
Abstract
Genome-wide association studies (GWAS) of body mass index (BMI) using large samples have yielded approximately a dozen robustly associated variants and implicated additional loci. Individually these variants have small effects and in aggregate explain a small proportion of the variance. As a result, replication attempts have limited power to achieve genome-wide significance, even with several thousand subjects. Since there is strong prior evidence for genetic influence on BMI for specific variants, alternative approaches to replication can be applied. Instead of testing individual loci sequentially, a genetic risk sum score (GRSS) summarizing the total number of risk alleles can be tested. In the current study, GRSS comprising 56 top variants catalogued from two large meta-analyses was tested for association with BMI in the Molecular Genetics of Schizophrenia controls (2,653 European-Americans, 973 African-Americans). After accounting for covariates known to influence BMI (ancestry, sex, age), GRSS was highly associated with BMI (p value = 3.19E−06) although explained a limited amount of the variance (0.66%). However, area under receiver operator criteria curve (AUC) estimates indicated that the GRSS and covariates significantly predicted overweight and obesity classification with maximum discriminative ability for predicting class III obesity (AUC = 0.697). The relative contributions of the individual loci to GRSS were examined post hoc and the results were not due to a few highly significant variants, but rather the result of numerous variants of small effect. This study provides evidence of the utility of a GRSS as an alternative approach to replication of common polygenic variation in complex traits.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Obesity is a general medical condition, defined clinically by a body mass index (BMI) greater than 30 kg/m2 and is associated with increased risk of cardiovascular disease, type II diabetes, cancer and poor quality of life (National Center for Health Statistics 2007; Ogden et al. 2007; Kopelman 2000). The National Center for Health Statistics reports over 34% of American adults are obese with another 34% meeting criteria for being overweight (National Center for Health Statistics 2007; Ogden et al. 2006). Although increase in energy intake with reduced physical activity contributes to the increase in obesity, genetic factors have consistently been demonstrated to influence individual differences in BMI, with twin and family studies estimating heritabilities of ~0.70 (Maes et al. 1997; Silventoinen and Kaprio 2009).
Genome-wide association studies (GWAS) have successfully identified polymorphisms that contribute to disease risk for numerous complex traits and diseases (Wellcome Trust Case Control Consortium 2007). GWAS for BMI and obesity using sample sizes in the tens of thousands have yielded many putative risk variants of individually small effect. The first common single nucleotide polymorphisms (SNPs) associated with BMI and common obesity were in the fat mass and obesity-associated (FTO) gene and near melanocortin 4 receptor (MC4R) and have been widely replicated (Frayling et al. 2007; Dina et al. 2007; Herbert et al. 2006; Scuteri et al. 2007; Hinney et al. 2007; Loos et al. 2008; Chambers et al. 2008). Additionally, two large-scale BMI meta-analyses, Thorleifsson et al. (2009) and Willer et al. (2009), yielded 13 genetic loci reaching genome-wide significance, including the previously implicated variants in or near FTO and MC4R. These variants were highly significant but had modest effects with 0.06–0.4 kg/m2 per allele change in BMI and modest obesity (BMI > 30 kg/m2) odds ratios ranging 1.03–1.3. Although many loci are expected to contribute to a complex trait like BMI, the large number implied by the current result was unexpected to many (Yang et al. 2005; Maher 2008). Despite the large sample size (n > 30,000), Willer et al. (2009) estimated 5–10% power to detect genome-wide significant variants with effect sizes of 0.06–0.1 BMI units per allele. Therefore, it is likely that many variants influencing BMI did not reach genome-wide significance in these meta-analyses.
Replication attempts using studies unselected for BMI have limited power to achieve genome-wide significance, even with thousands of subjects (Sebastiani et al. 2009). Since there is strong a priori evidence for genome-wide significant and suggestive variants from the large meta-analyses, alternative approaches to replication can be applied. Instead of testing individual loci sequentially, a genetic risk sum score (GRSS) summarizing the total number of risk alleles can be constructed and tested. The aggregate risk should be significant if a sufficient proportion of the variants have real effects. GRSS have been used to test the total impact of associated variants on complex traits and disease. For example, Aulchenko et al. (2009) used 54 variants in a GRSS which accounted for ~4% of the phenotypic variance in height. Risk scores incorporating 18–20 genome-wide significant variants have been shown to be associated and predictive of type II diabetes, though algorithms including family history and additional risk factors perform better (Meigs et al. 2008; Talmud et al. 2010). GRSS have also been applied to BMI and obesity in populations of European and Chinese descent which incorporated 8–15 variants and accounted for 0.5–1.12% of the phenotypic variance (Thorleifsson et al. 2009; Willer et al. 2009; Renstrm et al. 2009; Zhao et al. 2009; Li et al. 2010; Cheung et al. 2010). Presently, BMI GRSS have only incorporated genome-wide significant variants. However, research by Evans et al.(2009) suggests that in some cases, including bipolar disorder, coronary heart disease, hypertension and type II diabetes, using liberal thresholds (α = 0.5) for SNP selection in GRSS may improve predictive ability.
The purpose of this study was to test a GRSS comprising replicated genome-wide significant variants as well as additional variants with suggestive evidence catalogued from large-scale meta-analyses for association with BMI in 2,653 European-Americans and 973 African-Americans from the Molecular Genetics of Schizophrenia control sample (MGS-C). Based on the expected BMI effect sizes of 0.05–0.3 kg/m2 per allele change in BMI, the MGS-C sample would have limited power to detect genome-wide significant variants for individual loci. However, the aggregate risk should be adequate if a sufficient proportion of the reported variants are real. Therefore, these analyses serve as a replication attempt of top variants catalogued from large-scale meta-analyses via a sum score approach.
Materials and methods
Participants and phenotypes
The MGS-C sample has been previously described in detail (Sanders et al. 2008; Shi et al. 2009; Sanders et al. 2010). In summary, Knowledge Networks, Inc., a survey research company, recruited self-identified non-Hispanic European-American and African-Americans from a nationwide panel of survey participants, which was assembled by random digit dialing except 772 of the African-Americans were recruited through a subcontract to Survey Sampling International by internet banner advertisement recruitment. The institutional review board approval was obtained at NorthShore University HealthSystem and participants completed an online consent with an identical hard-copy consent signed at venipuncture. Participants completed an online questionnaire, available at http://nimhgenetics.org, which included items on height and current weight. BMI was calculated from respondents’ self-reported height and current weight. Participants were removed from data analysis if there were missing data on either height or weight or if calculated BMI was less than 15 or greater than 60 as values not in this range were likely data entry errors. There were 2,653 European-Americans and 973 African-Americans included in the present study. Phenotypic details are displayed in Table 1 with full sample characteristics found in Sanders et al. (2010).
Genotyping
Venipuncture for DNA extraction and establishment of lymphoblastoid cell lines was completed at Rutgers University Cell and DNA Repository. DNA samples were genotyped using the Affymetrix 6.0 array at the Broad Institute. There were 3,827 participants genotyped (n = 2,817 European-American, n = 1,010 African-American) of which 3,626 (95%) passed stringent quality control criteria. Principal component (PC) scores reflecting continental and within-Europe ancestries of each subject were computed and outliers were excluded. Genomic control λ values for autosomes after quality control procedures were 1.005 for African-American and 0.998 for the European-Americans.
Selection of 56 SNPs
Preliminary SNP selection identified 78 variants meeting criteria for genome-wide or suggestive significance in either of two large meta-analyses of BMI, 43 from Thorleifsson et al. (2009) and 35 from Willer et al. (Thorleifsson et al. 2009; Willer et al. 2009). Thorleifsson and colleagues report genome-wide significant (p < 1.6E−07) associations with 29 SNPs in 11 chromosomal regions, using a discovery sample of n = 34,416 and replication sample of n = 5,586. The Willer et al. meta-analysis detected 8 genome-wide significant (p < 5.0E−08) SNPs in first- and second-stage samples of n = 32,387 and n = 54,316, respectively. Only variants in or near FTO and MC4R were found to be genome-wide significant in both meta-analyses. The remaining genetic loci were suggestive in the opposing meta-analyses (p < 0.05) except rs7138803 on 12q13 (p = 0.14). Significance level for one SNP, rs10938397 on 4p12, could not be compared between meta-analyses because there was no corresponding proxy SNP. Of the 78 variants catalogued, 29 had matching SNPs on the Affymetrix 6.0 array. For the 49 SNPs not present, proxies (45 r 2 > 0.8; 4 r 2 > 0.7) were identified using SNP Annotation and Proxy Search V2.1 (Johnson et al. 2008). Following removal of seven duplicate proxies and six variants from Willer et al. for which no proxies were available (r 2 > 0.7), 65 SNPs remained. Haploview version 4.10 was used to determine phase and corresponding proxy alleles (Barrett et al. 2005; Barrett 2009). In order to avoid bias due to correlated effects, SNP pruning (r 2 > 0.8) was performed using PLINK v. 1.07p (Purcell et al. 2007). Of the 56 remaining SNPs, 19 met genome-wide significance criteria in the two meta-analyses. The additional 37 were included as they were the next top SNPs reported (p < 0.05). Although our SNP selection threshold was more liberal than the traditional genome-wide significance threshold, it was more conservative than other models of complex disease risk prediction (Evans et al. 2009; Purcell et al. 2009). Supplemental Table 1 details information on the 78 catalogued SNPs.
Genetic risk sum score
Under an additive model, 56 variants were used to construct the GRSS. The use of an additive model was chosen as specific non-additive effects have yet to be associated and confirmed in the literature. The GRSS was calculated by summation of the number of risk alleles across the 56 variants divided by the number of SNPs in the score to obtain an average number of risk alleles per locus. GRSS were calculated using the profile option in PLINK. If SNP information was missing in an individual then the scoring routine imputed expected values based on sample allele frequency. R version 2.20.0 was used to fit linear regression models using standard covariates and GRSS as predictors with BMI as the outcome variable. To facilitate interpretation of effects in linear models independent variables were centered.
Prediction of obesity
One method to assess diagnostic efficiency is to graph a receiver operator criteria (ROC) curve, which is a plot of the true positive rate (sensitivity) against the false positive rate (1 − specificity) and calculate the corresponding area under the curve (AUC). An AUC may range from 0.5, non-informative, to a maximum of 1.0, perfect discrimination between cases and controls. An AUC is the probability that the predictor is greater for cases than controls (Armitage and Colton 2005; Hanley and McNeil 1982). Generally, an AUC of 0.80 is suitable for screening while 0.99 is acceptable for diagnosis (Janssens et al. 2006). To test various BMI thresholds, current BMI was dichotomized to create categories of overweight and obesity class I, II and III which had corresponding ranges of BMI > 25, 30, 35 and 40 kg/m2, respectively. Binary logistic regression was used to calculate predicted probabilities of the models and was used as the predictor to generate ROC curves. Discriminative accuracy of the GRSS and covariates (molecularly derived ancestry, sex, age, ancestry by sex interactions) to predict BMI category was estimated by calculating the AUC from ROC curves using PASW Statistics version 17.0.
Results
Phenotypic detail
Descriptive statistics for age and BMI are presented by race and sex in Table 1. The mean age of participants was 48.8 and ranged from 18 to 90 and as depicted in Supplemental Fig. 1 produced a relatively normal distribution. BMI was not significantly associated with age (p = 0.135; Supplemental Fig. 2). Males were significantly older than females and European-American females and males were significantly older than African-American females and males (p < 0.0001). When partitioning the sample by clinically established BMI (kg/m2) categories, 29.0% was either under or normal weight (BMI < 25), 33.4% was overweight (25 ≤ BMI < 30), 20.4% was obese class 1 (30 ≤ BMI < 35), 9.5% was obese class II (35 ≤ BMI < 40) and 7.7% was obese class III (40 ≤ BMI). There was a significant ancestry by sex interaction with BMI. As expected, females had significantly greater BMI than males with African-American females having greater BMI than European-American females and African-American males having greater BMI than European-American males (p < 0.0001). Phenotypic findings in the MGS-C sample are consistent with cross-sectional data from the National Center for Health Statistics and National Health and Nutrition Examination Study (Wang et al. 2008), finding obesity more prevalent in women and African-Americans. Additional sample characteristics have been previously reported (Sanders et al. 2010).
Genetic risk sum score
Fifty-six variants catalogued from two large-scale BMI meta-analyses were used to construct the GRSS (Thorleifsson et al. 2009; Willer et al. 2009). These variants were summarized in the GRSS which was calculated by summation of the number of risk alleles across the SNPs for each individual divided by the number of SNPs in the score to achieve an average allele count. The frequencies of GRSSs are shown in Fig. 1 and produced a relatively normal distribution. The mean GRSS, or average number of risk alleles present per locus, was 0.494 (SD = 0.052) with a range from 0.318 to 0.691 which corresponds to an average of 55 risk alleles per person with a range from 35 to 77.

Frequencies of genetic risk sum score
Results from linear regression analyses are presented in Table 2. Standard covariates known to influence BMI (ancestry, sex and age) were included in the models. Described previously (Sanders et al. 2008; Shi et al. 2009), 224 ancestry informative markers were used to construct 10 PC scores designed to discriminate between European, African, Ameri-Indian and Asian ancestry. PC1 (distinguishes European from African ancestry) and PC4 (distinguishes Eastern and Western European ancestry) were significantly associated with BMI and therefore included as covariates. Interactions between the covariates were tested and significant interactions were found between PC1 and sex and PC4 and sex. No other interactions between the covariates were significant. Model 1, the base model, included the standard covariates and the significant interactions between ancestry PCs and sex and accounted for 3.5% of the variance in BMI. Model 2, which added the GRSS to the base model, fits significantly better [F (1,3027) = 21.8, p = 3.2E−06] and accounted for an additional 0.66% of phenotypic variance in BMI for a total of 4.1%. We note that the GRSS accounted for more of the variance in BMI than either sex or age. Interactions between the covariates and the GRSS were tested but no significant interactions were found (presented in Supplemental Table 2). Therefore, our results suggest that GRSS was equally associated with BMI in men and women, in European- and African-Americans and across all ages.
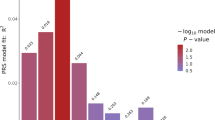
The relative contributions of the individual loci to the GRSS were examined post hoc by dropping the most significantly associated SNP from the score iteratively until the score was no longer statistically associated with BMI. As depicted in Fig. 2, the GRSS reached non-significance after dropping the top 23 variants.

Number of SNPs in genetic risk sum score by −log significance of score. GRSS genetic risk sum score, −log negative logarithm base 10, SNPs single nucleotide polymorphisms
Prediction of obesity
To test the discriminative accuracy of the GRSS and covariates (molecularly derived ancestry, sex, age, ancestry by sex interactions) to predict obesity, ROC curves were plotted and the corresponding AUC were calculated. To test various BMI thresholds, current BMI was dichotomized to create categories of overweight and obesity class I, II and III. Table 3 displays statistics from ROC curve analysis by BMI category. AUC estimates indicated that the model significantly predicted overweight and obesity classification with maximum discriminating ability when predicting class III obesity (AUC = 0.697, 95% CI = [0.663, 0.731]). We note that the clinical setting may prefer to use self-identified ancestry as opposed to molecularly derived ancestry in risk prediction because of genotyping cost. In the MGS-C data, the use of self-identified ancestry did not greatly change AUC estimates. For example, when predicting BMI > 30 kg/m2, an AUC = 0.588 was reported when using molecularly derived ancestry versus an AUC = 0.586 when using self-identified ancestry in the model (full data not shown).
Discussion
In this paper, we have constructed a GRSS comprising 56 common polygenic variants and shown its association with BMI in 2,653 European-Americans and 973 African-Americans from the MGS-C sample. The GRSS was highly associated with BMI (p value = 3.19E−06) and accounted for 0.66% of phenotypic variance in BMI. The association of the GRSS with BMI was comparable to sex, a known factor to influence body composition. The average effect of carrying 10 risk variants was an increase in BMI of 1.1 kg/m2. This corresponds to a weight increase in an average male (5 feet 9 inches, 180 lb) of 8 lb and an average female (5 feet 4 inches, 155 lb) of 7 lb. Further, we have shown the association of the GRSS with BMI was not the result of the few most significant SNPs but rather the aggregate of many SNPs of small effect. These results are consistent with the common disease common variants hypothesis indicating genetic variants common in the population with small effects contribute to the heritability of common traits and diseases.
ROC curves and the corresponding AUC estimates indicated statistical discriminative ability to predict obesity (BMI > 30 kg/m2, AUC = 0.588, 95% CI = [0.567, 0.610]). AUC estimates were similar to those found in previous studies. For example, Renstrom et al. (2009) used a genetic score of 9 SNPs and reported an AUC estimate of 0.575 in a sample of 353 obese and 1,370 normal weight diabetic and non-diabetic northern Swedes. Additionally, a study by Cheung et al. (2010) estimated an AUC of 0.582 with a genetic score including 13 SNPs in a Chinese sample of 470 obese cases and 700 normal weight controls. Although these AUC estimates were statistically significant, they were below 0.8, the threshold used in clinical practice for screening. In the MGS-C sample, however, the ability to predict morbid obesity (class III) was notably better and approached clinical criteria for a screening test (AUC = 0.697, 95% CI = [0.663, 0.731]).
In the MGS-C sample, 4.1% of the phenotypic variance in BMI was accounted for using a model including sex, ancestry based on molecular derived principal components, age, and a GRSS comprising 56 SNPs. Despite high heritability of BMI, much variance in BMI remains unaccounted for. Based on the progress in identifying loci influencing height, it is likely that a considerable portion of the ‘missing heritability’ resides in unidentified variants yet to be discovered by larger sample sizes (Yang et al. 2010). Large-scale international collaborative groups will be required to identify these additional variants with similar and smaller effect sizes.
Additionally, predictive models have yet to include other sources of variation known or hypothesized to influence BMI such as rare variants, gene–gene (G×G) or gene–environment (G×E) interactions, copy number variation, and epigenetic effects. For instance, rare variants which were not included in the current genetic risk profiles are likely to contribute to BMI heritability. For example, in a study by Blakemore et al. (2009), a rare variant in the visfatin gene was associated (p value = 8.0E−5, minor allele frequency 1.6% in control and 0.4% in obese subjects) with reduced risk for obesity. There is also evidence to support the influence of copy number variation (CNV) on BMI. In the Willer et al. (2009) meta-analysis, when examining CNV by SNP-CNV linkage disequilibrium, they found 10-kb and 45-kb deletion polymorphisms upstream of NEGR1 with the 45-kb deletion flanked by their two most associated BMI SNPs. The recent advent of SNP arrays designed for CNV detection may reveal additional genetic associations with BMI. Epigenetic variation, although more widely researched in syndromic obesity such as Prader–Willi, may also be linked to common obesity. Finally, G×G interactions have yet to be included in risk prediction of body composition. Twin studies support the role of non-additive genetic effects although most study designs have limited ability to detect them (Maes et al. 1997; Flint and Mackay 2009).
Since obesity has increased dramatically while the genome has arguably remained stable, future research needs to address moderation effects of the environment. Known obesogenic factors such as physical activity and food intake have been shown to account for a significant portion of the variance in BMI with estimates ranging 5–10% (Chambers and Swanson 2010; Newby et al. 2006; French et al. 1994; Jebb and Moore 1999). Additionally, research is beginning to elucidate G×E affecting BMI (Rampersaud et al. 2008; Lappalainen et al. 2009; Qi et al. 2008; Brandsttter et al. 2009; Razquin et al. 2010). At least two genes included in the current GRSS show evidence for G×E effects. For example, Rampersaud et al. (2008), in a study of 704 Old Order Amish, found the effects of FTO variants associated with elevated body weight were attenuated in subjects with higher physical activity levels. Additionally, interactions between MCR4 and dietary intake and selection have been shown in model organisms (Fan et al. 1997; Huszar et al. 1997; Marsh et al. 1999; Chen et al. 2000). For example, mice when given a 3-choice diet and administered a melanocortin agonist preferentially decreased fat consumption (Samama et al. 2003). Further, variation in human MCR4 has been associated with binge eating (Branson et al. 2003; Potoczna et al. 2004; Valladares et al. 2010) and with higher total energy intake and selection of foods high in dietary fat (Qi et al. 2008). BMI prediction models will benefit from incorporating known obesogenic environmental variables such as physical activity and food selection and intake.
The purpose of this study was to test a GRSS as an alternative approach to replication of association of common polygenic variation with BMI. As hypothesized the MGS-C sample had limited power to replicate individual loci when employing genome-wide significant thresholds even though there was strong a priori evidence of these variants to influence BMI. However, by constructing a GRSS summarizing the total number of risk alleles, the aggregate risk was found to be highly significantly associated with BMI. This study provides evidence of the utility of GRSS as an alternative approach to replication of common polygenic variation in complex traits. Furthermore, the results from the AUC analysis demonstrate meaningful progress towards a screening test that perhaps if used in conjunction with known obesogenic predictors such as physical activity and food selection and intake could identify persons for early environmental or medical intervention to prevent morbid obesity and the associated negative health consequences.
Abbreviations
- GWAS:
-
Genome-wide association study
- BMI:
-
Body mass index
- GRSS:
-
Genetic risk sum score
- AUC:
-
Area under the curve
- FTO:
-
Fat mass and obesity-associated gene
- MC4R:
-
Melanocortin 4 receptor
- kg:
-
Kilogram
- m:
-
Meter
- MGS-C:
-
Molecular Genetics of Schizophrenia control sample
- PC:
-
Principal component
- ROC:
-
Receiver operator criteria
- SNP:
-
Single nucleotide polymorphism
- CNV:
-
Copy number variation
- AA:
-
African-American
- EA:
-
European-American
References
Armitage P, Colton T (2005) Encyclopedia of biostatistics. Wiley, Chichester
Aulchenko YS, Struchalin MV, Belonogova NM, Axenovich TI, Weedon MN, Hofman A et al (2009) Predicting human height by victorian and genomic methods. Eur J Hum Genet 17(8):1070–1075
Barrett J (2009) Haploview: visualization and analysis of SNP genotype data. Cold Spring Harbor protocols 2009, pdb.ip71
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263
Blakemore AIF, Meyre D, Delplanque J, Vatin V, Lecoeur C, Marre M et al (2009) A rare variant in the visfatin gene (NAMPT/PBEF1) is associated with protection from obesity. Obesity 17(8):1549
Brandsttter A, Lingenhel A, Zwiauer K, Strobl W, Kronenberg F (2009) Decrease of Lp(a) during weight reduction in obese children is modified by the apo(a) kringle-IV copy number variation. Int J Obes 33:1136
Branson R, Potoczna N, Kral J, Lentes K, Hoehe M, Horber F (2003) Binge eating as a major phenotype of melanocortin 4 receptor gene mutations. N Engl J Med 348:1096–1103
Chambers J, Swanson V (2010) A health assessment tool for multiple risk factors for obesity: age and sex differences in the prediction of body mass index. Br J Nutr 104:298–307
Chambers J, Elliott P, Zabaneh D, Zhang W, Li Y, Froguel P, Balding D, Scott J, Kooner J (2008) Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat Genet 40:716
Chen AS, Metzger JM, Trumbauer ME, Guan XM, Yu H, Frazier EG, Marsh DJ, Forrest MJ, Gopal-Truter S, Fisher J, Camacho RE, Strack AM, Mellin TN, MacIntyre DE, Chen HY, Van der Ploeg LH (2000) Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res 9:145–154
Cheung CY, Tso AW, Cheung BM, Xu A, Ong KL, Fong CH, Wat NM, Janus ED, Sham PC, Lam KS (2010) Obesity susceptibility genetic variants identified from recent genome-wide association studies: implications in a Chinese population. J Clin Endocrinol Metab. doi:10.1210/jc.2009-1465
Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, Carlsson LM, Kiess W, Vatin V, Lecoeur C, Delplanque J, Vaillant E, Pattou F, Ruiz J, Weill J, Levy-Marchal C, Horber F, Potoczna N, Hercberg S, Le Stunff C, Bougneres P, Kovacs P, Marre M, Balkau B, Cauchi S, Chevre JC, Froguel P (2007) Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet 39:724–726. doi:10.1038/ng2048
Evans D, Visscher P, Wray N (2009) Harnessing the information contained within genome-wide association studies to improve individual prediction of complex disease risk. Hum Mol Genet 18:3525
Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD (1997) Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385:165–168
Flint J, Mackay TFC (2009) Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res 19:723–733
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJ, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894. doi:10.1126/science.1141634
French SA, Jeffery RW, Forster JL, McGovern PG, Kelder SH, Baxter JE (1994) Predictors of weight change over two years among a population of working adults: the Healthy Worker Project. Int J Obes 18:145–154
Hanley JA, McNeil BJ (1982) The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143:29–36
Herbert A, Gerry N, McQueen M, Heid I, Pfeufer A, Illig T, Wichmann H, Meitinger T, Hunter D, Hu F, Colditz G, Hinney A, Hebebrand J, Koberwitz K, Zhu X, Cooper R, Ardlie K, Lyon H, Hirschhorn J, Laird N, Lenburg M, Lange C, Christman M (2006) A common genetic variant is associated with adult and childhood obesity. Science 312:279
Hinney A, Nguyen T, Scherag A, Friedel S, Brönner G, Müller T, Grallert H, Illig T, Wichmann H, Rief W, Schäfer H, Hebebrand J (2007) Genome wide association (GWA) study for early onset extreme obesity supports the role of fat mass and obesity associated gene (FTO) variants. PloS ONE 2:e1361
Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F (1997) Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88:131–141
Janssens ACJW, Aulchenko Y, Elefante S, Borsboom GJJM, Steyerberg E, van Duijn C (2006) Predictive testing for complex diseases using multiple genes: fact or fiction? Genetics Med 8:395–400
Jebb SA, Moore MS (1999) Contribution of a sedentary lifestyle and inactivity to the etiology of overweight and obesity: current evidence and research issues. Med Sci Sports Exerc 31:S534–S541
Johnson A, Handsaker R, Pulit S, Nizzari M, O’Donnell C, de Bakker PIW (2008) SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24:2938–2939
Kopelman PG (2000) Obesity as a medical problem. Nature 404:635
Lappalainen T, Tolppanen A, Kolehmainen M, Schwab U, Lindstrm J, Tuomilehto J, Pulkkinen L, Eriksson J, Laakso M, Gylling H, Uusitupa M (2009) The common variant in the FTO gene did not modify the effect of lifestyle changes on body weight: the Finnish Diabetes Prevention Study. Obesity 17:832
Li S, Zhao J, Luan J, Luben R, Rodwell S, Khaw K, Ong K, Wareham N, Loos RJF (2010) Cumulative effects and predictive value of common obesity-susceptibility variants identified by genome-wide association studies. Am J Clin Nutr 91:184
Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, Inouye M, Freathy RM, Attwood AP (2008) Beckmann JS, Berndt SI, Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, Jacobs KB, Chanock SJ, Hayes RB, Bergmann S, Bennett AJ, Bingham SA, Bochud M, Brown M, Cauchi S, Connell JM, Cooper C, Smith GD, Day I, Dina C, De S, Dermitzakis ET, Doney AS, Elliott KS, Elliott P, Evans DM, Sadaf Farooqi I, Froguel P, Ghori J, Groves CJ, Gwilliam R, Hadley D, Hall AS, Hattersley AT, Hebebrand J, Heid IM, KORA, Lamina C, Gieger C, Illig T, Meitinger T, Wichmann HE, Herrera B, Hinney A, Hunt SE, Jarvelin MR, Johnson T, Jolley JD, Karpe F, Keniry A, Khaw KT, Luben RN, Mangino M, Marchini J, McArdle WL, McGinnis R, Meyre D, Munroe PB, Morris AD, Ness AR, Neville MJ, Nica AC, Ong KK, O’Rahilly S, Owen KR, Palmer CN, Papadakis K, Potter S, Pouta A, Qi L, Nurses’ Health Study, Randall JC, Rayner NW, Ring SM, Sandhu MS, Scherag A, Sims MA, Song K, Soranzo N, Speliotes EK, Diabetes Genetics Initiative, Syddall HE, Teichmann SA, Timpson NJ, Tobias JH, Uda M, SardiNIA Study, Vogel CI, Wallace C, Waterworth DM, Weedon MN, Wellcome Trust Case Control Consortium, Willer CJ, FUSION, Wraight, Yuan X, Zeggini E, Hirschhorn JN, Strachan DP, Ouwehand WH, Caulfield MJ, Samani NJ, Frayling TM, Vollenweider P, Waeber G, Mooser V, Deloukas P, McCarthy MI, Wareham NJ, Barroso I, Jacobs KB, Chanock SJ, Hayes RB, Lamina C, Gieger C, Illig T, Meitinger T, Wichmann HE, Kraft P, Hankinson SE, Hunter DJ, Hu FB, Lyon HN, Voight BF, Ridderstrale M, Groop L, Scheet P, Sanna S, Abecasis GR, Albai G, Nagaraja R, Schlessinger D, Jackson AU, Tuomilehto J, Collins FS, Boehnke M, Mohlke KL (2008) Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet 40:768-775. doi: 10.1038/ng.140
Maes HH, Neale MC, Eaves LJ (1997) Genetic and environmental factors in relative body weight and human adiposity. Behav Genet 27:325–351
Maher B (2008) Personal genomes: the case of the missing heritability. Nature 456:18–21
Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD (1999) Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet 21:119–122
Meigs J, Shrader P, Sullivan L, McAteer J, Fox C, Dupuis J, Manning A, Florez J, Wilson PWF, D’Agostino R, Cupples LA (2008) Genotype score in addition to common risk factors for prediction of type 2 diabetes. New Engl J Med 359:2208
National Center for Health Statistics NHANES (2007) Data sets and related documentation. http://www.cdc.gov
Newby PK, Weismayer C, Akesson A, Tucker K, Wolk A (2006) Longitudinal changes in food patterns predict changes in weight and body mass index and the effects are greatest in obese women. J Nutr 136:2580–2587
Ogden C, Carroll M, Curtin L, McDowell M, Tabak C, Flegal K (2006) Prevalence of overweight and obesity in the United States, 1999–2004. JAMA 295:1549
Ogden CL, Yanovski SZ, Carroll MD, Flegal KM (2007) The epidemiology of obesity. Gastroenterology 132:2087–2102. doi:10.1053/j.gastro.2007.03.052
Potoczna N, Branson R, Kral J, Piec G, Steffen R, Ricklin T, Hoehe M, Lentes K, Horber F (2004) Gene variants and binge eating as predictors of comorbidity and outcome of treatment in severe obesity. J Gastrointest Surg 8:971–981
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly M, Sham P (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559
Purcell S, Wray N, Stone J, Visscher P, O’Donovan M, Sullivan P, Sklar P (2009) Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460:748
Qi L, Kraft P, Hunter D, Hu F (2008) The common obesity variant near MC4R gene is associated with higher intakes of total energy and dietary fat, weight change and diabetes risk in women. Hum Mol Genet 17:3502
Rampersaud E, Mitchell B, Pollin T, Fu M, Shen H, O’Connell J, Ducharme J, Hines S, Sack P, Naglieri R, Shuldiner A, Snitker S (2008) Physical activity and the association of common FTO gene variants with body mass index and obesity. Arch Intern Med 168:1791
Razquin C, Martinez JA, Martinez-Gonzalez MA, Bes-Rastrollo M, Fernndez-Crehuet J, Marti A (2010) A 3-year intervention with a Mediterranean diet modified the association between the rs9939609 gene variant in FTO and body weight changes. Int J Obes 34:266
Renstrm F, Payne F, Nordstrm A, Brito E, Rolandsson O, Hallmans G, Barroso I, Nordstrm P, Franks P (2009) Replication and extension of genome-wide association study results for obesity in 4923 adults from northern Sweden. Hum Mol Genet 18:1489
Samama P, Rumennik L, Grippo J (2003) The melanocortin receptor MCR4 controls fat consumption. Regul Pept 113:85–88
Sanders A, Duan J, Levinson D, Shi J, He D, Hou C, Burrell G, Rice J, Nertney D, Olincy A, Rozic P, Vinogradov S, Buccola N, Mowry B, Freedman R, Amin F, Black D, Silverman J, Byerley W, Crowe R, Cloninger CR, Martinez M, Gejman P (2008) No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: implications for psychiatric genetics. Am J Psychiatry 165:497–506
Sanders A, Levinson D, Duan J, Dennis JM, Li R, Kendler K, Rice J, Shi J, Mowry B, Amin F, Silverman J, Buccola N, Byerley W, Black D, Freedman R, Cloninger CR, Gejman P (2010) The Internet-based MGS2 control sample: self report of mental illness. Am J Psychiatry 167:854–865
Scuteri A, Sanna S, Chen W, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orr M, Usala G, Dei M, Lai S, Maschio A, Busonero F, Mulas A, Ehret G, Fink A, Weder A, Cooper R, Galan P, Chakravarti A, Schlessinger D, Cao A, Lakatta E, Abecasis G (2007) Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genetics 3:e115
Sebastiani P, Timofeev N, Dworkis D, Perls T, Steinberg M (2009) Genome-wide association studies and the genetic dissection of complex traits. Am J Hematol 84:504–515
Shi J, Levinson D, Duan J, Sanders A, Zheng Y, Pe’er I, Dudbridge F, Holmans P, Whittemore A, Mowry B, Olincy A, Amin F, Cloninger CR, Silverman J, Buccola N, Byerley W, Black D, Crowe R, Oksenberg J, Mirel D, Kendler K, Freedman R, Gejman P (2009) Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460:753–757
Silventoinen K, Kaprio J (2009) Genetics of tracking of body mass index from birth to late middle age: evidence from twin and family studies. Obes Facts 2:196
Talmud P, Hingorani A, Cooper J, Marmot M, Brunner E, Kumari M, Kivimki M, Humphries S (2010) Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. BMJ (Clinical research ed.) 340:b4838
Thorleifsson G, Walters GB, Gudbjartsson D, Steinthorsdottir V, Sulem P, Helgadottir A, Styrkarsdottir U, Gretarsdottir S, Thorlacius S, Jonsdottir I, Jonsdottir T, Olafsdottir E, Olafsdottir G, Jonsson T, Jonsson F, Borch-Johnsen K, Hansen T, Andersen G, Jorgensen T, Lauritzen T, Aben K, Verbeek ALM, Roeleveld N, Kampman E, Yanek L, Becker L, Tryggvadottir L, Rafnar T, Becker D, Gulcher J, Kiemeney L, Pedersen O, Kong A, Thorsteinsdottir U, Stefansson K (2009) Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet 41:18
Valladares M, Domnguez-Vsquez P, Obregn AM, Weisstaub G, Burrows R, Maiz A, Santos JL (2010) Melanocortin-4 receptor gene variants in Chilean families: association with childhood obesity and eating behavior. Nutr Neurosci 13:71–78
Wang Y, Beydoun M, Liang L, Caballero B, Kumanyika S (2008) Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity 16:2323–2330
Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678. doi:10.1038/nature05911
Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, Berndt SI, Elliott AL, Jackson AU, Lamina C, Lettre G, Lim N, Lyon HN, McCarroll SA, Papadakis K, Qi L, Randall JC, Roccasecca RM, Sanna S, Scheet P, Weedon MN, Wheeler E, Zhao JH, Jacobs LC, Prokopenko I, Soranzo N, Tanaka T, Timpson NJ, Almgren P, Bennett A, Bergman RN, Bingham SA, Bonnycastle LL, Brown M, Burtt NP, Chines P, Coin L, Collins FS, Connell JM, Cooper C, Smith GD, Dennison EM, Deodhar P, Elliott P, Erdos MR, Estrada K, Evans DM, Gianniny L, Gieger C, Gillson CJ, Guiducci C, Hackett R, Hadley D, Hall AS, Havulinna AS, Hebebrand J, Hofman A, Isomaa B, Jacobs KB, Johnson T, Jousilahti P, Jovanovic Z, Khaw KT, Kraft P, Kuokkanen M, Kuusisto J, Laitinen J, Lakatta EG, Luan J, Luben RN, Mangino M, McArdle WL, Meitinger T, Mulas A, Munroe PB, Narisu N, Ness AR, Northstone K, O’Rahilly S, Purmann C, Rees MG, Ridderstrale M, Ring SM, Rivadeneira F, Ruokonen A, Sandhu MS, Saramies J, Scott LJ, Scuteri A, Silander K, Sims MA, Song K, Stephens J, Stevens S, Stringham HM, Tung YC, Valle TT, Van Duijn CM, Vimaleswaran KS, Vollenweider P, Waeber G, Wallace C, Watanabe RM, Waterworth DM, Watkins N, Wellcome Trust Case Control Consortium, Witteman JC, Zeggini E, Zhai G, Zillikens MC, Altshuler D, Caulfield MJ, Chanock SJ, Farooqi IS, Ferrucci L, Guralnik JM, Hattersley AT, Hu FB, Jarvelin MR, Laakso M, Mooser V, Ong KK, Ouwehand WH, Salomaa V, Samani NJ, Spector TD, Tuomi T, Tuomilehto J, Uda M, Uitterlinden AG, Wareham NJ, Deloukas P, Frayling TM, Groop LC, Hayes RB, Hunter DJ, Mohlke KL, Peltonen L, Schlessinger D, Strachan DP, Wichmann HE, McCarthy MI, Boehnke M, Barroso I, Abecasis GR, Hirschhorn JN, Genetic Investigation of ANthropometric Traits Consortium (2009) Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 41:25–34. doi:10.1038/ng.287
Yang Q, Khoury M, Friedman J, Little J, Flanders WD (2005) How many genes underlie the occurrence of common complex diseases in the population? Int J Epidemiol 34:1129–1137
Yang J, Benyamin B, McEvoy B, Gordon S, Henders A, Nyholt D, Madden P, Heath A, Martin N, Montgomery G, Goddard M, Visscher P (2010) Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42(7):565–569
Zhao J, Bradfield J, Li M, Wang K, Zhang H, Kim C, Annaiah K, Glessner J, Thomas K, Garris M, Frackelton E, Otieno FG, Shaner J, Smith R, Chiavacci R, Berkowitz R, Hakonarson H, Grant SFA (2009) The role of obesity-associated loci identified in genome-wide association studies in the determination of pediatric BMI. Obesity 17:2254
Acknowledgments
This work was supported by National Institute of Health [DA26119 to R.E.P. and H.H.M., DA018673, DA025109 to H.H.M.]. Data and biomaterials from the National Institute of Mental Health Genetics Initiative MGS-C sample were collected by NorthShore University HealthSystem, Evanston, IL, R01 MH59571, Pablo V. Gejman, MD (Collaboration Coordinator; PI) as part of a collaborative R01 application comprising ten sites. From 2003 to 2006, the principal investigators and co-investigators were: NorthShore University HealthSystem, Evanston, IL, R01 MH59571, Pablo V. Gejman, MD (Collaboration Coordinator; PI); Alan R. Sanders, MD, Emory University School of Medicine, Atlanta, GA, R01 MH59587; Farooq Amin, MD (PI), University of California, San Francisco, CA, R01 MH60870; William F. Byerley, MD (PI), University of Iowa, Iowa, IA, R01 MH59566; Donald W. Black, MD (PI); Raymond R. Crowe, MD, Washington University, St. Louis, MO, R01 MH60879; C. Robert Cloninger, MD (PI), University of Colorado, Denver, CO, R01 MH59565, Robert Freedman, MD (PI); Ann Olincy, MD, Stanford University, Palo Alto, CA, R01 MH61675; Douglas F. Levinson, MD (PI), Louisiana State University, New Orleans, LA, R01 MH67257; Nancy G. Buccola, APRN, BC, MSN (PI), University of Queensland, Brisbane, Queensland, Australia, R01 MH59588; Bryan J. Mowry, MD (PI), Mt. Sinai School of Medicine, New York, NY, R01 MH59586; Jeremy M. Silverman, PhD (PI). Our schizophrenia GWAS (genotyping and analyses) was supported by the Genetic Association Information Network (GAIN) and grants U01MH79469 (Pablo V. Gejman, MD), U01MH79470 (Douglas F. Levinson, MD), and R01MH81800 U01MH79469 (Pablo V. Gejman, MD). We thank the individuals who volunteered for the control sample for their participation. Special thanks to Charles O. Gardner for statistical consultation, Brien P. Riley for insightful comments on the manuscript and T. Bernard Bigdeli for recommendations on Fig. 2.
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
439_2010_917_MOESM3_ESM.xls
Supplemental Table 1 Displays the 78 SNPs catalogued, alleles, frequencies, proxy and association information (XLS 58 kb)
Rights and permissions
About this article
Cite this article
Peterson, R.E., Maes, H.H., Holmans, P. et al. Genetic risk sum score comprised of common polygenic variation is associated with body mass index. Hum Genet 129, 221–230 (2011). https://doi.org/10.1007/s00439-010-0917-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-010-0917-1