
Abstract
The success of genome-wide association studies (GWAS) to identify risk loci of complex diseases is now well-established. One persistent major hurdle is the cost of those studies, which make them beyond the reach of most research groups. Performing GWAS on pools of DNA samples may be an effective strategy to reduce the costs of these studies. In this study, we performed pooling-based GWAS with more than 550,000 SNPs in two case-control cohorts consisting of patients with Type II diabetes (T2DM) and with chronic rhinosinusitis (CRS). In the T2DM study, the results of the pooling experiment were compared to individual genotypes obtained from a previously published GWAS. TCF7L2 and HHEX SNPs associated with T2DM by the traditional GWAS were among the top ranked SNPs in the pooling experiment. This dataset was also used to refine the best strategy to correctly identify SNPs that will remain significant based on individual genotyping. In the CRS study, the top hits from the pooling-based GWAS located within ten kilobases of known genes were validated by individual genotyping of 1,536 SNPs. Forty-one percent (598 out of the 1,457 SNPs that passed quality control) were associated with CRS at a nominal P value of 0.05, confirming the potential of pooling-based GWAS to identify SNPs that differ in allele frequencies between two groups of subjects. Overall, our results demonstrate that a pooling experiment on high-density genotyping arrays can accurately determine the minor allelic frequency as compared to individual genotyping and produce a list of top ranked SNPs that captures genuine allelic differences between a group of cases and controls. The low cost associated with a pooling-based GWAS clearly justifies its use in screening for genetic determinants of complex diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2007, the identification of genes associated with complex diseases was considered one of the major breakthroughs in science (Pennisi 2007). This success story was attributable to the widespread application of genome-wide association studies (GWAS) to study complex diseases and physiologic traits. Using this approach, geneticists can identify genetic variants that co-occur with diseases by genotyping hundreds of thousands of SNPs that are selected to capture a large fraction of common variation in the human genome. This strategy has had considerable success in identifying reproducible susceptibility genes for complex traits and diseases in a hypothesis-free study design. This development was mostly attributable to our better comprehension of the structure of human genome variation (HapMap 2005; Frazer et al. 2007) and to the rapid progress in genotyping technologies (Syvanen 2005). As a result, unprecedented capabilities are currently available to interrogate the human genome for susceptibility loci and the benefits of this new approach can be observed for many complex diseases (Herbert et al. 2006; Klein et al. 2005; Rioux et al. 2007; Sladek et al. 2007; WTCCC 2007). However, major financial resources are required to conduct a GWAS, which include collecting and phenotyping the large number of subjects with and without a particular disease as well as the high cost of whole-genome genotyping technologies.
All together, GWAS necessitate millions of dollars to be completed. These resources are beyond the reach of most research groups and are inaccessible for diseases with limited funding support. A number of groups reported the development of an approach to reduce the overall cost of GWAS by replacing individual genotyping by pooled genomic DNA (allelotyping) (Meaburn et al. 2005; Pearson et al. 2007; Sham et al. 2002). This approach relies on constructing separate pools of patient and control DNA samples and hybridizing these pools on high-density genotyping arrays. SNP microarray probe intensities, which measure the allele frequencies in each pool rather than discrete genotypes, are then analyzed and ranked by likelihood of association with the disease. The effectiveness of the technique was successfully demonstrated by the identification of previously published as well as novel genetic susceptibility loci (Melquist et al. 2007; Pearson et al. 2007; Steer et al. 2007).
Motivated by these recent advances, we demonstrate a practical application of pooling-based GWAS. We compared the results of GWAS performed by allelotyping with the results of individual genotyping in two case-control populations characterized for type 2 diabetes mellitus (T2DM) or chronic rhinosinusitis (CRS). Data derived from these two case-control cohorts are used to assess particular aspects of pooling-based GWAS. Overall, the results indicate that DNA pooling on high-density genotyping arrays is a cost-effective alternative approach to performing GWAS on individual DNAs to identify susceptibility genes for complex diseases. Although a sizeable amount of genetic information is lost compared to traditional GWAS, the large reduction in cost, coupled with the substantial fraction of genetic association that is still captured, clearly justifies the utility of the approach.
Methods
Research subjects
T2DM study sample
The cases and controls represent the stage 1 sample described by Sladek et al. (2007). Briefly, diabetic subjects were recruited during medical examinations taking place at the Endocrinology-Diabetology Department of the Corbeil-Essonnes Hospital and at the UMR8090 unit in Lille. These subjects were diagnosed with T2DM according to the 1997 American Diabetes Association (ADA) criteria. To enrich for risk alleles and reduce phenotypic heterogeneity, only diabetic subjects with at least one affected first degree relative and with a body mass index (BMI) below 30 kg/m2 were included. Control subjects were selected from the Epidemiological Study on the Insulin Resistance syndrome (DESIR) program. Inclusion criteria include normal fasting glucose (<6.1 mM) and a BMI below 27 kg/m2. A total of 606 cases and 629 controls were available for the current DNA pooling experiment. Informed consent was obtained from all study participants, and the institutional review boards overseeing these sites approved the study protocol.
CRS study sample
The cases and controls were derived from an ongoing project that has been described previously (Al-Shemari et al. 2008). In the present study, 210 individuals with severe CRS and 189 controls were available for genetic analysis. Briefly, patients were recruited from three tertiary rhinology clinics from July 2005 to February 2006. Initial diagnoses were obtained from patient records, and classified according to the 2004 American Academy of Otolaryngology-Head and Neck Surgery guidelines (Meltzer et al. 2004). Severe CRS was defined as (1) persistent signs or symptoms of CRS despite previous endoscopic sinus surgery (ESS); or (2) a history of more than one ESS for CRS, regardless of outcome. The control population was recruited from either patients’ spouses, non-blood relatives living in the same household, or by random telephone screening matching patients’ postal code. The study was approved by the McGill University Health Centre Institutional Review Board and informed consent was obtained from all participants.
DNA extraction
For the T2DM study, genomic DNA was extracted from peripheral blood cells using PUREGENE D50K DNA isolation kits (Gentra Systems, Minneapolis, MN, USA) or DNeasy Blood & Tissue Kits (Qiagen, Hilden, Germany). Similarly, DNA from cases in the CRS cohort was extracted from peripheral blood leukocytes. DNA from CRS controls was extracted from saliva, using the Oragene DNA Purification Protocol (DNA Genotek, Ottawa, ON, USA). Isolated DNA from both blood and saliva was stored at −80°C prior to use.
DNA pooling
T2DM study sample
The T2DM cohort was first cleaned as previously described (Sladek et al. 2007). Briefly, population stratification was identified with the STRUCTURE program (Pritchard et al. 2000) and individuals showing evidence of intercontinental admixture were discarded. In addition, samples that failed individual genotyping on the Illumina Infinium Human1 or the HumanHap300 genotyping BeadChips were discarded from the pooling experiment. The remaining DNAs were pooled together in equivalent molar amounts using the following procedure. To minimize the total amount of DNA utilized, only 1 µl of DNA was taken from each sample. The DNA was diluted in 19 µl of water to reach a total volume of 20 µl. These dilutions were quantified in duplicate by fluorimetry (PicoGreen™). Samples with low DNA concentration (<2 ng/µl) or with more than 15% difference in the duplicates were discarded. The remaining DNAs included 606 cases and 629 controls that were placed on 9 and 8 96-well plates, respectively. Individual DNAs were first pooled together by plate in equivalent molar amounts by adjusting the volume to add 20 ng per sample. The plate pools were then quantified by fluorimetry and normalized to 5 ng/µl. These normalized plate pools were subsequently concentrated by evaporation (Turbo Vap® 96Concentration Workstation, Caliper Life Science) to reach a concentration of 50 ng/µl. Finally, the plate pools were pooled together according to their affection status (cases or controls) while adjusting the volume to account for the number of individuals in each plate pool. All the pippetting was performed manually.
CRS study sample
Pools were constructed to contain equal quantities of DNA from each individual sample. DNAs were first quantified in duplicate by fluorimetry (PicoGreen™). Samples with low DNA concentration (<20 ng/µl) were discarded. The remaining samples (209 cases and 184 controls) were adjusted by dilution or evaporation to reach a concentration of 50 ng/µl. DNA concentrations were re-estimated in duplicate by fluorimetry and a second round of adjustment and re-quantification was performed if required. All samples were then checked with PCR to identify those that did not yield a robust product. DNAs for which we were unable to reach a concentration close to 50 ng/µl as well as those that failed the PCR check were discarded. The remaining DNAs (173 cases and 130 controls) were pooled together according to their affection status by adjusting the volume for the remaining small difference in concentrations between samples. A total of 250 ng of DNA (5 µl for samples at 50 ng/µl) were added to the pools for each sample using the MultiPROBE® II HT EX liquid handling platform (PerkinElmer).
Pool validation in the CRS study sample
Once equi-molar amounts of each sample were combined, the genomic DNA pools were validated by comparing allele frequencies of six SNPs obtained by allelotyping and individual genotyping. This step was performed to insure concordance between DNA pools and individual samples before moving to large-scale genotyping. A number of SNPs have previously been genotyped in the current case/control population (Al-Shemari et al. 2008). Accordingly, six SNPs (rs2291418, rs7099684, rs3780901, rs7393696, rs1761212, rs4319601) were selected for allelotyping to cover a range of allele frequencies. Allele frequencies of genomic DNA pools were estimated and compared to the allele frequencies calculated from the individual genotyping. The allelotyping measurements were performed on the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF) (Sequenom, San Diego, CA, USA) as previously described (Oeth et al. 2005).
High-density SNP genotyping
Genomic DNA pools were assayed using the Illumina protocol for individual genotyping. Briefly, 750 ng of pooled genomic DNA was labeled and hybridized to the Illumina Sentrix® HumanHap550 Genotyping BeadChip, which interrogated 555,175 SNPs. For the T2DM cohort, five replicates were used for case and control pools (a total of ten arrays). Four replicates were used for case and control pools in the CRS cohort (a total of eight arrays). Supplementary Figure 1 illustrates the pooling design. The same CRS DNA pools were also hybridized on the Human1M BeadChip in four replicates (a total of eight arrays) for comparison purposes with the HumanHap550 BeadChip.
Individual genotyping and analyses
Individual genotyping for the T2DM cohort was comprehensively described by Sladek et al. (2007). Briefly, 750 ng of genomic DNA was labeled and hybridized on the Illumina Infinium Human1 and HumanHap300 BeadChips, which interrogated 109,365 and 317,503 SNPs, respectively.
In the CRS cohort, the individual genotyping was performed on 1536 SNPs using the Illumina GoldenGate assay (Shen et al. 2005). The allele-specific extension reaction was performed with 250 ng of genomic DNA. The reaction products then underwent ligation, PCR amplification and labeling before being hybridized on the Sentrix Array Matrix. The genotypes were called using BeadStudio. A total of 49 of the 1,536 SNPs failed the genotyping assay. Seven additional SNPs were excluded because of low call rate (<90%, n = 5) or because of low minor allele frequency (<0.01, n = 2). Markers showing deviation from Hardy–Weinberg equilibrium were also excluded (P < 0.01, n = 23). Hardy–Weinberg equilibrium for the 63 SNPs located on the X chromosome was calculated in women only. The average call rate for the remaining 1,457 SNPs was 99.87%. The same samples used in the pooling experiment were genotyped (n = 303). Samples successfully genotyped in less than 95% of markers were excluded from analysis (n = 3). The association tests were performed with a chi-squared test (1 df) using the software PLINK (v0.99s) (Purcell et al. 2007). These statistical tests were then adjusted for possible population stratification using the genomic control method (Devlin and Roeder 1999). Briefly, a correction factor was calculated from a total of 100 ancestry informative markers previously described by Price et al. (2008). These markers were typed using the SEQUENOM platform as previously described (Al-Shemari et al. 2008). Supplementary Figure 2 illustrates the overall experimental design.
Analyses of the pooling experiment
Three methods were used to rank SNPs based on their likelihood of being associated with the diseases (T2DM or CRS). First, the allelic frequency differences between case and control pools were measured and ranked for all SNPs. Allele frequencies for SNPs in each pool were calculated by BeadStudio (B allele frequency). Briefly, these allele frequencies estimations are obtained by interpolation of the known allele frequencies of three canonical genotype clusters represented on a polar coordinate view that originated from an individual genotyping training set (Peiffer et al. 2006). Second, SNPs were sorted with an F ratio, which is obtained by dividing the B allele frequency variance between groups by the variance within groups. A modification of the F ratio was also used for the CRS cohort. A filter was applied to keep only SNPs that had at least a 10% difference in allele frequencies between cases and controls. The remaining SNPs were sorted by the F ratio. We call this method the Filter-F ratio. Finally, the SNPs were also ranked using the silhouette score as described previously (Pearson et al. 2007). Briefly, this score is calculated from a scatter plot specific for each SNP with signal intensity fraction on the x-axis [signalAllele2/(SignalAllele1 + SignalAllele2)] and the logarithm of the signals from both alleles on the y-axis [log(SignalAllele1 + SignalAllele2)]. A quality score is then derived based on cluster analyses. A high silhouette score is observed when data points are tight within groups and distant between groups. These scores were calculated with the GenePool software using the files generated by BeadStation 500 that contain the red and green intensity values corresponding to the two SNP alleles (Pearson et al. 2007). Data were normalized (gpextract-n 1) and the lower limit of acceptable number of beads was set at 5 (gpanalyze-b 5). The silhouette scores were calculated with the modified Manhattan method (gpanalyze-p 0-d 2).
Results
Pooling results in the T2DM cohort
The true effectiveness of a pooling-based GWAS can best be appreciated by comparing the results with those obtained by individual GWAS genotyping. In 2007, the first GWAS on T2DM was published (Sladek et al. 2007). The scan was performed on a total of 409,927 SNPs typed on two Illumina platforms (Human1 and HumanHap300). A total of 28 and 43 SNPs passed the genome-wide significant threshold for association with T2DM on the Human1 and HumanHap300, respectively. In the current work, we took the same DNA samples and pooled them together as illustrated in Supplementary Figure 1. The genomic DNA pools were hybridized on the Illumina HumanHap550 and ranked based on three methods. Table 1 shows the ranking results of the three methods for the most significant SNPs based on individual genotyping. First, SNPs located within the TCF7L2 locus, the gene most strongly associated with T2DM, were among the top-ranked for all methods. This result suggest that genes conferring an effect size similar to TCF7L2 in T2DM (OR = 1.65 and 2.77 for heterozygotes and homozygotes, respectively) will be successfully identified by pooling-based GWAS regardless of the choice of method. More globally, the median ranking for all non-redundant significant SNPs in Table 2 (n = 47) is 4,952, 8,200, and 9,995 for the absolute difference, the F ratio and the silhouette ranking methods, respectively. These numbers are relatively impressive considering that any random subset of SNPs on these high-throughput arrays is expected to give a median ranking equal to approximately half the number of SNPs tested (i.e. around 275,000 for the HumanHap550). To test the significance of these results, permutation testing was performed on 10,000 sets of 47 SNPs randomly selected from ranking number 1 to 555,175. The probability of obtaining median values of 4,952, 8,200, and 9,995 were 2.77 × 10−12, 4.95 × 10−12, and 6.79 × 10−12, respectively. Thus, there is a clear enrichment of significant SNPs in the top ranking.
Based on the median values calculated in Table 1, the single best method of ranking seems to be the absolute difference in allele frequency between cases and controls. However, the best method of ranking and selecting SNPs that will undergo validation by individual genotyping is likely to be influenced by the resources available. Many research groups will proceed by genotyping tens or hundreds of SNPs following a pooling-based GWAS, while others may adopt an approach similar to the one that we used in the CRS cohort by genotyping the maximum number of SNPs on a custom multiplex assay that is commercially available. Using the individual genotyping results from the T2DM cohort, we evaluated the best method for selecting SNPs from the pooled arrays. A total of 47 SNPs assessed on the HumanHap550 array was previously associated with T2DM in a GWAS conducted by individual genotyping (Sladek et al. 2007). Table 2 indicates the number of these SNPs that rank in the top 10, 100, 200, 1,000, 1,536, 5,000, 10,000 and 20,000 using the different methods of ranking. The silhouette ranking method is the single best method to identify the highest number of SNPs validated by individual genotyping when the top 1,536 SNPs or less are considered. For example, among the 47 SNPs associated with T2DM identified by individual genotyping, 18 (38.3%) are in the top 1536 using the silhouette ranking method. The same number for the F ratio and the absolute difference in allele frequencies are 11 (23.4%) and 12 (25.5%), respectively. However, when more SNPs are considered for validation by individual genotyping, there is a shift from the ‘silhouette’ to the ‘absolute difference’ methods for the single best method of ranking (Fig. 1a). Accordingly, the best method of ranking depends on the number of SNPs that are considered for validation by individual genotyping. Based on the data derived from the T2DM cohort, the silhouette ranking method would be favored if less than 3,000 SNPs are considered (Fig. 1a). Otherwise, the absolute difference in allele frequencies between cases and controls seems to perform better.

Performance comparison between the different methods of ranking. The y-axis represents the 47 SNPs that were previously associated with T2DM using a traditional GWAS (Sladek et al. 2007). The x-axis represents the number of SNPs that are considered for validation by individual genotyping. a The ranking of the 47 SNPs are illustrated for the three different methods of ranking (solid lines). The vertical dash line represents the ranking number 3,000, where a shift in the single best method of ranking occurs between the silhouette score and the absolute difference in allele frequency. The dash lines represent the number of true genetic loci that are identified by combining the three methods of ranking. For example, 1,264 SNPs overlap among the top 4,380 SNPs for at least two methods of ranking. Twenty of them belong to the list of 47 true genetic loci (gray dotted lines). In comparison, 154 SNPs overlap among the top 4,354 SNPs for the three methods of ranking. Ten of them belong to the list of 47 true genetic loci (black dotted lines). b Comparison by combining only two methods of ranking. The dash line is the same as in a. The other solid lines combine the information of only two methods. For example, 249 SNPs overlap among the top 3,221 ranked SNPs between the absolute difference and silhouette ranking methods. Fifteen of these 249 SNPs belong to the list of 47 true genetic loci (dotted lines). The vertical dash line represents a threshold of 1,000, where a shift in the best method of ranking occurs. The figure suggests that the best method of ranking is conditional to the number of SNPs the investigators are willing to confirm by genotyping. If less than 1,000 SNPs are considered, the best strategy is to combine the absolute difference and the silhouette ranking methods (black solid line). In contrast, if more than 1,000 SNPs undergo validation by individual genotyping, the best strategy is to combine the absolute difference and the F ratio ranking methods (dark gray solid line)
We hypothesized that combining the methods of ranking would improve our ability to identify true genetic loci. Accordingly, we compared the performance of combining two or three methods of ranking to identify the 47 genetic loci found in the T2DM cohort by individual genotyping (Fig. 1). First we evaluated the number of SNPs that overlap for two or three methods. For example, if we take the top 5,000 SNPs for the three methods of ranking, a total of 200 SNPs overlap among the three methods, including ten (21.3%) SNPs associated with T2DM in the original GWAS (Sladek et al. 2007). In comparison, 1,547 SNPs overlap between at least two methods of ranking, of which 20 (42.6%) are included among the 47 SNPs associated with T2DM. Illustrated continuously, this later strategy seems to perform better compared to single methods taken individually (Fig. 1a, gray line). Next we evaluate the performance of combining only two methods (Fig. 1b). In this scenario only SNPs that overlap between the two methods would be selected for validation by individual genotyping. Combining the silhouette and the absolute difference ranking methods (pink line) provides the best strategy to identify genetic loci found by individual genotyping if less than 1,000 SNPs are considered for validation. Otherwise, combining the absolute difference and the F ratio (yellow line) performs better.
Pool validation in the CRS cohort
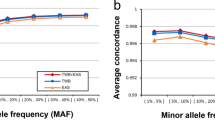
Prior to hybridizing genomic DNA pools on Illumina HumanHap550 arrays, a smaller scale experiment was conducted to assess the accuracy of the pools. The allele frequencies of six SNPs was estimated from genomic DNA pools and compared to the real allele frequencies calculated from individual genotyping. Figure 2 demonstrates the high concordance between the two methods and confirms the accuracy of allelotyping. These results also set the stage for a larger scale quantitative allelotyping experiment with the Illumina HumanHap550 arrays.

Validation of genomic DNA pools. Six SNPs were genotyped to derive the exact allele frequencies. The allele frequencies of these same SNPs were then estimated by allelotyping using the genomic DNA pools of cases and controls. The scatter plots show the tight correlation between the true allele frequencies (y-axis) and the one estimated from genomic DNA pools (x-axis) in both cases and controls. The gray and black dots represent cases and controls, respectively
Pooling results in the CRS cohort
A total of eight arrays were hybridized with the genomic DNA pools for cases and controls. The text files generated by the BeadStation 500 as well as the B allele frequencies calculated by BeadStudio were used to rank the SNPs with three different methods (see “Methods”). A genome-wide appreciation of the results for the silhouette ranking method is depicted in Fig. 3. For example, the top ranked SNP is located on the long arm of chromosome 15 (rs12437477). A subset of highly ranked SNPs was then selected for individual genotyping using the Illumina GoldenGate assay. This assay can evaluate a maximum of 1,536 SNPs. First, we used a gene-centric approach to choose SNPs located within or close to known genes. Then, SNPs were ranked on the three different methods described in the method section. The top 1,536 SNPs were obtained by each method. The overlap between the three methods of ranking is illustrated in Fig. 4. Any SNP ranked in the top 1,536 SNPs for at least two methods was selected for individuals genotyping (n = 741). The GoldenGate panel was then completed with the next best SNPs based on the silhouette ranking method.

Genome-wide plot of the silhouette score in the CRS cohort. The chromosome number is indicated on the top of each subgraph. The y-axis represents the ranking on a log scale and the x-axis indicates the location of the SNPs in physical distance

Venn diagram showing the number of overlapping SNPs in the CRS cohort that are ranked in the top 1,536 in each of the three methods of ranking. All of the SNPs on the HumanHap550 BeadChip were ranked by three methods: (1) the absolute difference in allele frequency between cases and controls, (2) the Filter-F ratio, and (3) the silhouette score. The top 1,536 SNPs were selected in each method and the number of overlapping SNPs is illustrated
The allele frequencies obtained by individual genotyping are concordant with a validated European-derived dataset (HapMap CEU) (Supplementary Figure 3). More interestingly, there is a tight correlation between the allele frequencies obtained by allelotyping compared to individual genotyping (Fig. 5). The results suggest that approximately 90% of the variability observed in the allele frequencies obtained by individual genotyping can be captured by allelotyping. Since allelotyping appears to robustly predict allele frequencies in the pooled samples, we sought to determine whether it also provides a reliable estimate of the absolute allele frequency difference between cases and controls (Fig. 6). We compared the frequencies obtained by allelotyping and genotyping for the 1,404 top-ranked SNPs identified in the CRS study. These results demonstrated that the effect size determined by allelotyping among the top ranked SNPs is clearly overestimated when compared to the individual genotyping results.

Allele frequencies between individual genotyping and allelotyping in the CRS cohort. A total of 1,404 SNPs are illustrated

Box plot showing the distribution of the observed difference in allele frequencies between cases and controls when calculated by allelotyping and individual genotyping. The figure is derived from the results of 1,404 SNPs taken from the CRS cohort
Obviously, the effectiveness of pooling-based GWAS will ultimately be determined by the number of SNPs that are associated with the disease using the individual genotypes. Accordingly, association between the CRS phenotype and individual genotypes were performed. Figure 7 (left panel) shows the frequency of P values for the 1,457 SNPs that passed quality control. There is a clear overrepresentation of SNPs with low P values, with 598 SNPs (41.0%) showing association with CRS at P values lower than 0.05. Genomic control was then used to adjust for potential inflated association tests arising from unknown sources such as population heterogeneity or cryptic relatedness. From a set of 100 null loci, the inflation factor was estimated to be 1.14 (see “Methods”). The distribution of P values adjusted for this inflation factor is shown in Fig. 7 (right panel). Although the number of low P values is decreased, there are still 492 SNPs with P values lower than 0.05. Taken together, the results suggest that a pooling-based GWAS can effectively identify SNPs that differ significantly in terms of allele frequencies between two groups of individuals.

Association test results for 1,457 SNPs successfully genotyped on an individual basis in the CRS cohort. The left and right panels show results without or with genomic control, respectively. Without genomic control, a total of 598 SNPs are associated with CRS at a P value lower than 0.05. This number is lowered to 492 with the use of genomic control. The horizontal dashed line represents the mean number of P values that is expected by chance (i.e. the heights of the bars are expected to fluctuate around that line). The figure clearly illustrates the overrepresentation of SNPs with lower P values
We were next interested in evaluating our strategy for selecting SNPs to be individually genotyped. A dichotomous variable was constructed in which SNPs were classified as true-positive (P ≤ 0.05) or false-positive (P > 0.05) based on individual genotypes. The percentage of true-positive SNPs selected for individual genotyping increases with the number of methods that rank the SNPs highly (Supplementary Figure 4). For example, 62.4% of true-positive SNPs are found among those that ranked within the top 1,536 for the three methods of ranking. This suggests that combining methods may be more effective than using the single best one. Nevertheless, using a logistic regression model, we evaluated the performance of each method to correctly discriminate the true- and false-positive SNPs. Table 3 presents the results of the single and multiple logistic regression models. Taken individually, the absolute difference in allele frequency between cases and controls as well as the Filter-F ratio significantly influence the outcome of the individual genotyping results. The average allele frequency estimated for the whole cohort, including cases and controls, is also significant when considered alone in the model (z value = 4.9). In multiple regression models, the ‘absolute difference’ is always the most significant variable. Interestingly, when all the variables are included in the same model, the absolute difference explains the largest part of the variance, but both the silhouette score and the Filter-F ratio are also significant predictors in the model (Table 3). Although the contribution of the absolute difference variable is convincing, it is, practically speaking, insufficient to predict the true- from the false-positives. Supplementary Figure 5 shows the distribution of SNPs based on their absolute difference in allele frequency and whether the SNP is a true-positive (blue) or a false-positive (red). Taken together, the results from the CRS cohort suggest that the single best allelotyping variable that predicts SNPs that end up significantly associated with the disease using the individual genotypes is the ‘absolute difference’. Similar to the observation made with the T2DM cohort, combining the methods may provide additional refinement. However, these results must be interpreted with caution considering that only SNPs selected from the top ranks of the pooling experiment were analyzed.
To verify the accuracy of the ranking with the Illumina BeadChips, we tested the same DNA genomic pools on the Human1M array. The 555,004 SNPs common to both platforms were compared in term of the difference in allele frequencies between cases and controls. Figure 8 shows the gradual decline in Pearson correlation coefficients with the increasing number of top ranked SNPs considered. Even though the correlation coefficient drops progressively from 0.94 for the first 10,000 SNPs to 0.57 when all the SNPs are considered (n = 555,004), stronger correlations are observed for the top rated SNPs, with correlation coefficients of 0.993 for the top 100 SNPs and 0.968 for the top 1,000 SNPs. These results clearly demonstrate the robustness of the allelotyping method on the Illumina genome-wide BeadChip platform.

Correlation in allele frequency difference between cases and controls obtained with the Illumina HumanHap550 and the Illumina Human1M platforms. SNPs were ranked based on the absolute difference between cases and controls obtained by allelotyping using the HumanHap550 array. The same genomic DNA pools were then tested by allelotyping using the Human1M array in order to compare the results between the two platforms. The correlation coefficients were calculated cumulatively at every 10,000 SNPs (i.e. the first 10,000, the first 20,000, …). A line was then drawn along the dots
Discussion
High-density genotyping arrays permit genome-wide genotyping of hundreds of thousands of SNPs in parallel. These GWAS are particularly suited to discovering previously unsuspected genes or pathways involved in a specific disease. The power of such hypothesis-free study designs to identify genetic factors of complex diseases is now well-established (Herbert et al. 2006; Klein et al. 2005; Rioux et al. 2007; Sladek et al. 2007; WTCCC 2007). However, the cost associated with traditional GWAS is still prohibitive for most research groups and the application is limited for diseases where large amounts of funding are inaccessible. In the current study, we investigate the effectiveness of a low-cost alternative based on pooled genomic DNA.
It should be emphasized that the only advantage of a pooling-based GWAS compared to traditional GWAS is the reduction in the cost of genotyping. However, the level of savings is substantial. An accurate estimate of cost reduction is difficult to perform considering the moving target price of the high-density genotyping arrays. At the time we performed our pooling experiments the cost of the arrays was just under $1,000. Accordingly, the pooling experiments performed in this study for the CRS and the T2DM cohorts cost on the order of $8,000–$10,000. This level of genotyping expense is similar to many genetic association studies looking at a specific candidate gene. Thus, this methodology opens up the possibility for any research group to use a genome-wide approach. The current study clearly demonstrates that the “low-hanging fruit” can be successfully identified by allelotyping on high-density genotyping arrays. In the T2DM cohort, SNPs located in the TCF7L2 gene were highly ranked for all ranking methods. Accordingly, SNPs with an effect size similar to or greater than those associated with T2DM in the TCF7L2 gene are likely to be found with a pooling experiment. The fraction of SNPs that are validated by individual genotyping can considerably influence the overall cost and power of a study. Therefore the cost effectiveness of a pooling-based GWAS must be judged considering the number of SNPs that will undergo validation by individual genotyping, and also the rapidly dropping price of high-density genotyping arrays. However, the current results suggest that successful identification of genes can be expected with a reasonable number of SNPs validated by individual genotyping. For example, in the T2DM cohort, if only the top 10 SNPs from the silhouette ranking would have undergone validation, two of the five genes identified by Sladek et al. (2007) would have been found. While this represents a significant reduction in the number of genes identified, the genotyping would have been performed for less than $15,000, compared to greater than one million dollars for scanning each sample individually.
The results we obtained with the CRS cohort are truly informative to help understand the genetic architecture of this disease. At the present time, little is known about the genetics of CRS. While familial aggregation has been observed in genetic epidemiology studies (Cohen et al. 2006; Drake-Lee 1992; Greisner and Settipane 1996; Qu et al. 2007), few candidate genes have been identified (Al-Shemari et al. 2008; Bussu et al. 2007; Cheng et al. 2006; Fajardo-Dolci et al. 2006; Kim et al. 2007; Luxenberger et al. 2000; Molnar-Gabor et al. 2000; Takeuchi et al. 2000; Wang et al. 2000; Zhai et al. 2007). We had initially begun studying the genetics of CRS by utilizing a candidate gene approach (Al-Shemari et al. 2008) without a predetermined concept of which areas of the genome to interrogate. However, by performing a pooling-based GWAS and validating the best 1,500 SNPs by individual genotyping, we identified approximately 600 SNPs from 445 genes that were potentially associated with CRS (P < 0.05). Each of these novel high-priority SNPs had allele frequency differences between cases and controls at a level worthy of additional investigation. The most significant SNP for each of the top 10 genes is shown in Supplementary Table 1. Overall, the CRS results clearly demonstrated that differences in allele frequency between cases and controls can be identified using pooling-based GWAS. However, it should be stressed that a substantial fraction of these differences in allele frequency will be the results of chance owing to the small sample size and the large number of genetic markers tested. Validation of these results in a second and larger CRS cohort will be required to separate the true results from the false positives. Nevertheless, we believe that we have made a substantial leap in prioritizing susceptibility genes for CRS, which is particularly impressive considering the relatively small cost of the genotyping.
With a pooling approach, the likelihood of false-positives is high when performing an initial screen with 550,000 markers. Accordingly, validation is essential to confirm the findings observed in the first stage of the study. First, individual genotyping of the putative loci needs to be performed to ensure that the construction of the genomic DNA pools did not inflate the true allelic frequency difference between cases and controls. Then, independent replication in a second population and/or functional studies will still be required.
Currently, the most effective use of DNA pooling consists of a two-stage design in which markers showing putative association are followed-up by individual genotyping (Sham et al. 2002). However, it is still unclear which statistical methods are most appropriate to prioritize SNPs for the second stage. By comparing many statistical tests, Pearson et al. (2007) demonstrated that the silhouette score was consistently the most effective method to rank SNPs. It had the greatest number of SNPs in the top ranks compared to individual genotyping on Affymetrix arrays. In the current study, we used three different methods of ranking to identify the most promising loci. In the T2DM cohort, the three statistics identify the TCF7L2 locus as top candidate region, suggesting that strong genetic effects are likely to be found irrespective of the methods of ranking. However, there is still a large difference between the ranking methods and their ability to identify the genetic loci found by individual genotyping. For example, if 1,000 SNPs are considered for validation by individual genotyping, combining the methods of ranking can double the number of loci identified compared to the absolute difference or F ratio alone (Fig. 1a). Results derived from the T2DM cohort suggest that the silhouette method is the single best method of ranking when less than 3,000 SNPs are considered for validation by individual genotyping. However, the superiority of the silhouette score decreases as the number of SNPs considered for validation increases.
Intuitively, combining the methods of ranking should increase the number of true genetic loci. A SNP with a high silhouette score, a large difference in allele frequency between cases and controls, and a large ratio of variance between vs within groups (large F ratio) is more likely to be a true genetic locus. In the T2DM data, we demonstrated that combining the methods of ranking surpasses any single method. The best strategy seems to be to combine two methods of ranking, which differ depending on the number of SNPs considered for validation. As a rule of thumb, if less than a 1,000 SNPs are considered for validation by individual genotyping, the top ranked SNPs that overlap between the silhouette and the absolute difference in allele frequency should be prioritized. Otherwise, the top ranked SNPs that overlap between the absolute difference and the F ratio ranking methods should perform better. It should be emphasized that these conclusions are reached by testing pooling-based results against those obtained from a novel GWAS. Although this GWAS was based on individual genotyping, it is likely to have missed or overrated some genetic loci implicated in T2DM and does not constitute a perfect standard to assess the pooling results. It is also unclear whether the results reached in the T2DM cohort can be directly applied to other cohorts. Our experimental results confirm that a pooling-based GWAS can detect susceptibility loci in these data. Further empirical studies will be required to confirm these results, and simulation studies are need to characterized the full parameter space of pooling-based designs.
DNA pooling methods clearly have limitations compared to individual genotyping. First, DNA pooling adds extra experimental error (e.g. pipetting for pool construction) to the allele frequency measurement that directly influences the power to detect small effect sizes. In addition, pooling results in a loss in the ability to study subphenotypes, haplotypes, or specific genetic models as well as to undertake gene-gene and gene-environment interaction studies. Lastly, DNA pooling does not allow detection of and adjustments for population stratification. It is thus important to examine population stratification using a limited number of individual genotypes before pooling DNAs. Nevertheless, despite these limitations, the pooled genome-wide screen technique provides an attractive alternative to currently expensive GWAS.
In conclusion, the current study demonstrates that a pooling-based GWAS is a cost-effective method to identify genes implicated in complex diseases. The method can be successfully applied to identify major genetic associations with disease. The method is effective in producing a list of top SNPs that is enriched with significant genetic associations found by individual genotyping. In addition, the approach substantially lowers the cost of GWAS that are conventionally too expensive for many orphan diseases, diseases specific to isolated populations, or diseases with limited funding, such as CRS. However, the results also highlight the importance of additional validation following the pooling-based GWAS.
References
Al-Shemari H, Bosse Y, Hudson TJ, Cabaluna M, Duval M, Lemire M, Vallee-Smedja S, Frenkiel S, Desrosiers M (2008) Influence of leukotriene gene polymorphisms on chronic rhinosinusitis. BMC Med Genet 9:21
Bussu F, Tiziano FD, Giorgio A, Pinto AM, De Corso E, Angelozzi C, Brahe C, Paludetti G (2007) Argl6gly polymorphism of the beta2-adrenoceptor gene (ADRBeta2) as a susceptibility factor for nasal polyposis. Am J Rhinol 21:378–382
Cheng YK, Lin CD, Chang WC, Hwang GY, Tsai SW, Wan L, Tsai MH, Tsai JJ, Tsai FJ (2006) Increased prevalence of interleukin-1 receptor antagonist gene polymorphism in patients with chronic rhinosinusitis. Arch Otolaryngol Head Neck Surg 132:285–290
Cohen NA, Widelitz JS, Chiu AG, Palmer JN, Kennedy DW (2006) Familial aggregation of sinonasal polyps correlates with severity of disease. Otolaryngol Head Neck Surg 134:601–604
Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004
Drake-Lee A (1992) Nasal polyps in identical twins. J Laryngol Otol 106:1084–1085
Fajardo-Dolci G, Solorio-Abreu J, Romero-Alvarez JC, Zavaleta-Villa B, Cerezo-Camacho O, Jimenez-Lucio R, Olivo-Diaz A (2006) DQA1 and DQB1 association and nasal polyposis. Otolaryngol Head Neck Surg 135:243–247
Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Shen Y, Sun W, Wang H, Wang Y, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallee C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y et al (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–861
Greisner WA 3rd, Settipane GA (1996) Hereditary factor for nasal polyps. Allergy Asthma Proc 17:283–286
HapMap (2005) A haplotype map of the human genome. Nature 437:1299–1320
Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, Illig T, Wichmann HE, Meitinger T, Hunter D, Hu FB, Colditz G, Hinney A, Hebebrand J, Koberwitz K, Zhu X, Cooper R, Ardlie K, Lyon H, Hirschhorn JN, Laird NM, Lenburg ME, Lange C, Christman MF (2006) A common genetic variant is associated with adult and childhood obesity. Science 312:279–283
Kim SH, Park HS, Holloway JW, Shin HD, Park CS (2007) Association between a TGFbeta1 promoter polymorphism and rhinosinusitis in aspirin-intolerant asthmatic patients. Respir Med 101:490–495
Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308:385–389
Luxenberger W, Posch U, Berghold A, Hofmann T, Lang-Loidolt D (2000) HLA patterns in patients with nasal polyposis. Eur Arch Otorhinolaryngol 257:137–139
Meaburn E, Butcher LM, Liu L, Fernandes C, Hansen V, Al-Chalabi A, Plomin R, Craig I, Schalkwyk LC (2005) Genotyping DNA pools on microarrays: tackling the QTL problem of large samples and large numbers of SNPs. BMC Genomics 6:52
Melquist S, Craig DW, Huentelman MJ, Crook R, Pearson JV, Baker M, Zismann VL, Gass J, Adamson J, Szelinger S, Corneveaux J, Cannon A, Coon KD, Lincoln S, Adler C, Tuite P, Calne DB, Bigio EH, Uitti RJ, Wszolek ZK, Golbe LI, Caselli RJ, Graff-Radford N, Litvan I, Farrer MJ, Dickson DW, Hutton M, Stephan DA (2007) Identification of a novel risk locus for progressive supranuclear palsy by a pooled genomewide scan of 500, 288 single-nucleotide polymorphisms. Am J Hum Genet 80:769–778
Meltzer EO, Hamilos DL, Hadley JA, Lanza DC, Marple BF, Nicklas RA, Bachert C, Baraniuk J, Baroody FM, Benninger MS, Brook I, Chowdhury BA, Druce HM, Durham S, Ferguson B, Gwaltney JM Jr, Kaliner M, Kennedy DW, Lund V, Naclerio R, Pawankar R, Piccirillo JF, Rohane P, Simon R, Slavin RG, Togias A, Wald ER, Zinreich SJ (2004) Rhinosinusitis: establishing definitions for clinical research and patient care. Otolaryngol Head Neck Surg 131:S1–S62
Molnar-Gabor E, Endreffy E, Rozsasi A (2000) HLA-DRB1, -DQA1, and -DQB1 genotypes in patients with nasal polyposis. Laryngoscope 110:422–425
Oeth P, Beaulieu M, Park C, Kosman D, del Mistro G, van den Boom D, Jurinke C (2005) iPLEX Assay: increased plexing efficiency and flexibility for MassARRAY system through single base primer extension with mass-modified terminators. SEQUENOM Application Note
Pearson JV, Huentelman MJ, Halperin RF, Tembe WD, Melquist S, Homer N, Brun M, Szelinger S, Coon KD, Zismann VL, Webster JA, Beach T, Sando SB, Aasly JO, Heun R, Jessen F, Kolsch H, Tsolaki M, Daniilidou M, Reiman EM, Papassotiropoulos A, Hutton ML, Stephan DA, Craig DW (2007) Identification of the genetic basis for complex disorders by use of pooling-based genomewide single-nucleotide-polymorphism association studies. Am J Hum Genet 80:126–139
Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J, Cheung SW, Shen RM, Barker DL, Gunderson KL (2006) High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res 16:1136–1148
Pennisi E (2007) Breakthrough of the year. Human genetic variation. Science 318:1842–1843
Price AL, Butler J, Patterson N, Capelli C, Pascali VL, Scarnicci F, Ruiz-Linares A, Groop L, Saetta AA, Korkolopoulou P, Seligsohn U, Waliszewska A, Schirmer C, Ardlie K, Ramos A, Nemesh J, Arbeitman L, Goldstein DB, Reich D, Hirschhorn JN (2008) Discerning the ancestry of European Americans in genetic association studies. PLoS Genet 4:e236
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Qu SH, Li TY, Li M, Shi JB, Wen WP, Wen WH (2007) Genetic epidemiologic study on nasal polyps. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 42:608–611
Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ, Brant SR (2007) Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39:596–604
Sham P, Bader JS, Craig I, O’Donovan M, Owen M (2002) DNA pooling: a tool for large-scale association studies. Nat Rev Genet 3:862–871
Shen R, Fan JB, Campbell D, Chang W, Chen J, Doucet D, Yeakley J, Bibikova M, Wickham Garcia E, McBride C, Steemers F, Garcia F, Kermani BG, Gunderson K, Oliphant A (2005) High-throughput SNP genotyping on universal bead arrays. Mutat Res 573:70–82
Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445:881–885
Steer S, Abkevich V, Gutin A, Cordell HJ, Gendall KL, Merriman ME, Rodger RA, Rowley KA, Chapman P, Gow P, Harrison AA, Highton J, Jones PB, O’Donnell J, Stamp L, Fitzgerald L, Iliev D, Kouzmine A, Tran T, Skolnick MH, Timms KM, Lanchbury JS, Merriman TR (2007) Genomic DNA pooling for whole-genome association scans in complex disease: empirical demonstration of efficacy in rheumatoid arthritis. Genes Immun 8:57–68
Syvanen AC (2005) Toward genome-wide SNP genotyping. Nat Genet 37(Suppl):S5–S10
Takeuchi K, Majima Y, Sakakura Y (2000) Tumor necrosis factor gene polymorphism in chronic sinusitis. Laryngoscope 110:1711–1714
Wang X, Moylan B, Leopold DA, Kim J, Rubenstein RC, Togias A, Proud D, Zeitlin PL, Cutting GR (2000) Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhinosinusitis in the general population. JAMA 284:1814–1819
WTCCC (2007) Genome-wide association study of 14, 000 cases of seven common diseases and 3, 000 shared controls. Nature 447:661–678
Zhai L, Sun Y, Tang L, Liu H (2007) Polymorphism between loci for human leukocyte antigens DR and DQ in patients with nasal polyps. Ann Otol Rhinol Laryngol 116:66–68
Acknowledgments
Y. Bossé is a research scholar of the Heart and Stroke Foundation of Canada. R. Sladek and J.C. Engert are research scholars of the ‘Fonds de la Recherche en Santé du Québec’. Funding for the CRS studies was provided by the Fondation Antoine Turmel, a non-profit philanthropic organisation. Funding for T2DM studies was provided by Génome Québec and Genome Canada. We would like to acknowledge the bioinformatics research staff at the Translational Genomics Research Institute (TGEN) for their help with the GenePool software. We also want to recognize the bioinformatics staff at the McGill University and Genome Quebec Innovation Center for automating the pooling-based GWAS analyses.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Bossé, Y., Bacot, F., Montpetit, A. et al. Identification of susceptibility genes for complex diseases using pooling-based genome-wide association scans. Hum Genet 125, 305–318 (2009). https://doi.org/10.1007/s00439-009-0626-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-009-0626-9