients with KS presented with long palpebral fissures, a hallmark of the facial phenotype (Table 1, Adam and Hudgins 2005). These findings clearly indicate a certain clinical variability regarding facial features in Kabuki patients and make clinical evaluation more difficult. The overall mutation detection rate in our study was 56%. Consideration only of patients with typical facial gestalt in addition to other clinical features would have raised the detection rate to approximately 70% (19 of 27) (Tables 1, 2), but in that case at least one individual would have been missed. Quantitative testing of MLL2 for larger deletions or duplications, e.g., by MLPA, might increase the mutation detection rate. In a recent MLL2 mutation screen the detection rate was comparable with approximately 75% (Paulussen et al. 2011).

Faces of MLL2 mutation-positive patients with Kabuki syndrome. Facial appearance of 12 patients with Kabuki syndrome harboring heterozygous MLL2 mutations. Typical characteristics include long palpebral fissures with lateral eversion of the lower eyelid, long eyelashes, arched eyebrows with lateral thinning, depressed nasal tip, and large, prominent ears with dysplastic helices
In contrast to both the previous studies, we did find a higher percentage of putative causative missense mutations in MLL2 (7/19 = 37%) compared to the studies by Paulussen et al. (2/34 = 6%) and Ng et al. (5/32 = 16%). Taking into consideration that the causative nature of identified missense mutations in our study and the study by Ng et al. could not be finally proven by de novo occurrence or inheritance from an affected parent for all missense mutations, definite frequencies of missense mutations could be much lower in both studies (3/19 = 16% and 3/32 = 9%, respectively). In prior studies, only truncating mutations were detected in the N-terminal region of MLL2 and missense mutations were located only within functionally important domains of the C-terminal part of MLL2. Interestingly, two de novo missense mutations, p.V1192M and p.H1453R, we detected in our study are located more in the N-terminus of the MLL2 protein not affecting a conserved functional protein domain of MLL2 suggesting a different underlying functional effect of these mutations. It will be important in future studies to elucidate the effect of these mutations on protein structure and function.
One patient, K1771, who carried the maternally transmitted p.S543L missense variant, showed most but not all of the key features of KS, while his mother was phenotypically unremarkable. Although the causative nature of this mutation remains uncertain, it might point toward incomplete penetrance of dominantly inherited MLL2 mutations, a finding that has already been described in other autosomal dominant developmental syndromes (Dodé and Hardelin 2010, Francis and Antzelevitch 2005).
Postnatal short stature is a common finding in KS, which we observed in 58% of patients with MLL2 mutations. Microcephaly has been reported to be present in 25–30% of patients with KS (Matsumoto and Niikawa 2003). We discovered the prevalence to be as high as 58% among our patients with MLL2 mutations, which suggests that this feature might have been underestimated in the past. Muscular hypotonia and feeding problems are frequent in KS and were seen in 65 and 68% of our patients. 44% of our patients in the MLL2 mutation-positive group who did undergo brain imaging showed brain abnormalities, namely white matter anomalies (20%), ventricular dilatation (19%), cerebral atrophy (13%) and corpus callosum a-/dysgenesis (13%). Brain anomalies had a higher prevalence in the MLL2 mutation-negative group (60%), as did epilepsy (47 vs. 5%), suggesting the presence of alternative entities in this group. Renal anomalies have been reported to occur in over 25% of patients with KS (Adam and Hudgins 2005). In this study, renal anomalies were present in 41% of our patients with MLL2 mutations versus 7% in the mutation-negative group, signifying that renal malformations are frequent in KS and should be looked for in KS patients who have not had a renal sonogram. The incidence of congenital heart defects in KS has been estimated at 40–50% (Adam and Hudgins 2005). We confirm this frequency, having found congenital heart defects in 47% of our MLL2-positive patients.
Other frequent findings in KS patients include persistent fingerpads, an indicative diagnostic criterion which we found in 90% of patients with MLL2 mutations, and a susceptibility to middle ear infections, which 47% of our patients in the MLL2 mutation-positive group suffered from. In the future, it will be interesting to specify epigenetic alterations and altered expression of genes responsible for the various symptoms seen in patients with KS.
We performed statistical analysis of clinical symptoms observed in MLL2 mutation carriers versus non-carriers. Although the number of patients in our cohort was limited, short stature and renal anomalies were more frequently present in MLL2 mutation carriers (p = 0.026 and 0.031, respectively; Fisher’s exact test) (Table 3). In addition, MLL2 mutation carriers obviously presented more often with a typical facial gestalt (17/19) compared with non-carriers (9/15). However, this finding was not statistically significant (p = 0.1; Fisher’s exact test). Our data are in agreement with the study by Paulussen et al. describing statistically significant differences between MLL2 mutation carriers and non-carriers in the presence of growth retardation and distinct facial appearance.
Our candidate gene screen did not provide convincing evidence for a second Kabuki gene, although our data show that further genetic heterogeneity is likely. We identified a single alteration in MLL with a yet unclear causative role, but this variation is predicted to be benign and was inherited from the unaffected mother. Although unlikely, we cannot completely rule out a recessive inheritance and a second mutation inherited from the father which was not identified by conventional Sanger sequencing of MLL coding exons. Use of whole exome sequencing in MLL2-negative patients will likely prove genetic heterogeneity in Kabuki syndrome in the near future.
Taken together, our results confirm that MLL2 is the major gene for Kabuki syndrome with a wide spectrum of de novo mutations.
References
Adam MP, Hudgins L (2005) Kabuki syndrome: a review. Clin Genet 67:209–219
Armstrong L, Abd El Moneim A, Aleck K, Aughton DJ, Baumann C, Braddock SR, Gillessen-Kaesbach G, Graham JM Jr, Grebe TA, Gripp KW, Hall BD, Hennekam R, Hunter A, Keppler-Noreuil K, Lacombe D, Lin AE, Ming JE, Kokitsu-Nakata NM, Nikkel SM, Philip N, Raas-Rothschild A, Sommer A, Verloes A, Walter C, Wieczorek D, Williams MS, Zackai E, Allanson JE (2005) Further delineation of Kabuki syndrome in 48 well-defined new individuals. Am J Med Genet A 132A:265–272
Chang PY, Hom RA, Musselman CA, Zhu L, Kuo A, Gozani O, Kutateladze TG, Cleary ML (2010) Binding of the MLL PHD3 finger to histone H3K4me3 is required for MLL-dependent gene transcription. J Mol Biol 400:137–144
Choufani S, Shuman C, Weksberg R (2010) Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 154C:343–354
Dodé C, Hardelin JP (2010) Clinical genetics of Kallmann syndrome. Ann Endocrinol (Paris) 71:149–157
Eggermann T, Begemann M, Spengler S, Schröder C, Kordass U, Binder G (2010) Genetic and epigenetic findings in Silver-Russel syndrome. Pediatr Endocrinol Rev 8:86–93
Fan Z, Yamaza T, Lee JS, Yu J, Wang S, Fan G, Shi S, Wang CY (2009) BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat Cell Biol 11:1002–1009
Francis J, Antzelevitch C (2005) Brugada syndrome. Int J Cardiol 101:173–178
Gibney ER, Nolan CM (2010) Epigenetics and gene expression. Heredity 105:4–13
Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, van Lier B, Steehouwer M, van Reeuwijk J, Kant SG, Roepman R, Knoers NV, Veltman JA, Brunner HG (2010) Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet 87:418–423
Ho HH, Eaves LC (1997) Kabuki make-up (Niikawa-Kuroki) syndrome: cognitive abilities and autistic features. Dev Med Child Neurol 39:487–490
Hoischen A, van Bon BW, Gilissen C, Arts P, van Lier B, Steehouwer M, de Vries P, de Reuver R, Wieskamp N, Mortier G, Devriendt K, Amorim MZ, Revencu N, Kidd A, Barbosa M, Turner A, Smith J, Oley C, Henderson A, Hayes IM, Thompson EM, Brunner HG, de Vries BB, Veltman JA (2010) De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet 42:483–485
Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, Canaani E (2007) Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol 27:1889–1903
Kalay E, Yigit G, Aslan Y, Brown KE, Pohl E, Bicknell LS, Kayserili H, Li Y, Tüysüz B, Nürnberg G, Kiess W, Koegl M, Baessmann I, Buruk K, Toraman B, Kayipmaz S, Kul S, Ikbal M, Turner DJ, Taylor MS, Aerts J, Scott C, Milstein K, Dollfus H, Wieczorek D, Brunner HG, Hurles M, Jackson AP, Rauch A, Nürnberg P, Karagüzel A, Wollnik B (2011) CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet 43:23–26
Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I (1981) A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr 99:570–573
Matsumoto N, Niikawa N (2003) Kabuki make-up syndrome: a review. Am J Med Genet 11:57–65
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ (2010a) Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42:30–35
Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, Rieder MJ, Yoshiura K, Matsumoto N, Ohta T, Niikawa N, Nickerson DA, Bamshad MJ, Shendure J (2010b) Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42:790–793
Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajiim T (1981) Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr 99:565–569
Niikawa N, Kuroki Y, Kajii T et al (1988) Kabuki makeup (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet 31:565–589
Paulussen AD, Stegmann AP, Blok MJ, Tserpelis D, Posma-Velter C, Detisch Y, Smeets EE, Wagemans A, Schrander JJ, van den Boogaard MJ, van der Smagt J, van Haeringen A, Stolte-Dijkstra I, Kerstjens-Frederikse WS, Mancini GM, Wessels MW, Hennekam RC, Vreeburg M, Geraedts J, de Ravel T, Fryns JP, Smeets HJ, Devriendt K, Schrander-Stumpel CT (2011) MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat 32:E2018–E2025
Utine GE, Alanay Y, Aktaş D, Boduroğlu K, Alikaşifoğlu M, Tunçbilek E (2008) Kabuki syndrome and trisomy 10p. Genet Couns 19:291–300
Acknowledgments
We thank all family members who participated in this study and Karin Boss for critically reading the manuscript. This work was supported by the German Federal Ministry of Education and Research (BMBF) by grant number 01GM0880 (SKELNET) and 01GM0801 (E-RARE network CRANIRARE) to BW and 01GM0802 (E-RARE network CRANIRARE) to DW.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2011_1004_MOESM1_ESM.ppt
MLL2 mutations identified in patients with Kabuki syndrome. Electropherograms of identified heterozygous MLL2 mutations compared to wild-type sequences. The last-row cell of the right-most column shows the MLL variant identified in patient K1772 (PPT 555 kb)
Rights and permissions
About this article
Cite this article
Li, Y., Bögershausen, N., Alanay, Y. et al. A mutation screen in patients with Kabuki syndrome. Hum Genet 130, 715–724 (2011). https://doi.org/10.1007/s00439-011-1004-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-1004-y