
Abstract
In vitro studies have shown that p53 mediates a protective response against DNA damage by causing either cell-cycle arrest and DNA repair, or apoptosis. These responses have not yet been demonstrated in humans. A common source of DNA damage in humans is cigarette smoke, which should activate p53 repair mechanisms. As the level of p53 is regulated by MDM2, which targets p53 for degradation, the G-allele of a polymorphism in intron 1 of MDM2 (rs2279744:G/T), that results in higher MDM2 levels, should be associated with a reduced p53 response and hence more DNA damage and corresponding tissue destruction. Similarly, the alleles of rs1042522 in TP53 that encode arginine (G-allele) or proline (C-allele) at codon 72, which cause increased pro-apoptotic (G-allele) or cell-cycle arrest activities (C-allele), respectively, may moderate p53’s ability to prevent DNA damage. To test these hypotheses, we examined lung function in relation to cumulative history of smoking in a population-based cohort. The G-alleles in MDM2 and TP53 were found to be associated with accelerated smoking-related decline in lung function. These data support the hypothesis that p53 protects from DNA damage in humans and provides a potential explanation for the variation in lung function impairment amongst smokers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The protein product of the p53 gene (TP53 in humans) is important for preventing cancers (Malkin et al. 1990; Donehower et al. 1992; Lang et al. 2004), although precisely how it does so is not entirely clear. p53 protein is a transcription factor that responds to many different cellular stresses, including DNA damage (Braithwaite and Prives 2006; Vousden and Lu 2002). p53 is present at very low concentrations in normal cells, but upon stress, p53 concentration increases several fold due to phosphorylation, which prevents it from being targeted for degradation by the E3 ubiquitin ligase, MDM2 (Bond et al. 2005; Momand et al. 1992). Once stabilised, p53 can transactivate several downstream genes which leads to (i) induction of apoptosis to eliminate DNA damaged cells, (ii) transient cell-cycle arrest allowing DNA repair to occur, or (iii) permanent cell-cycle arrest (senescence) (Braithwaite and Prives 2006; Vousden and Lu 2002). Thus, in these ways, p53 protects the organism from exposure to environmental insults, including DNA damage. These protective responses of p53 are all likely to contribute to tumour suppression to a greater or lesser extent, depending on tissue type and the nature of the stress.
In humans, TP53 and MDM2 exist as polymorphic variants (Murphy 2006). The G-allele of a polymorphism in intron 1 of MDM2 (rs2279744:G/T) is known to result in increased levels of MDM2 due to the creation of a binding site for the common transcription factor Sp1 (Bond et al. 2004). This leads to reduced p53 protein levels and an attenuated p53 response. In some cases, individuals homozygous for this allele are more tumour prone (Bond et al. 2005). In TP53, the alleles of rs1042522 that encode arginine (G-allele) or proline (C-allele) at codon 72, are thought to influence the nature of the biological response of p53 to stress (Chipuk et al. 2004; Dumont et al. 2003; Leu et al. 2004; Pim and Banks 2004; Thomas et al. 1999; Yu et al. 2000). p53 protein harbouring the arginine residue (p53R72) has been reported to strongly interact with MDM2, resulting in enhanced nuclear export and association with the mitochondria. This appears to promote the non-transcriptional apoptotic activity of p53. In contrast, there are data suggesting that p53P72 is more likely to induce cell-cycle arrest in response to DNA damage. Consistent with these polymorphisms conferring different biological activities on p53, their allele frequencies vary with both ethnicity and latitude (Beckman et al. 1994).
Although the ability of p53 to respond to environmental stresses in cell culture and in animal models is well-documented (Braithwaite and Prives 2006; Vousden and Lu 2002), there are few data demonstrating that this occurs in humans. A common environmental source of DNA damage in humans is cigarette smoke which contains many mutagenic compounds. If p53 is protecting cells from DNA damage caused by exposure to mutagens, such as those in cigarette smoke, the degree of protection should vary according to the strength and nature of the p53 response. Thus individuals with the G-allele of the MDM2 polymorphism (rs2279744:G/T) with a weaker p53 response, may be less able to deal with exposure to cigarette smoke than individuals with the C-allele. Similarly, the degree of protection from exposure to cigarette smoke will vary with the biological nature of the p53 response and therefore individuals differing in the rs1042522 polymorphism should exhibit different degrees of protection. The hallmarks of smoking-induced airways damage is a reduction in lung function as measured by Forced Expiratory Volume in 1 second (FEV1), and in the ratio of FEV1 to Forced Vital Capacity (FEV1/FVC), which persist despite inhalation of a bronchodilator, indicating “irreversible” impairment (Rabe et al. 2007). To test the above hypotheses, these lung function measurements were made in a population-based cohort between ages 18 and 32 years in relation to cumulative history of smoking. We find that the extent of lung function decline after exposure to cigarette smoke exposure does indeed vary with the strength and nature of the p53 response.
Materials and methods
The cohort
The Dunedin Multidisciplinary Health and Development Study is described in detail elsewhere (Hancox et al. 2007; Rasmussen et al. 2002; Sears et al. 2003). Briefly this is a longitudinal study of an unselected birth cohort of 1,037 individuals (52% male) born in Dunedin in 1972/1973. The cohort represents the full range of socioeconomic status in New Zealand’s South Island. This analysis was restricted to the 863 Study members who reported that all four of their grandparents were of European origin (based on questions at the age 26 assessment). Women who were pregnant at either age 18 or 32 were also excluded (n = 29). 679 of the eligible participants had post-bronchodilator lung function measured at both ages 18 and 32 years of whom 668 provided a DNA sample for analysis. The Otago Ethics Committees approved the study and written informed consent was obtained at each assessment.
Lung function
Post-bronchodilator spirometry was measured at ages 18 and 32 years using an Ohio computerised spirometer (Ohio instruments) and a SensorMedics body plethysmograph (Yorba Linda, CA), respectively. At least three acceptable manoeuvres were obtained with the best FEV1 and FVC from any of the tests reported and used for calculation of FEV1/FVC (Standardization of Spirometry 1995). Spirometry was measured 10 min after inhalation of 5 mg/ml salbutamol nebulised for 2 min at age 18 and after 200 μg salbutamol via a metered dose inhaler using a large volume spacer at age 32. A portable spirometer (Spiropro, Sensormedics, Yorba Linda CA) was used to test Study members who were unable to sit in the plethysmograph or were unable to attend the research unit (n = 10 in this analysis). At each age, standing height was measured to the nearest millimetre.
Cigarette smoking
Personal smoking history was obtained from the Study members at each ages 18, 21, 26 and 32. Cumulative smoking history to age 32 was calculated from these assessments. One pack-year is defined as the equivalent of 20 cigarettes a day for 1 year.
Genotyping
Both polymorphisms were genotyped using primer extension-mass spectrometry (SEQUENOM, San Diego, CA). All PCR and MassEXTEND™ reactions were conducted utilising standard conditions using 2.5 ng of genomic DNA per sample (Bansal et al. 2002). Primers used to genotype the 309G/T polymorphism in intron 1 of MDM2 (c.−5 + 309G > T; rs2279744:g.G > T) were -5′-ACGTTGGATGTCGGAGGTCTCCGCGG-3′ (forward PCR primer), 5′-ACGTTGGATGCCGACAGGCACCTGCGATC-3′ (reverse PCR primer) and 5′-TCCGGACCTCCCGCGCCG-3′ (extension primer). The primers used to genotype the R72P polymorphism in TP53 (c.215G > C; rs1042522:g.G > C) were 5′-ACGTTGGATGGGCCGCCGGTGTAGGAGC-3′ (forward PCR primer), ACGTTGGATGCCAGGTCCAGATGAAGCTCC-3′ (reverse PCR primer) and 5′-GCCAGAGGCTGCTCCCC-3′ (extension primer). Automated analysis of these samples by matrix-assisted laser desorption ionisation/time-of-flight mass spectrometry was performed on a SEQUENOM–Bruker MassARRAY mass spectrometer. A randomly selected 10% of PCR amplified products from the cohort were re-genotyped using restriction fragment length polymorphism analysis with BstUI (TP53) or MspA1I (MDM2). Results demonstrated exact concordance with those obtained with mass spectrometry.
Statistical analysis
The influence of smoking (in pack-years) and MDM2 or TP53 genotype (coded as 0, 1, or 2 risk alleles, respectively) on spirometric lung function was assessed using linear regression with adjustments for lung function at age 18 (to adjust for any pre-existing differences in lung function), sex, and standing heights at age 18 and 32 (to adjust for sex- and height-related differences in expected lung function at each age). Interactions between genotype and the airway response to smoking were assessed by computing multiplicative genotype × smoking terms in these regression analyses. Separate analyses were repeated for each MDM2 and TP53 genotype. The effect of smoking on lung function was assessed amongst those with different combinations of these polymorphisms by repeating the analyses amongst those with different numbers of “risk” alleles (MDM2 rs2279744 G-alleles and TP53 rs1042522 G-alleles). The possibility that there may be a gene-environment correlation such that the genetic polymorphisms of MDM2 or TP53 influenced smoking behaviour was assessed by comparing the smoking histories for the different genotypes. Initial analyses found no evidence that the effects of smoking on lung function differed for men and women. Hence, in subsequent analyses, sexes were grouped together with an adjustment in the models. All analyses were performed using Stata version 10 (College Station, TX). Both outcome measures (FEV1 and FEV1/FVC) were approximately normally distributed. Plots of the residuals from the regression versus fitted values identified one clear outlier for the analyses of FEV1, but removal of this individual made no material difference to the regression analyses.
Results
HDM2 G-allele is associated with accelerated decline in lung function after cigarette smoke exposure
As expected, there was a significant main effect of cigarette smoke exposure on lung function change between ages 18 and 32 years. The number of pack-years smoked was significantly associated with lower post-bronchodilator FEV1 values and lower FEV1/FVC ratios at age 32 after adjustment for these measures at age 18 (p = 0.018 and p = 0.001, respectively). However, there was no relationship with genotype at either of the MDM2 or TP53 polymorphisms on either measure of lung function at either age 18 or 32 years (all p values > 0.2).
To determine whether there is a relationship between the MDM2 polymorphism ( rs2279744:G/T) and lung function when smoking is taken into consideration, the cumulative history of smoking (in ‘pack-years’) was obtained between these ages. Amongst individuals homozygous for the G-allele, cumulative smoking history was associated with lower post-bronchodilator FEV1 values, whereas this association was not significant among individuals with the T-allele (Table 1), yielding a statistically significant interaction between the number of MDM2 G-alleles and smoking (p int = 0.004). Thus a weaker p53 response in cigarette smokers is associated with an accelerated decline in lung function. The association between smoking history and post-bronchodilator FEV1/FVC ratios also appeared to be stronger among individuals homozygous for the G-allele than among T carriers, but the interaction between smoking and the number of MDM2 G-alleles did not reach statistical significance (p int = 0.15).
The TP53 G-allele is associated with accelerated lung function decline in smokers
The above results suggest that p53 does protect lung tissue from cigarette smoke exposure as hypothesised. We, therefore, asked whether smoking-related impairment of lung function varied according to the nature of the p53 response. Thus, lung function measurements in relation to smoking history were analysed with respect to the TP53 rs1042522 polymorphism. The data show that individuals homozygous for the G-allele (encoding p53R72) are more susceptible to smoking-induced lung function impairment (Table 2). Among the G-allele homozygotes, smoking history was significantly associated with lower values for FEV1 and FEV1/FVC, whereas these associations were not significant for carriers of at least one C-allele. The interactions between the number of G-alleles and smoking were statistically significant for both post-bronchodilator FEV1 and the FEV1/FVC ratio (p int = 0.020 and 0.037, respectively). Thus the degree of protection from lung function decline after cigarette smoke exposure appears to vary according to both the strength and nature of the p53 response.
Accelerated decline in lung function after cigarette smoking varies with the number of risk alleles
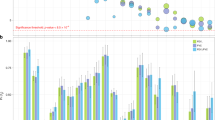
To test whether combinations of the MDM2 and TP53 polymorphisms strengthened the association with smoking-related lung function decline, we calculated a score based on the number of “risk” alleles (G-alleles for MDM2, and G-alleles for TP53). There was no significant association between the MDM2 and TP53 variants, (χ2 = 4.54, p = 0.34). Thus, an individual homozygous for both T-alleles for rs2279744 and C-alleles for rs1042522 has 0 risk alleles, whereas an individual homozygous for G-alleles at both loci has four risk alleles. Regression analyses of the association between smoking and lung function amongst these subgroups are shown in Table 3. There were significant interactions between smoking and the number of risk alleles for post-bronchodilator FEV1 and FEV1/FVC (p int = 0.001 and 0.020, respectively). The association between heavy smoking and decline in post-bronchodilator FEV1 was stronger in those with more risk alleles (Fig. 1). Thus, individuals with combinations of the rs2279744 G-allele in MDM2 and the rs1042522 G-allele in TP53 appear to be at greater risk of developing smoking-related impairment of lung function.

Mean percent change in FEV1 between ages 18 and 32 according to smoking history and the number of risk alleles. White bars indicate non-smokers (no pack-years, n = 350), grey bars indicate light smokers (less than 5 pack-years, n = 97), black bars indicate heavy smokers (at least 5 pack-years, n = 221). The trend across risk allele groups is significant for moderate-heavy smokers (p = 0.005) but not for non- or light smokers (p = 0.83 and 0.12, respectively). Error bars represent standard errors of the mean
There was an unexpected trend to improved lung function amongst smokers with no risk alleles (Table 3). It seems most likely that this is a chance finding since the sample was small (n = 29) and the trend was not statistically significant. If this group was removed from the analysis, the interaction between the number of risk alleles and smoking remained statistically significant for FEV1 but not for FEV1/FVC (p int = 0.008 and 0.21, respectively).
Discussion
Mechanism
The observation of greater smoking-induced impairment of FEV1 in individuals who are homozygous for the MDM2 G-allele is consistent with its role in attenuating the p53 response. Similarly the difference in the liability of the TP53 rs1042522 alleles to confer susceptibility to smoking-induced lung damage may relate to their differential ability to promote the apoptotic and cell-cycle arrest functions of p53. p53R72 (encoded by the G-allele) binds more tightly to MDM2 than p53P72 (encoded by the C-allele) (Dumont et al. 2003), and this interaction promotes export of p53 from the nucleus (Yu et al. 2000) to the mitochondrial membrane where it mediates non-transcriptional apoptosis (Chipuk et al. 2004; Leu et al. 2004). p53P72 by contrast is reported to be more effective than p53R72 at causing cell-cycle arrest (Pim and Banks 2004; Thomas et al. 1999). Thus, our data showing that individuals with p53P72 alleles appear to be more resistant to smoking-induced lung damage suggest that cell-cycle arrest and DNA repair are more important than apoptosis in protecting lung tissue from smoke-induced DNA damage. Although ultimately less efficient than apoptosis at removing DNA damaged cells, activation of repair by p53P72 would afford considerable protection from DNA damage but at the same time, minimise tissue damage after exposure to cigarette smoke. In addition, as p53P72 binds less well to MDM2, therefore, being less susceptible to ubiquitin mediated degradation, it would have a higher steady-state level than p53R72 and therefore, should be more responsive to DNA damage. This could provide an advantage to cells continually exposed to cigarette smoke.
Relationship with advanced disease
Lung function naturally declines slowly after reaching a peak in young adulthood. Our data suggest that the extent to which smoking accelerates this decline depends on the strength of the p53 response. However, age 32 is too young to detect serious smoking-related lung damage and the relationship between our observations and advanced smoking-related diseases such as chronic obstructive pulmonary disease and lung cancer remains unclear. Indeed, two recent case–control studies indicate that p53P72 alleles are more common amongst patients with chronic obstructive pulmonary disease than amongst healthy smokers (Arif et al. 2008; Lee et al. 2006). Reconciling these observations with our findings is difficult, although these studies often involve Asian populations with very different allele frequencies to our European population. Some increased association between p53P72 and lung cancer has also been reported (Fan et al. 2000), although this is not a consistent finding (Matakidou et al. 2003). Of interest, however, are the observations that p53P72 has been found to be associated with increased longevity in the general population (van Heemst et al. 2005) and increased survival after diagnosis of cancer or other serious diseases (Bojesen and Nordestgaard 2008; Orsted et al. 2007), suggesting that p53P72 may contribute to general good health. As suggested above, this could also be due to the higher steady-state level of p53P72 conferring a protective advantage against chronic exposure to agents leading to genetic damage.
Potential limitations of the study
A limitation of this research is the small numbers in some of the genotype groups—particularly when combining MDM2 and TP53 genotypes. Another is the relatively short (14 year) duration of follow-up in the context of the many years of heavy smoke exposure usually required to induce clinically important lung damage. Despite these issues, we identified significant interactions between these genotypes and lung function. The finding that functional genetic polymorphisms in the p53 pathway moderate the effect of cigarette smoking on lung function requires replication in independent cohorts. However, we think it is unlikely that our findings are due to chance. First, the findings are biologically plausible as p53 plays a key role in regulating the cellular response to genotoxic stress. Second, the nature of the p53 responses associated with these MDM2 and TP53 genotypes are biologically consistent with each other, and in combination, showed the strongest association. Third, we were able to rule out the presence of gene-environment correlations; that is, there were no significant differences in the pack-year smoking histories of individuals as a function of the number of risk alleles in MDM2 (p = 0.51), TP53 (p = 0.43) or the sum of MDM2 and TP53 risk alleles (p = 0.95). Fourth, measurements of lung function were made 14 years apart, which allowed us to document that genetic polymorphisms involved in p53 function moderated the effect of smoking on ‘within-individual’ changes in lung function over the course of young adulthood. Fifth, smoking exposure was ascertained via repeated, prospective assessments, largely negating the problems associated with retrospective recall.
Conclusion
We have found that common polymorphisms of the gene for p53, and its principal regulator MDM2, are associated with accelerated smoking-related decline in lung function in young adults. These data may provide an explanation for differences in the susceptibility of individuals to the adverse effects of smoking. Furthermore, they provide novel evidence that p53 can mediate physiologically adaptive responses to genotoxic insults in humans.
References
Arif E, Vibhuti A, Deepak D, Singh B, Siddiqui MS, Pasha MAQ (2008) COX2 and p53 risk-alleles coexist in COPD. Clin Chim Acta 397:46–50
Bansal A, van den Boom D, Kammerer S, Honisch C, Adam G, Cantor CR, Kleyn P, Braun A (2002) Association testing by DNA pooling: an effective initial screen. Proc Natl Acad Sci USA 99:16871–16874
Beckman G, Birgander R, Sjalander A, Saha N, Holmberg PA, Kivela A, Beckman L (1994) Is p53 polymorphism maintained by natural selection? Hum Hered 44:266–270
Bojesen SE, Nordestgaard BG (2008) The common germline Arg72Pro polymorphism of p53 and increased longevity in humans. Cell Cycle 7:158–163
Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, Hwang SJ, Strong LC, Lozano G, Levine AJ (2004) A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 119:591–602
Bond GL, Hu W, Levine AJ (2005) MDM2 is a central node in the p53 pathway: 12 years and counting. Curr Cancer Drug Targets 5:3–8
Braithwaite AW, Prives CL (2006) p53: more research and more questions. Cell Death Differ 13:877–880
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010–1014
Donehower LA, Harvey M, Slagle BL, Mcarthur MJ, Montgomery CA, Butel JS, Bradley A (1992) Mice deficient for P53 are developmentally normal but susceptible to spontaneous tumors. Nature 356:215–221
Dumont P, Leu JI, Della PAIII, George DL, Murphy M (2003) The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 33:357–365
Fan R, Wu MT, Miller D, Wain JC, Kelsey KT, Wiencke JK, Christiani DC (2000) The p53 codon 72 polymorphism and lung cancer risk. Cancer Epidemiol Biomark Prevent 9:1037–1042
Hancox RJ, Poulton R, Greene JM, Filsell S, McLachlan CR, Rasmussen F, Taylor DR, Williams MJ, Williamson A, Sears MR (2007) Systemic inflammation and lung function in young adults. Thorax 62:1064–1068
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El Naggar AK, Lozano G (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119:861–872
Lee YL, Chen W, Tsai WK, Lee JC, Chiou HL, Shih CM, Wang YC (2006) Polymorphisms of p53 and p21 genes in chronic obstructive pulmonary disease. J Lab Clin Med 147:228–233
Leu JI, Dumont P, Hafey M, Murphy ME, George DL (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 6:443–450
Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, Friend SH (1990) Germ line P53 mutations in a familial syndrome of breast-cancer, sarcomas, and other neoplasms. Science 250:1233–1238
Matakidou A, Eisen T, Houlston RS (2003) TP53 polymorphisms and lung cancer risk: a systematic review and meta-analysis. Mutagenesis 18:377–385
Momand J, Zambetti GP, Olson DC, George D, Levine AJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237–1245
Murphy ME (2006) Polymorphic variants in the p53 pathway. Cell Death Differ 13:916–920
Orsted DD, Bojesen SE, Tybjaerg-Hansen A, Nordestgaard BG (2007) Tumor suppressor p53 Arg72Pro polymorphism and longevity, cancer survival, and risk of cancer in the general population. J Exp Med 204:1295–1301
Pim D, Banks L (2004) p53 polymorphic variants at codon 72 exert different effects on cell cycle progression. Int J Cancer 108:196–199
Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R, van Weel C, Zielinski J (2007) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 176:532–555
Rasmussen F, Taylor DR, Flannery EM, Cowan JO, Greene JM, Herbison GP, Sears MR (2002) Risk factors for airway remodeling in asthma manifested by a low postbronchodilator FEV1/vital capacity ratio: a longitudinal population study from childhood to adulthood. Am J Respir Crit Care Med 165:1480–1488
Sears MR, Greene JM, Willan AR, Wiecek EM, Taylor DR, Flannery EM, Cowan JO, Herbison GP, Silva PA, Poulton R (2003) A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. N Engl J Med 349:1414–1422
Standardization of Spirometry (1995) 1994 Update. Am J Respir Critical Care Med 152:1107–1136
Thomas M, Kalita A, Labrecque S, Pim D, Banks L, Matlashewski G (1999) Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol Cell Biol 19:1092–1100
van Heemst D, Mooijaart SP, Beekman M, Schreuder J, de Craen AJM, Brandt BW, Slagboom PE, Westendorp RGJ (2005) Variation in the human TP53 gene affects old age survival and cancer mortality. Exp Gerontol 40:11–15
Vousden KH, Lu X (2002) Live or let die: the cell’s response to p53. Nat Rev Cancer 2:594–604
Yu ZK, Geyer RK, Maki CG (2000) MDM2-dependent ubiquitination of nuclear and cytoplasmic P53. Oncogene 19:5892–5897
Acknowledgments
This work was supported by the Health Research Council of New Zealand (03/27); Dunedin School of Medicine Strategic Research Initiative Grant; the US National Institute of Mental Health (grants MH45070, MH49414 and MH077874), and the UK Medical Research Council (G0100527). DNA collection and extraction was funded by the University of Wisconsin. Dr. Sears holds the AstraZeneca Chair in Respiratory Epidemiology at McMaster University. Avshalom Caspi holds a Royal Society Wolfson Merit Award. Professor Braithwaite is a Cancer Institute NSW Programme Leader. We are grateful to the Study members and their parents for their continued support. We also thank Dr. Phil A. Silva, the study founder. Mr. T. Manley is thanked for assistance with the genotyping. The authors have no conflicting financial interests.
These studies were carried out according to current New Zealand standards with ethical approval and informed individual consents.
Author information
Authors and Affiliations
Corresponding author
Additional information
Christene R. McLachlan is deceased.
Rights and permissions
About this article
Cite this article
Hancox, R.J., Poulton, R., Welch, D. et al. Accelerated decline in lung function in cigarette smokers is associated with TP53/MDM2 polymorphisms. Hum Genet 126, 559–565 (2009). https://doi.org/10.1007/s00439-009-0704-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-009-0704-z