
Abstract
While the number of success stories for mapping genes associated with complex diseases using genome-wide association approaches is growing, there is still much work to be done in developing methods for such studies when the samples are collected from a population, which may not be homogeneous. Here we report the first genome-wide association study to identify genes associated with asthma in an admixed population. We genotyped 96 Puerto Rican moderate to severe asthma cases and 88 controls as well as 109 samples representing Puerto Rico’s founding populations using the Affymetrix GeneChip Human Mapping 100K array sets. The data from samples representing Puerto Rico’s founding populations was used to identify ancestry informative markers for admixture mapping analyses. In addition, a genome-wide association analysis using logistic regression was performed on the data. Although neither admixture mapping nor regression analysis gave any significant association with asthma after correction for multiple testing, an overlap analysis using the top scoring SNPs from different methods suggested chromosomal regions 5q23.3 and 13q13.3 as potential regions harboring genes for asthma in Puerto Ricans. The validation analysis of these two regions in 284 Puerto Rican asthma trios gave significant association for the 5q23.3 region. Our results provide strong evidence that the previously linked 5q23 region is associated with asthma in Puerto Ricans. The detection of causative variants in this region will require fine mapping and functional validation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the USA, asthma prevalence is highest in Puerto Ricans (26%) and lowest in Mexicans (10%) (Carter-Pokras and Gergen 1993; Homa et al. 2000). This is paradoxical since both groups are considered “Hispanic” or “Latino”. Although there are many potential explanations for this observation, including environmental and socioeconomic factors, one likely explanation is that the genetic predisposition to asthma differs among subgroups within the Latino population. Latinos are admixed and share varying proportions of West African, Native American and European ancestry (Choudhry et al. 2006; Hanis et al. 1991). The mixed ancestry of Latinos provides unique opportunities in epidemiological and genetic studies and may be useful in untangling complex gene–gene and gene–environment interactions in disease susceptibility (Burchard et al. 2003; Choudhry et al. 2007).
Several recent advances in statistical methods and genotyping techniques have resulted in a paradigm shift in genetic association studies, making it possible to perform a genome-wide association analysis, which does not require a priori knowledge of disease associated genes (Hirschhorn and Daly 2005; Kennedy et al. 2003; Matsuzaki et al. 2004; Risch 2000). An alternative yet complementary approach to genome-wide association analysis is admixture mapping (Smith and O’Brien 2005). Admixture mapping is a method for localizing disease causing genetic variants that differ in frequency across populations. It is most advantageous to apply this approach to admixed populations such as Latinos, which descended from a recent mix of three ancestral groups that have been geographically isolated for thousands of years. The approach assumes that near a disease causing gene there will be enhanced ancestry from the population that has greater risk of getting the disease. Thus if one can calculate the ancestry along the genome for an admixed sample set, one could use that to identify disease causing gene variants (Chakraborty and Weiss 1988; McKeigue 1997; Pfaff et al. 2001; Stephens et al. 1994). Admixture mapping requires the genotyping of several thousand markers (Smith et al. 2004) while genome-wide association requires genotyping of hundreds of thousands of markers (Hinds et al. 2005). The Affymetrix GeneChip Human Mapping 100K array set can genotype 116,204 SNPs in a given individual with a single genotyping assay (Kennedy et al. 2003). The ease and cost effectiveness of the GeneChip arrays for large-scale genotyping make them far more attractive than conventional genotyping platforms for genome-wide studies.
We used the Affymetrix 100K arrays to perform genome-wide association and admixture mapping analyses to identify loci associated with asthma in Puerto Ricans. First, we selected ancestry informative markers (AIMs) from the 100K arrays by screening Puerto Rican ancestral populations. We then performed three different analyses: (1) genome-wide association analysis testing association between individual SNPs and asthma disease status, (2) admixture mapping comparing cases and controls using the program Admixmap and (3) admixture mapping using likelihood ratio test on locus-specific ancestry estimates determined using the program Structure. In all three analyses, we incorporated adjustments to correct for confounding due to population stratification. By combining results from these three different analytical methods, we determined a set of most promising candidate regions for asthma in Puerto Ricans. We then selected the top SNPs from these candidate regions and genotyped them in a second sample of Puerto Rican families with asthma for validation analysis.
Materials and methods
Study participants
A total of 380 Puerto Rican subjects with asthma and 88 ethnically matched controls were included in this study. The genome-wide analyses included 96 subjects with moderate to severe asthma and 88 healthy controls. The moderate to severe asthma was defined based on baseline lung function (Pre-FEV1) of the asthmatic subject. Subjects with Pre-FEV1 less than 80% of predicted were categorized as having “moderate-severe” asthma. The validation analysis included 284 Puerto Rican asthma trios (father, mother and affected child). All subjects were recruited as part of the Genetics of Asthma in Latino Americans (GALA) study. Recruitment and patient characteristics were described in detail elsewhere (Burchard et al. 2004; Choudhry et al. 2005; Lind et al. 2003) but will be briefly described here. Ethnicity and national origin were self-reported and were ascertained using standardized questions. Puerto Rican subjects were enrolled only if both biological parents and all four biological grandparents were reported to be of Puerto Rican ethnicity. Interviews with children were conducted in the presence of parents. Eligible subjects with asthma had physician-diagnosed asthma and had experienced two or more asthma symptoms (among wheezing, coughing, and shortness of breath) in the previous two years. All control subjects were screened and considered to be eligible to participate if they did not have clinical evidence of asthma, allergies, atopy or any other allergic or pulmonary disease. All subjects (asthmatics and healthy controls) were between the ages of 8 and 40 years and were interviewed by bilingual and bicultural field workers and physicians specialized in asthma.
Genotyping using Affymetrix 100K arrays
We genotyped 37 West African, 42 European and 30 Native American samples using the Affymetrix GeneChip Human Mapping 100K array set to find AIMs relevant to Puerto Rico’s founding populations. The 37 West African samples are from individuals living in London, UK and South Carolina, USA, who are either non-admixed or have very low levels of admixture. The 42 European samples are from Coriell’s North American Caucasian panel. The Native American samples (Mayan, n = 15 and Nahua, n = 15) were recruited from villages in remote areas of Mexico. Genotyping of Puerto Rican asthma cases and controls was also performed using the Affymetrix 100K arrays. The genotyping was done following standard Affymetrix protocols and the data was processed using the Affymetrix-provided Genotyping Console Software (GCOS) and GeneChip DNA Analysis Software (GDAS) (Affymetrix Inc., Santa Clara, CA, USA). Ten samples were run in duplicate to assess for concordance between runs. The concordance rate was >99.9% and the overall genotyping success rate was >98.5%. Markers were assessed for Hardy–Weinberg equilibrium using a χ2 goodness-of-fit test. After excluding markers on the X chromosome, markers with minor allele frequency (MAF) <5%, extreme deviation from Hardy–Weinberg equilibrium (χ2 > 10) or study-wide genotyping call rates <95%, we retained 97,112 markers for further association analysis.
Selection of ancestry informative markers (AIMs)
The genotype data from the parental population samples was used to identify AIMs, which were then used to perform admixture mapping and to adjust the analyses for population stratification. Since the contemporary Puerto Rican population is a mixture of three parental populations, West Africans, Europeans and Native Americans, we used an iterative process for selecting our AIMs. For each of the three possible pairs of ancestral populations, we identified markers where the difference in allele frequency (δ) was at least 0.5 between any two ancestral populations. Once we identified such markers, we selected a subset that was adequately distributed across the genome, with the markers being far enough apart that they were in linkage equilibrium in the ancestral populations. These markers formed our set of 2,730 AIMs, which were then used to estimate individual and locus specific ancestry.
Estimation of individual ancestry
The individual ancestry estimates (IAE) were calculated using two different programs, Admixmap (Hoggart et al. 2003, 2004) and Structure (Falush et al. 2003; Pritchard et al. 2000), and the 2,730 AIMs. The IAE from these two programs were highly correlated (r > 0.9) (results not shown). The IAE from the Structure program were used to adjust for population stratification in the 100K association analysis.
Estimation of locus specific ancestry and admixture mapping analysis
The admixture mapping analyses were performed using the panel of 2,730 AIMs as described above and using two different programs, Admixmap and Structure. Admixmap uses a Bayesian probability model fit using Markov chain Monte Carlo to estimate locus specific ancestry (Hoggart et al. 2004). To run Admixmap, we provided the program with the genotypes of the 2,730 AIMs for our case and control subjects, as well as for the ancestral representatives. Admixmap performs a test for association between ancestral status and disease status and returns a p value for each marker. We also estimated the ancestral proportion at each of the 2,730 AIMs using the program Structure (Montana and Pritchard 2004). At each marker, a likelihood ratio statistic was computed, testing the null hypothesis of no association between ancestry and disease status while taking each individual’s overall ancestry estimates into account. Under the null hypothesis, the statistics follow a χ2-distribution and indicates the strength of deviation in ancestry between cases and controls at each locus. Statistical significance was assessed using a permutation test.
Genome-wide association analysis: regression method
We used a logistic regression model to test for association between genotype and disease status for 97,112 markers on the 100K arrays assuming an additive model for the disease. In addition to age and gender, IAE estimates using the program Structure were included as covariates in all the regression models to control the inflation of type I error rate due to population stratification.
Correction for multiple testing
The admixture mapping and genome-wide association analyses were corrected for multiple testing using the multtest package in the statistical language R. We used adjusted p values as calculated by multtest under the Benjamini & Yekutieli step-up false discovery rate (FDR) controlling procedure (Benjamini and Yekutieli 2001).
Identification of follow-up regions by overlap analysis
To identify areas of potential interest we used the combined strength of the different analytical methods: (1) individual SNP association, (2) admixture mapping using a case–control approach with the program Admixmap and (3) admixture mapping using likelihood ratio test on locus specific ancestry estimates from the program Structure. We identified regions for subsequent analysis by selecting markers that were highly ranked (based on unadjusted p value or likelihood ratio test score) in at least two of the three analytical methods.
Validation
To test the validity of the regions identified through the overlap analysis, we performed validation analysis on the most promising markers in these regions on a sample of 284 Puerto Rican asthma trios. The genotyping was performed using the fluorescent polarization (FP) method as directed by the manufacturer (PerkinElmer, Waltham, MA, USA) (Chen et al. 1999). The association of the markers with asthma disease status was tested using the transmission-disequilibrium test as implemented in the program FBAT (Laird et al. 2000).
Results
Identification of AIMs
Table 1 shows the percent of markers on the Affymetrix 100K arrays informative for West African-European, West African-Native American and European-Native American ancestries at different levels of ancestry informativeness as measured by difference in allele frequency (delta, δ). Most markers were only informative for one pair of ancestral populations, while some were informative for more than one pair. Among the markers on the 100K arrays, there were more than 10,000 markers with δ > 0.5 for West African-European, West African-Native American or European-Native American ancestry (Table 1). We selected a panel of 2,730 AIMs which had a δ value of >0.5 and were evenly spaced across the genome (inter-marker distance mean: 1 cM, median: 0.79 cM, first quartile: 0.67 cM and third quartile: 1.05 cM and the overall inter-marker range: 0.01–26.07 cM) for estimation of individual ancestry estimates and admixture mapping analysis for asthma (see supporting material, Table 1S).
Evidence of population stratification in Puerto Rican Asthma cases and controls
The estimated average European and West African ancestry was different between our Puerto Rican cases and controls. Figure 1 shows box plots demonstrating differences in estimated European and West African ancestral proportions between cases (with medians of 62.0 and 22.3%, respectively) and controls (medians of 58.1 and 26.0%, respectively). The Native American ancestry was similar between the two groups (medians of 15.7% in cases and 15.9% in controls) (Fig. 1). The summary χ2 test using 2,730 AIMs also gave significant results (p = 0.01) suggesting that there are systematic differences in ancestry between our asthma cases and controls that could cause spurious genetic associations if measures of ancestry were not included in our analyses.

Boxplots showing distribution of ancestry estimates in Puerto Rican asthma cases and controls
Admixture mapping analysis
Neither of the admixture mapping analyses (Admixmap or likelihood ratio test based on Structure output) gave any significant results after correction for multiple testing using the FDR approach (Figs. 2, 3). There were 56 markers that had an unadjusted p value of <0.01 in the Admixmap analysis (Table 2) and 21 markers that gave a score of ≥10 in the likelihood ratio tests before correction for multiple testing (Table 3).

Distribution of p values for 2730 AIMs across the chromosomes from admixture mapping analysis using the program Admixmap

Distribution of likelihood ratio test scores for 2,730 AIMs across the chromosomes based on locus-specific ancestry estimates from the program Structure
Individual SNP analyses
Eight SNPs had p value of less than 10−4 in the regression analysis and were located on chromosomal regions 1p22.3, 1p32.2, 4q31.1, 10q23.31, 11q14.1, 13q13.3 and 13q22.3 (Table 4). The smallest unadjusted p value was 1.3 × 10−5 for a marker in chromosomal region 4q31.1. Taking into account the fact that 97,112 simultaneous hypothesis tests were conducted, none of the markers showed statistically significant association with disease status after correction for multiple testing (Fig. 4). However, it is still possible that some of the more significant markers did not have low enough p values simply due to the low power of the study, and to further explore this possibility we ranked the markers based on the relative strength of their association from the regression and admixture mapping analyses, and used these rankings for the overlap analysis.

Distribution of p values for 97,112 markers across the chromosomes from asthma case–control regression analysis
Overlap analysis
For the overlap analysis, we selected the highest ranked SNPs from each of the three analytical methods. We selected 81 SNPs with p values ≤0.001 from the regression analysis, 56 SNPs with p values ≤0.01 from the Admixmap analysis and 21 SNPs with a combined score of ≥10 from the likelihood ratio test. We then performed an overlap analysis on these SNPs and selected regions, which were identified as being highly ranked by at least two of the three methods employed. We considered loci to be overlapping if at least two methods had highly ranked SNPs within 50 kilobases (kb) of each other. From these analyses, we identified five “overlap regions” where the markers showed higher significance or were closer together than in the other overlap regions (Table 5).
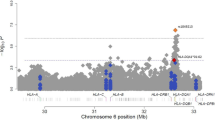
To test for further significance within these candidate regions, we selected windows on either side of the overlap SNPs, and tested for enhanced significance in these windows. Windows consisted of sets of 500 markers from the 97,112 marker set for each of the five overlap regions, with markers chosen so that the overlap SNP was at the center of the window in terms of physical distance. Therefore, the window sizes varied (9–12 Mb) according to the density of the markers on the 100K arrays. We plotted the histograms of the p values from the regression analysis for the markers in these windows, as well as quantile–quantile (QQ) plots to test for deviation from the empirical p value distribution of all 97,112 markers tested. These plots indicated that the largest deviations from the empirical distribution were in the 5q23.3 and 13q13.3 regions (Fig. 5). These deviations could either be due to enhanced association between SNPs and disease status in these regions or could be due to local differences in ancestry since our regression and admixture mapping analyses were adjusted only for ancestry on a genome-wide level.

Plots showing histograms of p values for the markers from the top five regions from the overlap analysis and quantile-quantile (QQ) plots of these p values against all p values from the 100K regression analysis
Validation
To validate our findings from the overlap analysis, we selected one marker each from the 500-marker window in 5q23.3 and 13q13.3 overlap regions. The markers, rs1496348 and rs817737, had the best p value in the regression analysis in the 500 marker window in 5q23.3 and 13q13.3 overlap regions, respectively, and therefore were selected for validation. The two markers were genotyped in a sample of 284 Puerto Rican family trios with asthma. Since the initial genome-wide association analysis was performed on moderate to severe asthmatics, the transmission-disequilibrium test (TDT) was performed on all trios and also on trios with moderate to severe asthma. The TDT analysis suggested positive association between the SNP rs1496348 (5q23.3 region) and moderate to severe asthma (p value = 0.02, Table 6). Allele A of this marker was over-transmitted among trios in which the proband had moderate to severe asthma. The same allele was associated with moderate to severe asthma in the initial genome-wide association analysis with a p value of 0.0007 (Table 6). The result of the TDT analysis for SNP rs817737 in 13q13.3 region was not significant for both asthma and moderate to severe asthma (Table 6).
Discussion
Genome-wide association studies are desirable since they do not require a priori knowledge of the genes involved in a disease or disease-related phenotypes and may also provide greater power than linkage-based methods for identifying common variants conferring modest risk (Hirschhorn and Daly 2005; Risch 2000). However, a potential downside to performing genetic association studies in recently admixed populations, such as Puerto Ricans, is the possibility for spurious associations (confounding) due to population stratification (Cardon and Palmer 2003; Devlin and Roeder 1999; Marchini et al. 2004; Ziv and Burchard 2003). To date, there have been several successful attempts at gene mapping for complex diseases, including asthma, by genome-wide association analysis, but none has done so in an admixed population (WTCCC 2007; Buch et al. 2007; Gudmundsson et al. 2007; Hafler et al. 2007; Hakonarson et al. 2007; McPherson et al. 2007; Moffatt et al. 2007; Samani et al. 2007; Saxena et al. 2007; Scott et al. 2007; Steinthorsdottir et al. 2007; Tomlinson et al. 2007; Winkelmann et al. 2007; Yeager et al. 2007; Zanke et al. 2007; Zeggini et al. 2007).
This is the first report of genome-wide association and admixture mapping analysis for asthma in Puerto Ricans. Although our initial efforts at identifying disease associated loci based on marker-by-marker test statistics were limited by our small sample size, our validation analysis indicates that we could identify and validate regions that have been previously associated with asthma disease status by combining the strength from distinct but complementary analytical methods—direct genome wide association and admixture mapping. While the direct genome association analysis can be performed on any population to identify genetic variants (both ethnic-specific and cosmopolitan) associated with a disease, admixture-mapping methodology detects genetic variants in recently admixed populations that are responsible for racial differences in disease risk. Compared to direct genome-wide association study designs, admixture mapping exploits long-range linkage disequilibrium that exists in recently admixed populations and therefore requires fewer markers (McKeigue 1998; Montana and Pritchard 2004), and is more robust to allelic heterogeneity (Terwilliger and Weiss 1998). Although none of our admixture mapping or genome-wide association analysis showed significant association after correction for multiple testing, it is likely that some of the top scoring markers from different analyses are true associations that did not reach statistical significance due to low power of our study. The overlap analysis combining the results of admixture mapping and genome-wide association analysis did identify two regions, 5q23.3 and 13q13.3, as the most promising candidates for asthma in Puerto Ricans. But it is possible that this analysis missed other regions that could be better captured using one of the analytical methods than the other.
For a complex disease like asthma successful identification of causative factors will require large and well characterized samples, as have been used in other successful genome-wide association studies for complex diseases. In addition, it should be noted that many different factors (genetic or environmental) may exist that contribute to the development and severity of asthma. While every effort was made to ensure homogeneity of the phenotype, the possibility that different individuals in our study may have different genetic factors contributing to disease still exists.
One lesson our analysis demonstrates is that association studies in admixed populations should include adjustments for ancestry. We have previously shown that there is evidence of substantial confounding from population stratification in association studies of asthma in Latino populations (Choudhry et al. 2006). Here, we confirm our previous findings using a much larger set of AIMs (n = 2,730) suggesting that any association study of asthma in Latino populations should be tested and corrected for population stratification. In addition, we have identified AIMs on the Affymetrix 100K arrays for Latino populations, which may be helpful for future investigators intending to perform admixture mapping or genome-wide association studies in Latino populations, complementing three recent reports of Latino Admixture Mapping panels (Mao et al. 2007; Price et al. 2007; Tian et al. 2007).
The excess of lower p values than expected by chance in the 5q23.2 and 13q13.3 chromosomal regions may suggest the existence of asthma susceptibility loci in these regions in Puerto Rican population (Fig. 5). Multiple genome-wide linkage studies for asthma and related atopic phenotypes have been performed in ethnically diverse populations. Although, the results of these linkage mappings varied across studies and across racial and ethnic groups, regions 5q and 13q are two of the few most frequently reproduced regions in these studies (Koppelman et al. 2002; Ober et al. 1998, 2000; Wiltshire et al. 1998; Xu et al. 2000). It is interesting to note that these regions contain the asthma candidate genes IL4, IL13, and TGFβ, which have been studied extensively (Bartram and Speer 2004; Basehore et al. 2004; Battle et al. 2007; Camoretti-Mercado and Solway 2005; Howard et al. 2002; Li et al. 2007; Marsh et al. 1994). In addition, these regions contain several new potential asthma candidate genes including IL3, IL5, IL9, SMAD5, IRF1, FBN2 and SMAD9 that warrant further investigation. In addition, our genome-wide association analysis suggests some other regions with low p values that may be associated with asthma including 1p22.3, 1p32.2, 4q31.1, 10q23.31, 11q14.1 and 13q22.3 (Table 4). Some of these regions have been shown to be linked with asthma in previous studies (Colilla et al. 2003; Haagerup et al. 2004; Hoffjan and Ober 2002; Huang et al. 2003; Mathias et al. 2006; Pillai et al. 2006; Postma et al. 2005). The detection of causative variants in these regions will require fine mapping and functional validation.
Our results provide strong evidence that the previously linked 5q23 region is associated with asthma in Puerto Ricans. By finding an association in a previously identified region we provide a “proof-of-principle” for genome-wide association studies in admixed populations. Our study underscores the value of applying complementary analytical techniques to admixed populations to uncover genetic risk factors for complex traits.
References
Bartram U, Speer CP (2004) The role of transforming growth factor beta in lung development and disease. Chest 125:754–765
Basehore MJ, Howard TD, Lange LA, Moore WC, Hawkins GA, Marshik PL, Harkins MS, Meyers DA, Bleecker ER (2004) A comprehensive evaluation of IL4 variants in ethnically diverse populations: association of total serum IgE levels and asthma in white subjects. J Allergy Clin Immunol 114:80–87
Battle N, Choudhry S, Tsai HJ, Eng C, Kumar G, Beckman K, Naqvi M, Meade K, Watson HG, LeNoir M, González Burchard E (2007) Ethnic specific gene–gene interaction between IL-13 and IL4R among African American Asthmatics. Am J Resp Crit Care Med (in press)
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29:1165–1188
Buch S, Schafmayer C, Volzke H, Becker C, Franke A, von Eller-Eberstein H, Kluck C, Bassmann I, Brosch M, Lammert F, Miquel JF, Nervi F, Wittig M, Rosskopf D, Timm B, Holl C, Seeger M, ElSharawy A, Lu T, Egberts J, Fandrich F, Folsch UR, Krawczak M, Schreiber S, Nurnberg P, Tepel J, Hampe J (2007) A genome-wide association scan identifies the hepatic cholesterol transporter ABCG8 as a susceptibility factor for human gallstone disease. Nat Genet 39:995–999
Burchard EG, Avila PC, Nazario S, Casal J, Torres A, Rodriguez-Santana JR, Toscano M, Sylvia JS, Alioto M, Salazar M, Gomez I, Fagan JK, Salas J, Lilly C, Matallana H, Ziv E, Castro R, Selman M, Chapela R, Sheppard D, Weiss ST, Ford JG, Boushey HA, Rodriguez-Cintron W, Drazen JM, Silverman EK (2004) Lower bronchodilator responsiveness in Puerto Rican than in Mexican subjects with asthma. Am J Respir Crit Care Med 169:386–392
Burchard EG, Ziv E, Coyle N, Gomez SL, Tang H, Karter AJ, Mountain JL, Perez-Stable EJ, Sheppard D, Risch N (2003) The importance of race and ethnic background in biomedical research and clinical practice. N Engl J Med 348:1170–1175
Camoretti-Mercado B, Solway J (2005) Transforming growth factor-beta1 and disorders of the lung. Cell Biochem Biophys 43:131–148
Cardon LR, Palmer LJ (2003) Population stratification and spurious allelic association. Lancet 361:598–604
Carter-Pokras OD, Gergen PJ (1993) Reported asthma among Puerto Rican, Mexican-American, and Cuban children, 1982 through 1984. Am J Public Health 83:580–582
Chakraborty R, Weiss KM (1988) Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc Natl Acad Sci USA 85:9119–9123
Chen X, Levine L, Kwok PY (1999) Fluorescence polarization in homogeneous nucleic acid analysis. Genome Res 9:492–498
Choudhry S, Coyle NE, Tang H, Salari K, Lind D, Clark SL, Tsai HJ, Naqvi M, Phong A, Ung N, Matallana H, Avila PC, Casal J, Torres A, Nazario S, Castro R, Battle NC, Perez-Stable EJ, Kwok PY, Sheppard D, Shriver MD, Rodriguez-Cintron W, Risch N, Ziv E, Burchard EG (2006) Population stratification confounds genetic association studies among Latinos. Hum Genet 118:652–664
Choudhry S, Seibold MA, Borrell LN, Tang H, Serebrisky D, Chapela R, Rodriguez-Santana JR, Avila PC, Ziv E, Rodriguez-Cintron W, Risch NJ, Burchard EG (2007) Dissecting complex diseases in complex populations: asthma in latino americans. Proc Am Thorac Soc 4:226–233
Choudhry S, Ung N, Avila PC, Ziv E, Nazario S, Casal J, Torres A, Gorman JD, Salari K, Rodriguez-Santana JR, Toscano M, Sylvia JS, Alioto M, Castro RA, Salazar M, Gomez I, Fagan JK, Salas J, Clark S, Lilly C, Matallana H, Selman M, Chapela R, Sheppard D, Weiss ST, Ford JG, Boushey HA, Drazen JM, Rodriguez-Cintron W, Silverman EK, Burchard EG (2005) Pharmacogenetic differences in response to albuterol between Puerto Ricans and Mexicans with asthma. Am J Respir Crit Care Med 171:563–570
Colilla S, Nicolae D, Pluzhnikov A, Blumenthal MN, Beaty TH, Bleecker ER, Lange EM, Rich SS, Meyers DA, Ober C, Cox NJ (2003) Evidence for gene-environment interactions in a linkage study of asthma and smoking exposure. J Allergy Clin Immunol 111:840–846
Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Xu J, Blondal T, Kostic J, Sun J, Ghosh S, Stacey SN, Mouy M, Saemundsdottir J, Backman VM, Kristjansson K, Tres A, Partin AW, Albers-Akkers MT, Godino-Ivan Marcos J, Walsh PC, Swinkels DW, Navarrete S, Isaacs SD, Aben KK, Graif T, Cashy J, Ruiz-Echarri M, Wiley KE, Suarez BK, Witjes JA, Frigge M, Ober C, Jonsson E, Einarsson GV, Mayordomo JI, Kiemeney LA, Isaacs WB, Catalona WJ, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2007) Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 39:631–637
Haagerup A, Borglum AD, Binderup HG, Kruse TA (2004) Fine-scale mapping of type I allergy candidate loci suggests central susceptibility genes on chromosomes 3q, 4q and Xp. Allergy 59:88–94
Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR, Hauser SL (2007) Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 357:851–862
Hakonarson H, Grant SF, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Casalunovo T, Taback SP, Frackelton EC, Lawson ML, Robinson LJ, Skraban R, Lu Y, Chiavacci RM, Stanley CA, Kirsch SE, Rappaport EF, Orange JS, Monos DS, Devoto M, Qu HQ, Polychronakos C (2007) A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 448:591–594
Hanis CL, Hewett-Emmett D, Bertin TK, Schull WJ (1991) Origins of U.S. Hispanics. Implications for diabetes. Diabetes Care 14:618–627
Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR (2005) Whole-genome patterns of common DNA variation in three human populations. Science 307:1072–1079
Hirschhorn JN, Daly MJ (2005) Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6:95–108
Hoffjan S, Ober C (2002) Present status on the genetic studies of asthma. Curr Opin Immunol 14:709–717
Hoggart CJ, Parra EJ, Shriver MD, Bonilla C, Kittles RA, Clayton DG, McKeigue PM (2003) Control of confounding of genetic associations in stratified populations. Am J Hum Genet 72:1492–1504
Hoggart CJ, Shriver MD, Kittles RA, Clayton DG, McKeigue PM (2004) Design and analysis of admixture mapping studies. Am J Hum Genet 74:965–978
Homa DM, Mannino DM, Lara M (2000) Asthma mortality in U.S. Hispanics of Mexican, Puerto Rican, and Cuban heritage, 1990–1995. Am J Respir Crit Care Med 161:504–509
Howard TD, Koppelman GH, Xu J, Zheng SL, Postma DS, Meyers DA, Bleecker ER (2002) Gene-gene interaction in asthma: IL4RA and IL13 in a Dutch population with asthma. Am J Hum Genet 70:230–206
Huang SK, Mathias RA, Ehrlich E, Plunkett B, Liu X, Cutting GR, Wang XJ, Li XD, Togias A, Barnes KC, Malveaux F, Rich S, Mellen B, Lange E, Beaty TH, Asthma CSotGo (2003) Evidence for asthma susceptibility genes on chromosome 11 in an African-American population. Hum Genet 113:71–75
Kennedy GC, Matsuzaki H, Dong S, Liu WM, Huang J, Liu G, Su X, Cao M, Chen W, Zhang J, Liu W, Yang G, Di X, Ryder T, He Z, Surti U, Phillips MS, Boyce-Jacino MT, Fodor SP, Jones KW (2003) Large-scale genotyping of complex DNA. Nat Biotechnol 21:1233–1237
Koppelman GH, Stine OC, Xu J, Howard TD, Zheng SL, Kauffman HF, Bleecker ER, Meyers DA, Postma DS (2002) Genome-wide search for atopy susceptibility genes in Dutch families with asthma. J Allergy Clin Immunol 109:498–506
Laird NM, Horvath S, Xu X (2000) Implementing a unified approach to family-based tests of association. Genet Epidemiol 19 Suppl 1: S36–S42
Li H, Romieu I, Wu H, Sienra-Monge JJ, Ramirez-Aguilar M, Del Rio-Navarro BE, Del Lara-Sanchez IC, Kistner EO, Gjessing HK, London SJ (2007) Genetic polymorphisms in transforming growth factor beta-1 (TGFB1) and childhood asthma and atopy. Hum Genet (in press)
Lind DL, Choudhry S, Ung N, Ziv E, Avila PC, Salari K, Ha C, Lovins EG, Coyle NE, Nazario S, Casal J, Torres A, Rodriguez-Santana JR, Matallana H, Lilly CM, Salas J, Selman M, Boushey HA, Weiss ST, Chapela R, Ford JG, Rodriguez-Cintron W, Silverman EK, Sheppard D, Kwok PY, Gonzalez Burchard E (2003) ADAM33 is not associated with asthma in Puerto Rican or Mexican populations. Am J Respir Crit Care Med 168:1312–1316
Mao X, Bigham AW, Mei R, Gutierrez G, Weiss KM, Brutsaert TD, Leon-Velarde F, Moore LG, Vargas E, McKeigue PM, Shriver MD, Parra EJ (2007) A genomewide admixture mapping panel for Hispanic/Latino populations. Am J Hum Genet 80:1171–1178
Marchini J, Cardon LR, Phillips MS, Donnelly P (2004) The effects of human population structure on large genetic association studies. Nat Genet 36:512–517
Marsh DG, Neely JD, Breazeale DR, Ghosh B, Freidhoff LR, Ehrlich-Kautzky E, Schou C, Krishnaswamy G, Beaty TH (1994) Linkage analysis of IL4 and other chromosome 5q31.1 markers and total serum immunoglobulin E concentrations. Science 264:1152–1156
Mathias RA, Gao P, Goldstein JL, Wilson AF, Pugh EW, Furbert-Harris P, Dunston GM, Malveaux FJ, Togias A, Barnes KC, Beaty TH, Huang SK (2006) A graphical assessment of p-values from sliding window haplotype tests of association to identify asthma susceptibility loci on chromosome 11q. BMC Genet 7:38
Matsuzaki H, Dong S, Loi H, Di X, Liu G, Hubbell E, Law J, Berntsen T, Chadha M, Hui H, Yang G, Kennedy GC, Webster TA, Cawley S, Walsh PS, Jones KW, Fodor SP, Mei R (2004) Genotyping over 100,000 SNPs on a pair of oligonucleotide arrays. Nat Methods 1:109–111
McKeigue PM (1997) Mapping genes underlying ethnic differences in disease risk by linkage disequilibrium in recently admixed populations. Am J Hum Genet 60:188–196
McKeigue PM (1998) Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations, by conditioning on parental admixture. Am J Hum Genet 63:241–251
McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC (2007) A common allele on chromosome 9 associated with coronary heart disease. Science 316:1488–1491
Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, Depner M, von Berg A, Bufe A, Rietschel E, Heinzmann A, Simma B, Frischer T, Willis-Owen SA, Wong KC, Illig T, Vogelberg C, Weiland SK, von Mutius E, Abecasis GR, Farrall M, Gut IG, Lathrop GM, Cookson WO (2007) Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448:470–473
Montana G, Pritchard JK (2004) Statistical tests for admixture mapping with case-control and cases-only data. Am J Hum Genet 75:771–789
Ober C, Cox NJ, Abney M, Di Rienzo A, Lander ES, Changyaleket B, Gidley H, Kurtz B, Lee J, Nance M, Pettersson A, Prescott J, Richardson A, Schlenker E, Summerhill E, Willadsen S, Parry R (1998) Genome-wide search for asthma susceptibility loci in a founder population. The Collaborative Study on the Genetics of Asthma. Hum Mol Genet 7:1393–1398
Ober C, Tsalenko A, Parry R, Cox NJ (2000) A second-generation genomewide screen for asthma-susceptibility alleles in a founder population. Am J Hum Genet 67:1154–1162
Pfaff CL, Parra EJ, Bonilla C, Hiester K, McKeigue PM, Kamboh MI, Hutchinson RG, Ferrell RE, Boerwinkle E, Shriver MD (2001) Population structure in admixed populations: effect of admixture dynamics on the pattern of linkage disequilibrium. Am J Hum Genet 68:198–207
Pillai SG, Chiano MN, White NJ, Speer M, Barnes KC, Carlsen K, Gerritsen J, Helms P, Lenney W, Silverman M, Sly P, Sundy J, Tsanakas J, von Berg A, Whyte M, Varsani S, Skelding P, Hauser M, Vance J, Pericak-Vance M, Burns DK, Middleton LT, Brewster SR, Anderson WH, Riley JH (2006) A genome-wide search for linkage to asthma phenotypes in the genetics of asthma international network families: evidence for a major susceptibility locus on chromosome 2p. Eur J Hum Genet 14:307–316
Postma DS, Meyers DA, Jongepier H, Howard TD, Koppelman GH, Bleecker ER (2005) Genomewide screen for pulmonary function in 200 families ascertained for asthma. Am J Respir Crit Care Med 172:446–452
Price AL, Patterson N, Yu F, Cox DR, Waliszewska A, McDonald GJ, Tandon A, Schirmer C, Neubauer J, Bedoya G, Duque C, Villegas A, Bortolini MC, Salzano FM, Gallo C, Mazzotti G, Tello-Ruiz M, Riba L, Aguilar-Salinas CA, Canizales-Quinteros S, Menjivar M, Klitz W, Henderson B, Haiman CA, Winkler C, Tusie-Luna T, Ruiz-Linares A, Reich D (2007) A genomewide admixture map for Latino populations. Am J Hum Genet 80:1024–1036
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Risch NJ (2000) Searching for genetic determinants in the new millennium. Nature 405:847–856
Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, Konig IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H (2007) Genomewide association analysis of coronary artery disease. N Engl J Med 357:443–453
Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Bostrom K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Rastam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjogren M, Sterner M, Surti A, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M (2007) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316:1341–1345
Smith MW, O’Brien SJ (2005) Mapping by admixture linkage disequilibrium: advances, limitations and guidelines. Nat Rev Genet 6:623–632
Smith MW, Patterson N, Lautenberger JA, Truelove AL, McDonald GJ, Waliszewska A, Kessing BD, Malasky MJ, Scafe C, Le E, De Jager PL, Mignault AA, Yi Z, De The G, Essex M, Sankale JL, Moore JH, Poku K, Phair JP, Goedert JJ, Vlahov D, Williams SM, Tishkoff SA, Winkler CA, De La Vega FM, Woodage T, Sninsky JJ, Hafler DA, Altshuler D, Gilbert DA, O’Brien SJ, Reich D (2004) A high-density admixture map for disease gene discovery in african americans. Am J Hum Genet 74:1001–1013
Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S, Baker A, Snorradottir S, Bjarnason H, Ng MC, Hansen T, Bagger Y, Wilensky RL, Reilly MP, Adeyemo A, Chen Y, Zhou J, Gudnason V, Chen G, Huang H, Lashley K, Doumatey A, So WY, Ma RC, Andersen G, Borch-Johnsen K, Jorgensen T, van Vliet-Ostaptchouk JV, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Rotimi C, Gurney M, Chan JC, Pedersen O, Sigurdsson G, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2007) A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 39:770–775
Stephens JC, Briscoe D, O’Brien SJ (1994) Mapping by admixture linkage disequilibrium in human populations: limits and guidelines. Am J Hum Genet 55:809–824
Terwilliger JD, Weiss KM (1998) Linkage disequilibrium mapping of complex disease: fantasy or reality? Curr Opin Biotechnol 9:578–594
Tian C, Hinds DA, Shigeta R, Adler SG, Lee A, Pahl MV, Silva G, Belmont JW, Hanson RL, Knowler WC, Gregersen PK, Ballinger DG, Seldin MF (2007) A genomewide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am J Hum Genet 80:1014–1023
Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, Wild R, Rowan A, Fielding S, Howarth K, Silver A, Atkin W, Muir K, Logan R, Kerr D, Johnstone E, Sieber O, Gray R, Thomas H, Peto J, Cazier JB, Houlston R (2007) A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet 39:984–988
Wellcome Trust Care Control Consortium (WTCCC) (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678
Wiltshire S, Bhattacharyya S, Faux JA, Leaves NI, Daniels SE, Moffatt MF, James A, Musk AW, Cookson WO (1998) A genome scan for loci influencing total serum immunoglobulin levels: possible linkage of IgA to the chromosome 13 atopy locus. Hum Mol Genet 7:27–31
Winkelmann J, Schormair B, Lichtner P, Ripke S, Xiong L, Jalilzadeh S, Fulda S, Putz B, Eckstein G, Hauk S, Trenkwalder C, Zimprich A, Stiasny-Kolster K, Oertel W, Bachmann CG, Paulus W, Peglau I, Eisensehr I, Montplaisir J, Turecki G, Rouleau G, Gieger C, Illig T, Wichmann HE, Holsboer F, Muller-Myhsok B, Meitinger T (2007) Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat Genet 39:1000–1006
Xu J, Postma DS, Howard TD, Koppelman GH, Zheng SL, Stine OC, Bleecker ER, Meyers DA (2000) Major genes regulating total serum immunoglobulin E levels in families with asthma. Am J Hum Genet 67:1163–1173
Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hunter DJ, Chanock SJ, Thomas G (2007) Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 39:645–649
Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, Ferretti V, Laflamme P, Sundararajan S, Roumy S, Olivier JF, Robidoux F, Sladek R, Montpetit A, Campbell P, Bezieau S, O’Shea AM, Zogopoulos G, Cotterchio M, Newcomb P, McLaughlin J, Younghusband B, Green R, Green J, Porteous ME, Campbell H, Blanche H, Sahbatou M, Tubacher E, Bonaiti-Pellie C, Buecher B, Riboli E, Kury S, Chanock SJ, Potter J, Thomas G, Gallinger S, Hudson TJ, Dunlop MG (2007) Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet 39:989–994
Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, McCarthy MI, Hattersley AT (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316:1336–1341
Ziv E, Burchard EG (2003) Human population structure and genetic association studies. Pharmacogenomics 4:431–441
Acknowledgments
The authors would like to acknowledge the families and the patients for their participation. We would also like to thank the numerous health care providers and community clinics for their support and participation in the GALA Study. Finally, we would like to especially thank Jeffrey M. Drazen, M.D., Scott Weiss, M.D., Ed Silverman, M.D., Ph.D., Homer A. Boushey, M.D., Jean G. Ford, M.D. and Dean Sheppard, M.D. for all of their effort towards the creation of the GALA Study. This work was supported by National Institutes of Health (HL078885), RWJ Amos Medical Faculty Development Award, NCMHD Health Disparities Scholar, Extramural Clinical Research, Loan Repayment Program for Individuals from Disadvantaged Backgrounds, 2001–2003, to EGB), American Thoracic Society “Breakthrough Opportunities in Lung Disease” (BOLD) Award and Tobacco-Related Disease Research Program New Investigator Award (15KT-0008) to SC, and the Sandler Center for Basic Research in Asthma, the Sandler Family Supporting Foundation and the Flight Attendant Medical Research Institute (FAMRI).
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Choudhry and M. Taub contributed equally to the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Choudhry, S., Taub, M., Mei, R. et al. Genome-wide screen for asthma in Puerto Ricans: evidence for association with 5q23 region. Hum Genet 123, 455–468 (2008). https://doi.org/10.1007/s00439-008-0495-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-008-0495-7