
Abstract
Human trisomy 21, the chromosomal basis of Down syndrome (DS), is the most common genetic cause of heart defects. Regions on human chromosome 21 (Hsa21) are syntenically conserved with three regions located on mouse chromosome 10 (Mmu10), Mmu16 and Mmu17. In this study, we have analyzed the impact of duplications of each syntenic region on cardiovascular development in mice and have found that only the duplication on Mmu16, i.e., Dp(16)1Yey, is associated with heart defects. Furthermore, we generated two novel mouse models carrying a 5.43-Mb duplication and a reciprocal deletion between Tiam1 and Kcnj6 using chromosome engineering, Dp(16Tiam1-Kcnj6)Yey/+ and Df(16Tiam1-Kcnj6)Yey/+, respectively, within the 22.9-Mb syntenic region on Mmu16. We found that Dp(16Tiam1-Kcnj6)Yey/+, but not Dp(16)1Yey/Df(16Tiam1-Kcnj6)Yey, resulted in heart defects, indicating that triplication of the Tiam1-Knj6 region is necessary and sufficient to cause DS-associated heart defects. Our transcriptional analysis of Dp(16Tiam1-Kcnj6)Yey/+ embryos confirmed elevated expression levels for the genes located in the Tiam-Kcnj6 region. Therefore, we established the smallest critical genomic region for DS-associated heart defects to lay the foundation for identifying the causative gene(s) for this phenotype.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital heart disease is a serious public health challenge. The survival rate of patients with heart defects has increased drastically with improved medical care (Bedard et al. 2008; Marelli et al. 2009; Pillutla et al. 2009; Williams et al. 2006). Nevertheless, their life expectancy only reaches ~50 years of age (Daliento et al. 2002; Pillutla et al. 2009; Verheugt et al. 2010). A major investigative effort on congenital heart defects is to understand the molecular genetic mechanisms underlying abnormal cardiovascular development, which may lead to a paradigm shift for developing novel therapies for patients with congenital heart disease.
DS is the most common genetic cause of heart defects, which are detected in 40–60% of children with DS (Abbag 2006; Chaoui et al. 2005; Goodship et al. 1998; Roizen and Patterson 2003; Roofthooft et al. 2008; Rowe and Uchida 1961; Torfs and Christianson 1998). Experimental evidence supports the hypothesis that some DS phenotypes are caused by the triplication of specific genes on Hsa21 (Epstein 1990). To identify genomic regions that contain the critical genes associated with DS phenotypes, several groups have examined human segmental trisomies. In these studies, data generated from patients segmentally trisomic for Hsa21 were used to map genomic regions associated with DS phenotypes, including heart defects. Unfortunately, some patients carried additional chromosomal anomalies (Korbel et al. 2009; Korenberg et al. 1994; Lyle et al. 2009; Sinet et al. 1994), which complicates interpretation of the genotype–phenotype correlations. Because of these difficulties, mice have been used in genetic studies of DS because the regions on Hsa21 are syntenically conserved with three regions in the mouse genome located on Mmu10, Mmu16 and Mmu17 (Fig. 1). One group of models consists of mosaic mouse mutants carrying Hsa21 or a fragment of it (O’Doherty et al. 2005; Shinohara et al. 2001). The Tc1 mouse model, which carries Hsa21 with only two small deletions in which approximately 8% of Hsa21 genes are deleted, exhibits heart defects (Dunlevy et al. 2010; O’Doherty et al. 2005). The other group of models carries three exact copies of mouse syntenic regions of Hsa21 in various sizes. Ts65Dn, the most widely used model (Davisson et al. 1990; Reeves et al. 1995), is trisomic for 13.4 Mb of the 22.9 Mb Hsa21 syntenic region on Mmu16 (Fig. 1) (Akeson et al. 2001; Kahlem et al. 2004) and exhibits some major DS phenotypes, including heart defects (Moore 2006; Williams et al. 2008). However, Ts65Dn is also trisomic for a larger than 5.8 Mb subcentromeric region on Mmu17, which is not syntenic to any region on Hsa21 and may contribute to the cardiovascular phenotype (Li et al. 2007). To better mimic DS using mouse segmental trisomies, we have recently developed new mouse models, Dp(10)1Yey/+, Dp(16)1Yey/+ and Dp(17)1Yey/+, carrying individual duplications spanning the entire Hsa21 syntenic regions on Mmu10, Mmu16 and Mmu17, respectively (Fig. 1) (Li et al. 2007; Yu et al. 2010). We have showed that both Dp(16)1Yey/+ and Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+ genotypes led to heart defects with a similar frequency (Li et al. 2007; Yu et al. 2010).

Genomic dissection of DS-associated heart defects in mice
In the current project, we are focused on identifying a minimal critical genomic region for DS-associated heart defects. We examined the impact of Dp(10)1Yey/+ and Dp(17)1Yey/+ on heart development and after we found that Mmu16 is the only mouse chromosome associated with heart defects in DS, we generated and analyzed Dp(16Tiam1-Kcnj6)Yey/+ and Dp(16)1Yey/Df(16Tiam1-Kcnj6)Yey embryos. After we observed heart defects in Dp(16Tiam1-Kcnj6)Yey/+ embryos, we performed expression analysis on the mutant embryos and identified the genes with elevated expression in the duplicated region.
Materials and methods
Generation of mouse mutants carrying Dp(16Tiam1-Kcnj6)Yey or Df(16Tiam1-Kcnj6)Yey
We generated Dp(16Tiam1-Kcnj6)Yey/Df(16Tiam1-Kcnj6)Yey ES cells using Cre/loxP-mediated chromosome engineering (Yu and Bradley 2001). MICER vectors (Adams et al. 2004) were used as the targeting vectors (pTVTiam1 and pTVKcnj6) for inserting loxP to the endpoint 1 (EP1) and EP2, which are 403-kb proximal and 155-kb distal to the coding regions of Tiam1 and Kcnj6, respectively, in AB2.2 ES cells (Bradley et al. 1998). pTVTiam1 and pTVKcnj6 were linearized with restriction enzymes EcoNI and BaeI at the mouse genome homologous regions in the vectors, respectively, before electroporations. Eight double-targeted ES cell clones were isolated. A Cre-expression vector, pOG231 (O’Gorman et al. 1997), was transfected into double-targeted ES cells to induce recombination between targeted loxP sites, which led to duplication and reciprocal deletion. The duplication was designated as Dp(16Tiam1-Kcnj6)Yey, abbreviated as Dp(16)2Yey or Ts4Yey. The deletion was designated as Df(16Tiam1-Kcnj6)Yey, abbreviated as Df(16)2Yey or Ms3Yey. Gene targeting as well as chromosomal rearrangements was confirmed by Southern blot analysis of ES cell DNA. The presence of Dp(16)2Yey and Df(16)2Yey was also confirmed by FISH (see below). The ES cells carrying the desired genomic rearrangements were microinjected into blastocysts that were isolated from albino C57B6/J-Tyr c-Brd females to generate germ-line transmitting chimeras. The procedural details of ES cell culture, gene targeting and induction of Cre/loxP-mediated recombination, Southern blot analysis and injection of ES cells into blastocysts were described previously (Bradley 1987; Bradley et al. 1998; Ramirez-Solis et al. 1993, 1995).
Fluorescent in situ hybridization
FISH analysis was performed, as described previously (Yu et al. 2006). The metaphase chromosome spreads and interphase nuclei of ES cells were prepared, as described previously (Robertson 1987). To detect the chromosomal deletion and duplication between Tiam1 and Kcnj6, BAC clone RP23-280L21 was labeled with digoxigenin and detected with anti-digoxigenin-rhodamine antibody. BAC clone RP23-81D13 was used to identify Mmu16 and labeled with biotin and detected with fluorescin isothiocyanate-avidin (Fig. 2c, d). Chromosomes were counter-stained with DAPI (4′,6′-diamidino-2-2phenylindole) (Fig. 2d).

Development of new mouse models using chromosome engineering (Yu and Bradley 2001). a Schematic of the strategy to generate Dp(16)2Yey and Df(16)2Yey. The genome coordinates of the endpoints are shown. K, KpnI; B, BamHI; 5′, 5′HPRT fragment; 3′, 3′HPRT fragment; N, neomycin-resistance gene; P, puromycin-resistance gene; Ty, tyrosinase transgene; Ag, Agouti transgene; arrowhead, loxP site. b Southern blot analysis of ES cell DNA. Lanes 1 and 2, the ES cells targeted at Tiam1 and Kcnj6 hybridized with probes A and B, respectively; Lanes 3 and 4, the ES cells carrying the duplication and the reciprocal deletion hybridized with probes B and C, respectively. c–e FISH analysis of ES cells. c The genomic locations of BAC probes for FISH analysis are shown. FISH analysis of the metaphase chromosomes (d) and interphase nucleus (e) prepared from Dp(16)2Yey/Df(16)2Yey ES cells
Mice
The mutant mice and their wild-type littermates were maintained at a temperature- and humidity-controlled animal facility. The experimental procedures were approved by the Institutional Animal Care and Use Committee.
RNA extraction
RNA was extracted from the pharyngeal arch region and heart of E10.5 embryos using PureLink RNA Micro kit (Invitrogen Corp., Carlsbad, CA) as per the manufacturer’s instructions. The boundaries of the pharyngeal arch region were defined as previously described (Prescott et al. 2005). Prior to the RNA extraction, the embryos were genotyped using yolk sac DNA. After the elution step, RNA samples were concentrated by precipitation and resuspended in DEPC-treated nuclease-free water. The quality of the RNA samples was assessed by a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
Real-time quantitative reverse transcriptase PCR
Real-time quantitative PCR was used to analyze RNA levels of the selected genes. Gapdh is located on Mmu6 and served as a reference gene of the disomic state for all the mice examined. Total RNAs were isolated from the pharyngeal arch regions and hearts of E10.5 embryos, as described above. 1 μg of RNA from each embryo was used to generate cDNA using Superscript version III reverse transcriptase (Invitrogen Corp., Carlsbad, CA). The specific primers and probes for the genes were obtained from the TagMan® Gene Expression Assays System of Applied Biosystems, Inc. A 0.5 μg of cDNA from each embryo was analyzed by ABI 7900HT Real-Time Thermocycler (Applied Biosystems, Foster City, CA) with the following amplification conditions: an initial activation and denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and primer annealing and extension at 60°C for 1 min.
RNA labeling and microarray hybridization
To perform genome-wide expression profiling, Illumina MouseRef-8 v2 BeadChips (Illumina, Inc., San Diego, CA) were utilized. Total RNA isolated from the pharyngeal arch region and heart in the E10.5 Dp(16)2Yey/+ and wild-type embryos (250 ng for each embryo) was converted to cDNA, followed by an in vitro transcription step to generate labeled cRNA using the Ambion Illumina TotalPrep RNA Amplification Kit (Ambion, Austin, TX). The labeled probes were mixed with hybridization reagents and hybridized overnight to the BeadChips. After washing and staining, the BeadChips were imaged using the Illumina BeadArray Reader to measure the fluorescence intensity of the probes.
Microarray data analysis
Illumina BeadScan software was used to scan and extract the intensity of Illumina MouseRef-8 v2 gene expression array, and the data were corrected by background subtraction using the GenomeStudio module. The expression intensity was transformed into log2 scale by the lumi module in the R-based Bioconductor Package, which was then normalized using the quantile normalization algorithm. The quantile normalized data sets were used for the subsequent analysis. We used a fold-change filter (1.3-fold increase or decrease in the test group in relation to the wild-type control) in combination with two other conditions: i.e., the genes in the group with higher expression with a detection P value (over the background) not more than 5% and group average signal not less than 55 in the group. With the quantile normalized gene data sets for individual samples, we also tested the use of other statistical methods for identifying differentially expressed genes, including the Student’s t test and significant analysis of microarray (SAM), and obtained a shorter list of genes that overlap with the first list. However, since these shorter lists of genes fail to generate any statistically significant over-representation of the gene ontology terms, we decided to stay with the fold-change list for the rest of the analysis. Hierarchical clustering (HCL) and heat maps were generated using MeV (http://www.tm4.org/mev/) (Saeed et al. 2006). For the heat map of gene expression, the average signals of genes for each array were normalized as a ratio against the average signals of the wild-type controls. For gene ontology analysis, statistically significant enrichment of gene ontology terms represented by the differentially expressed genes was performed using the on-line DAVID bioinformatics tools at http://david.abcc.ncifcrf.gov/ (Dennis et al. 2003; Huang da et al. 2009).
Results
Development of mouse models carrying Dp(16)2Yey or Df(16)2Yey using chromosome engineering
To facilitate genetic analysis of DS-associated phenotypes, we generated a 5.43-Mb duplication and the reciprocal deletion between Tiam1 and Kcnj6 within the Hsa21 syntenic region on Mmu16 for three reasons: First, this region contains a sufficient number of genes, and the functions of many of these genes are unknown. Second, the duplicated gene at the proximal endpoint would contain Tiam1, which affects the functions of endothelial cells (Birukova et al. 2007a, b; Singleton et al. 2005). Third, the duplicated gene in the distal endpoint would be Kcnj6, which has been implicated in affecting heart rate (Lignon et al. 2008). We generated these models using Cre/loxP-mediated genomic engineering (Yu and Bradley 2001). MICER vectors (Adams et al. 2004) were used as pTVTiam1 and pTVKcnj6 for targeting loxP to the regions 403-kb proximal and 155-kb distal to the coding regions of Tiam1 and Kcnj6, respectively, in AB2.2 ES cells (Fig. 2a) (Bradley et al. 1998). A duplication and the reciprocal deletion were induced in ES cells by a transfection with a Cre-expression vector as described (Liu et al. 1998; Ramirez-Solis et al. 1995) and were confirmed by Southern blot analysis (Fig. 2b) and fluorescent in situ hybridization (FISH) (Fig. 2c–e). We used these ES cell clones to generate chimeras. The germ-line transmission of the duplication after crossing C57BL6J and 129Sv females with chimeric males was confirmed by Southern blot strategy, as shown in Fig. 2b. However, we could not obtain any Df(16)2Yey/+ mice from these crosses. One possibility is that the genotype of Df(16)2Yey/+ may have led to embryonic lethality. To test this possibility, we crossed the chimeric males with the Dp(16)1Yey/+ females. As predicted, we obtained Dp(16)1Yey/Df(16)2Yey progeny from this cross, providing evidence that the Tiam1-Kcnj6 region contains a gene(s) associated with haploinsufficient lethality. This gene(s) may underlie the embryonic lethality associated with human monosomy 21 (Chang et al. 2001; Joosten et al. 1997).
Establishment of the smallest critical genomic region for DS-associated heart defects by genetic dissection in mice
In the process of genetic analysis of DS-associated heart defects, we first examined the cardiovascular phenotypes of embryos carrying Dp(10)1Yey/+ (n = 24) or Dp(17)1Yey/+ (n = 23) at E18.5 and found no heart defects. These results indicate that the Hsa21 syntenic regions on Mmu10 and Mmu17 do not significantly contribute to heart defects and that the causative gene(s) is therefore located only on Mmu16.
Examination of Dp(16)2Yey/+ embryos at E18.5 showed that these embryos exhibit heart defects similar to Dp(16)1Yey/+ and Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+ embryos with a similar frequency in either the 129 Sv background or after crossing to C57BL6/J mice (Fig. 3; Table 1) (Li et al. 2007). We then generated Dp(16)1/Df(16)2 by crossing Dp(16)1/+ mice to the chimeras generated using Dp(16)2Yey/Df(16)2Yey ES cells. The analysis of this compound mutant at E18.5 found no heart defects (n = 24), indicating that the presence of three copies of the Tiam1-Kcnj6 region is necessary and sufficient to cause DS-associated heart defects in mice. Therefore, the causative gene(s) for this phenotype is located in the Tiam1-Kcnj6 region.

Cardiovascular abnormalities observed in Dp(16)2Yey/+ embryos at E18.5. a A dorsal view of the great arteries of a mutant embryo shows that the right subclavian artery aberrantly connects to the descending aorta to form a vascular ring around the trachea and the esophagus. b A ventral view of the heart and lung of a mutant embryo shows transposition of the great arteries, in which the anterior positioned aorta and the posterior pulmonary artery arise from the right and left ventricles, respectively. c A superior view of the ventricles of a mutant after the atria were removed. The orientation of the great arteries is normal but the aorta has a bicuspid aortic valve. d A ventral view of the atria of a mutant after the ventricles were removed shows atrial septal defect, with the arrowheads indicating the foramen of the defect. e A superior view of the ventricles of a mutant after the atria were removed. The mutant ventricles show a common AV valve; a bar with double arrowheads indicates AV orifice. f An intracardiac view of the left ventricle of a mutant. Arrows indicate two foramina of VSD, perimembranous type and larger inlet type. g The heart of a mutant embryo shows double outlet right ventricle with both the aorta and pulmonary artery rising from the right ventricle. The arrowheads indicate the position of the ventricular septum at the cardiac surface. h An intracardiac view of the same mutant embryo in g shows VSD as well as the aorta and pulmonary artery connected to the right ventricle. Ao, aorta; Ao valve, aortic valve; des. Ao, descending aorta; AV valve, atrioventricular valve; AVC, anterior vena cava; E, esophagus; LAA, left atrial appendage; LAVC, left anterior vena cava; LCA, left carotid artery; LSCA, left subclavian artery; LV, left ventricle; LV free wall, left ventricular free wall; MV, mitral valve; PA, pulmonary artery; PVC, posterior vena cava; RAA, right atrial appendage; RCA, right carotid artery; RSCA, right subclavian artery; RV, right ventricle; RV free wall, right ventricular free wall; RV sept, right ventricular septum; T, trachea; TV, tricuspid valve; VSD, ventricular septal defect. Scale bar 1 mm
We also analyzed Ts1Rhr embryos at E18.5 (n = 28), which is trisomic for the 4.12-Mb Cbr1-Fam3b region on Mmu16 (Fig. 1) (Olson et al. 2004), and found no heart defects, suggesting the duplication of the Cbr1-Fam3b region alone is not sufficient to cause DS-associated heart defects. A similar result was recently reported (Dunlevy et al. 2010).
Analysis of gene expression of Dp(16)2Yey/+ embryos
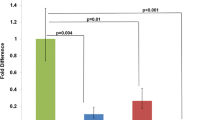
To examine the impact of Dp(16)2Yey on gene expression, we performed microarray-based genome-wide transcriptional profiling using RNA isolated from the pharyngeal arch region and the heart in E10.5 Dp(16)2Yey/+ and wild-type embryos as the templates. This included the analysis of the expression of genes located within the Tiam1-Kcnj6 region and genes located immediately external to this region. To examine the impact of Dp(16)2Yey on genome-wide expression patterns, we performed unsupervised clustering of the samples based on the expression profile of 5812 genes expressed at above-threshold levels. However, we were unable to obtain a separation of the seven arrays based on the genotypes. Only when the genes on Mmu16 or all the genes showing altered expression in Dp(16)1Yey/+ embryos were selected for analysis could the two genotypes be separated into two groups (Fig. 4). These results indicate that the Dp(16)2Yey does not lead to global alterations of the transcriptome but to alterations of expression for a defined set of genes. Based on the criteria described in “Materials and methods”, a total of 154 genes showed altered expression in Dp(16)1Yey/+ embryos. These genes can be divided into groups showing increased expression (89 genes) or decreased expression (65 genes) in Dp(16)2Yey/+ embryos (Supplemental Tables 1, 2). We also observed increased expression for 17 genes located in the Tiam1-Kcnj6 region (Fig. 4; Supplemental Table 1). However, for the genes located immediately proximal and distal to the duplicated region, no changes in expression levels were detected in the Dp(16)2Yey/+ embryos (Supplemental Table 3), suggesting that Dp(16)2Yey apparently did not alter the expression of neighboring genes surrounding the duplication in cis or in trans. We also performed real-time RT-PCR analysis for eight genes located within the Tiam1-Kcnj6 region using the same RNA samples. For the five genes present in the microarray, the RT-PCR data confirm the microarray result (Table 2; Supplemental Table 1). For Hunk, Synj1 and Urb1, which are not present in the microarray, the RT-PCR data show significant increases in the expression levels in Dp(16)2Yey/+ embryos, reflecting the consequences of the gene dosage alterations (Table 2). The analysis of microarray expression data using DAVID (http://david.abcc.nifcrf.gov/home.jsp) with Benjamini adjustment for multiple testing (Dennis et al. 2003; Huang da et al. 2009) showed that muscle-related proteins and heat shock proteins are over-represented among the differentially expressed genes (Benjamini < 0.05), which include Ankrd1, Csrp3, Mybpc3, Cryab, Hspb1, Hspb2 and Hspb3.

A heat map of the microarray-based gene expression profile around the Tiam1-Kcnj6 region with the sample clustering tree on top indicates that the presence of Dp(16)2Yey alters the transcriptional levels of the genes located within the duplicated region. The gene expression signals were normalized based on average of the control samples. The samples were clustered using the hierarchical clustering method based on all genes on Mmu16 expressed at above-threshold levels. Clustering of genes was not performed in order to preserve the gene order on the chromosome. All four Dp(16)2Yey/+ embryos show elevated expression for 17 genes located within the duplicated region (indicated by the red rectangle)
Discussion
The Hsa21 syntenic regions in the mouse genome are ~26.30 Mb in size and contain ~175 orthologous genes (Fig. 1) (Yu et al. 2010). The results from this study suggest that we can reduce the critical genomic region for DS-associated heart defects to 5.43 Mb with 52 candidate genes (Fig. 1; Supplemental Table 3). Based on this and other studies, the duplication between Cbr1 and Fam3b on Mmu16 in the Ts1Rhr model did not lead to heart defects (Dunlevy et al. 2010). Since there is a 1.61-Mb overlapping duplicated region between Dp(16)2Yey and Ts1Rhr carrying 14 orthologous genes (Fig. 1; Supplemental Table 3), two possibilities exist for the locations of the causative gene(s) for heart defects. First, the causative gene(s) may be located within the Tiam1-Cbr1 region (Fig. 1). Second, a causative gene(s) may be located in the Tiam1-Cbr1 region and another causative gene(s) may be located in the Cbr1-Kcnj6 region (Fig. 1). In the latter scenario, the heart defects are the consequence of the combined effect of simultaneous triplications of the causative genes in both regions, and triplications of the causative gene(s) in either region alone are not sufficient to cause heart defects. These possibilities could be examined by generating and analyzing mouse mutants carrying a duplication and the reciprocal deletion between Tiam1 and Cbr1. If the causative gene(s) is located in the Tiam1-Cbr1 region, Dp(16Tiam1-Cbr1)/+, but not Dp(16)1Yey/Df(16Tiam1-Cbr1), should result in heart defects. If the causative genes are located in both the Tiam1-Cbr1 and Cbr1-Kcnj6 regions and the simultaneous duplications of these genes are required to cause heart defects, neither Dp(16Taim1-Cbr1)/+ nor Dp(16)1Yey/Df(16Tiam1-Cbr1) should result in heart defects. This combination of strategies using both duplications and deletions for examining different possibilities can also be employed to dissect the Tiam1-Cbr1 and Cbr1-Kcnj6 regions to further narrow down the critical genomic regions.
Analysis of expression profiles has been utilized in DS research to explore the relationship between over-expression of individual trisomic genes and phenotypes (Kahlem et al. 2004; Lyle et al. 2004). Our results showed that the heart defects in Dp(16)2Yey/+ embryos are correlated with over-expression of 20 genes within the Tiam1-Kcnj6 region and, therefore, these genes should be analyzed first among the 52 genes located within the Tiam1-Kcnj6 region for their role in causing this phenotype (Fig. 4; Table 2; Supplemental Table 3). Among them, Rcan1 and Dyrk1a have been proposed as the causative genes for DS-associated heart defects based on a transgenic study in which a β-actin promoter was used as a regulatory element of the transgenes (Arron et al. 2006). However, because the regulatory element of these transgenes is not derived from the endogenous loci, their expression levels as well as spatial and temporal expression patterns may be different from those of the endogenous genes. Heart defects have been observed in Tc1 mice in which the human RCAN1 gene was deleted in the transchromosome (Dunlevy et al. 2010), suggesting three copies of the RCAN1 ortholog are not required to cause heart defects.
In addition to causing abnormalities in the cardiovascular system, trisomy 21 also causes abnormalities in almost all other organ systems (Roizen and Patterson 2003). Trisomy 21 is the most common genetic cause of intellectual disability from childhood to young adulthood and a leading genetic cause of gastrointestinal anomalies, childhood leukemia and early-onset Alzheimer-type neurodegeneration systems (Roizen and Patterson 2003). To perform genetic analysis of a specific DS phenotype, mouse models carrying defined duplications or deletions in different Hsa21 syntenic regions are required. Unfortunately, this type of model is currently available only for very few segments of the syntenic regions (Fig. 1). Therefore, the new mouse models developed from this study will undoubtedly become essential tools for genomic dissection of other phenotypes of DS, particularly for understanding the contribution of the Tiam1-Kcnj6 region to those phenotypes.
Our current genetic dissection project has led to the establishment of the smallest genomic region for DS-associated heart defects. This result should set the stage for future efforts to characterize the Tiam1-Kcnj6 genomic region in order to identify the critical gene(s) associated with this phenotype. The establishment of the causative gene(s) should unravel the entry points to the mechanistic details leading to congenital cardiovascular malformations in DS and may yield rare insights on cardiac development, which, in turn, may result in novel strategies for the prevention, diagnosis and treatment of congenital heart disease in children and adults regardless of their states of ploidy.
References
Abbag FI (2006) Congenital heart diseases and other major anomalies in patients with Down syndrome. Saudi Med J 27:219–222
Adams DJ, Biggs PJ, Cox T, Davies R, van der Weyden L, Jonkers J, Smith J, Plumb B, Taylor R, Nishijima I, Yu Y, Rogers J, Bradley A (2004) Mutagenic insertion and chromosome engineering resource (MICER). Nat Genet 36:867–871
Akeson EC, Lambert JP, Narayanswami S, Gardiner K, Bechtel LJ, Davisson MT (2001) Ts65Dn-localization of the translocation breakpoint and trisomic gene content in a mouse model for Down syndrome. Cytogenet Cell Genet 93:270–276
Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N, Miyakawa T, Francke U, Graef IA, Crabtree GR (2006) NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441:595–600
Bedard E, Shore DF, Gatzoulis MA (2008) Adult congenital heart disease: a 2008 overview. Br Med Bull 85:151–180
Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG (2007a) HGF attenuates thrombin-induced endothelial permeability by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the Rho pathway. FASEB J 21:2776–2786
Birukova AA, Malyukova I, Mikaelyan A, Fu P, Birukov KG (2007b) Tiam1 and betaPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. J Cell Physiol 211:608–617
Bradley A (1987) Production and analysis of chimaeric mice. In: Robertson E (ed) Teratocarcinomas and embryonic stem cells—a practical approach. IRL Press, Oxford, pp 113–151
Bradley A, Zheng B, Liu P (1998) Thirteen years of manipulating the mouse genome: a personal history. Int J Dev Biol 42:943–950
Chang LW, Chen PY, Kuo PL, Chang FM (2001) Prenatal diagnosis of a fetus with megacystis and monosomy 21. Prenat Diagn 21:512–513
Chaoui R, Heling KS, Sarioglu N, Schwabe M, Dankof A, Bollmann R (2005) Aberrant right subclavian artery as a new cardiac sign in second- and third-trimester fetuses with Down syndrome. Am J Obstet Gynecol 192:257–263
Daliento L, Mazzotti E, Mongillo E, Rotundo M, Dalla Volta S (2002) Life expectancy and quality of life in adult patients with congenital heart disease. Ital Heart J 3:339–347
Davisson MT, Schmidt C, Akeson EC (1990) Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog Clin Biol Res 360:263–280
Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA (2003) DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 4:3
Dunlevy L, Bennett M, Slender A, Lana-Elola E, Tybulewicz VL, Fisher EM, Mohun T (2010) Down syndrome-like cardiac developmental defects in embryos of the transchromosomic Tc1 mouse. Cardiovasc Res 88:287–295
Epstein CJ (1990) The consequences of chromosome imbalance. Am J Med Genet Suppl 7:31–37
Goodship J, Cross I, LiLing J, Wren C (1998) A population study of chromosome 22q11 deletions in infancy. Arch Dis Child 79:348–351
Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Joosten AM, De Vos S, Van Opstal D, Brandenburg H, Gaillard JL, Vermeij-Keers C (1997) Full monosomy 21, prenatally diagnosed by fluorescent in situ hybridization. Prenat Diagn 17:271–275
Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, Eppens B, Saran NG, Pletcher MT, South ST, Stetten G, Lehrach H, Reeves RH, Yaspo ML (2004) Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of Down syndrome. Genome Res 14:1258–1267
Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, Sobel EM, Barlow GM, Aylsworth AS, Carpenter NJ, Clark RD, Cohen MY, Doran E, Falik-Zaccai T, Lewin SO, Lott IT, McGillivray BC, Moeschler JB, Pettenati MJ, Pueschel SM, Rao KW, Shaffer LG, Shohat M, Van Riper AJ, Warburton D, Weissman S, Gerstein MB, Snyder M, Korenberg JR (2009) The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci USA 106:12031–12036
Korenberg JR, Chen XN, Schipper R, Sun Z, Gonsky R, Gerwehr S, Carpenter N, Daumer C, Dignan P, Disteche C (1994) Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc Natl Acad Sci USA 91:4997–5001
Li Z, Yu T, Morishima M, Pao A, LaDuca J, Conroy J, Nowak N, Matsui S, Shiraishi I, Yu Y (2007) Duplication of the entire 22.9-Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet 16:1359–1366
Lignon JM, Bichler Z, Hivert B, Gannier FE, Cosnay P, del Rio JA, Migliore-Samour D, Malecot CO (2008) Altered heart rate control in transgenic mice carrying the KCNJ6 gene of the human chromosome 21. Physiol Genomics 33:230–239
Liu P, Zhang H, McLellan A, Vogel H, Bradley A (1998) Embryonic lethality and tumorigenesis caused by segmental aneuploidy on mouse chromosome 11. Genetics 150:1155–1168
Lyle R, Gehrig C, Neergaard-Henrichsen C, Deutsch S, Antonarakis SE (2004) Gene expression from the aneuploid chromosome in a trisomy mouse model of down syndrome. Genome Res 14:1268–1274
Lyle R, Bena F, Gagos S, Gehrig C, Lopez G, Schinzel A, Lespinasse J, Bottani A, Dahoun S, Taine L, Doco-Fenzy M, Cornillet-Lefebvre P, Pelet A, Lyonnet S, Toutain A, Colleaux L, Horst J, Kennerknecht I, Wakamatsu N, Descartes M, Franklin JC, Florentin-Arar L, Kitsiou S, Ait Yahya-Graison E, Costantine M, Sinet PM, Delabar JM, Antonarakis SE (2009) Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur J Hum Genet 17:454–466
Marelli AJ, Therrien J, Mackie AS, Ionescu-Ittu R, Pilote L (2009) Planning the specialized care of adult congenital heart disease patients: from numbers to guidelines; an epidemiologic approach. Am Heart J 157:1–8
Moore CS (2006) Postnatal lethality and cardiac anomalies in the Ts65Dn Down syndrome mouse model. Mamm Genome 17:1005–1012
O’Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, Sesay A, Modino S, Vanes L, Hernandez D, Linehan JM, Sharpe PT, Brandner S, Bliss TV, Henderson DJ, Nizetic D, Tybulewicz VL, Fisher EM (2005) An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science 309:2033–2037
O’Gorman S, Dagenais NA, Qian M, Marchuk Y (1997) Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci USA 94:14602–14607
Olson LE, Richtsmeier JT, Leszl J, Reeves RH (2004) A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science 306:687–690
Pillutla P, Shetty KD, Foster E (2009) Mortality associated with adult congenital heart disease: trends in the US population from 1979 to 2005. Am Heart J 158:874–879
Prescott K, Ivins S, Hubank M, Lindsay E, Baldini A, Scambler P (2005) Microarray analysis of the Df1 mouse model of the 22q11 deletion syndrome. Hum Genet 116:486–496
Ramirez-Solis R, Davis AC, Bradley A (1993) Gene targeting in embryonic stem cells. Methods Enzymol 225:855–878
Ramirez-Solis R, Liu P, Bradley A (1995) Chromosome engineering in mice. Nature 378:720–724
Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT (1995) A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet 11:177–184
Robertson E (1987) Embryo-derived stem cell lines. In: Robertson E (ed) Teratocarcinomas and embryonic stem cells—a practical approach. IRL Press, Oxford, pp 77–112
Roizen NJ, Patterson D (2003) Down’s syndrome. Lancet 361:1281–1289
Roofthooft MT, van Meer H, Rietman WG, Ebels T, Berger RM (2008) Down syndrome and aberrant right subclavian artery. Eur J Pediatr 167:1033–1036
Rowe RD, Uchida IA (1961) Cardiac malformation in mongolism: a prospective study of 184 mongoloid children. Am J Med 31:726–735
Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J (2006) TM4 microarray software suite. Methods Enzymol 411:134–193
Shinohara T, Tomizuka K, Miyabara S, Takehara S, Kazuki Y, Inoue J, Katoh M, Nakane H, Iino A, Ohguma A, Ikegami S, Inokuchi K, Ishida I, Reeves RH, Oshimura M (2001) Mice containing a human chromosome 21 model behavioral impairment and cardiac anomalies of Down’s syndrome. Hum Mol Genet 10:1163–1175
Sinet PM, Theophile D, Rahmani Z, Chettouh Z, Blouin JL, Prieur M, Noel B, Delabar JM (1994) Mapping of the Down syndrome phenotype on chromosome 21 at the molecular level. Biomed Pharmacother 48:247–252
Singleton PA, Dudek SM, Chiang ET, Garcia JG (2005) Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J 19:1646–1656
Torfs CP, Christianson RE (1998) Anomalies in Down syndrome individuals in a large population-based registry. Am J Med Genet 77:431–438
Verheugt CL, Uiterwaal CS, van der Velde ET, Meijboom FJ, Pieper PG, van Dijk AP, Vliegen HW, Grobbee DE, Mulder BJ (2010) Mortality in adult congenital heart disease. Eur Heart J 31:1220–1229
Williams RG, Pearson GD, Barst RJ, Child JS, del Nido P, Gersony WM, Kuehl KS, Landzberg MJ, Myerson M, Neish SR, Sahn DJ, Verstappen A, Warnes CA, Webb CL (2006) Report of the National Heart, Lung, and Blood Institute Working Group on research in adult congenital heart disease. J Am Coll Cardiol 47:701–707
Williams AD, Mjaatvedt CH, Moore CS (2008) Characterization of the cardiac phenotype in neonatal Ts65Dn mice. Dev Dyn 237:426–435
Yu Y, Bradley A (2001) Engineering chromosomal rearrangements in mice. Nat Rev Genet 2:780–790
Yu YE, Morishima M, Pao A, Wang DY, Wen XY, Baldini A, Bradley A (2006) A deficiency in the region homologous to human 17q21.33-q23.2 causes heart defects in mice. Genetics 173:297–307
Yu T, Li Z, Jia Z, Clapcote SJ, Liu C, Li S, Asrar S, Pao A, Chen R, Fan N, Carattini-Rivera S, Bechard AR, Spring S, Henkelman RM, Stoica G, Matsui S, Nowak NJ, Roder JC, Chen C, Bradley A, Yu YE (2010) A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet 19:2780–2791
Acknowledgments
We thank Zhongyou Li, Jeffrey Conroy and Jeffrey LaDuca for their assistance and Richard DiCioccio and Paula Jones for their helpful suggestions on the manuscript. This study is supported in part by grants to Y.E. Yu from the Children’s Guild Foundation and the NIH (R01HL091519).
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Liu, M. Morishima and T. Yu contributed equally to this work.
An erratum to this article can be found at http://dx.doi.org/10.1007/s00439-011-0982-0
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Liu, C., Morishima, M., Yu, T. et al. Genetic analysis of Down syndrome-associated heart defects in mice. Hum Genet 130, 623–632 (2011). https://doi.org/10.1007/s00439-011-0980-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-0980-2