
Abstract
Single nucleotide polymorphisms (SNPs) are likely in the near future to have a fundamental role both in human identification and description. However, because allele frequencies can vary greatly among populations, a critical issue is the population genetics underlying calculation of the probabilities of unrelated individuals having identical multi-locus genotypes. Here we report on progress in identifying SNPs that show little allele frequency variation among a worldwide sample of 40 populations, i.e., have a low Fst, while remaining highly informative. Such markers have match probabilities that are nearly uniform irrespective of population and become candidates for a universally applicable individual identification panel applicable in forensics and paternity testing. They are also immediately useful for efficient sample identification/tagging in large biomedical, association, and epidemiologic studies. Using our previously described strategy for both identifying and characterizing such SNPs (Kidd et al. in Forensic Sci Int 164:20–32, 2006), we have now screened a total of 432 SNPs likely a priori to have high heterozygosity and low allele frequency variation and from these have selected the markers with the lowest Fst in our set of 40 populations to produce a panel of 40 low Fst, high heterozygosity SNPs. Collectively these SNPs give average match probabilities of less than 10−16 in most of the 40 populations and less than 10−14 in all but one small isolated population; the range is 2.02 × 10−17 to 1.29 × 10−13. These 40 SNPs constitute excellent candidates for the global forensic community to consider for a universally applicable SNP panel for human identification. The relative ease with which these markers could be identified also provides a cautionary lesson for investigations of possible balancing selection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In a previous paper we discussed the value of SNPs in forensics and presented our strategy for identifying a set of SNPs that are highly informative around the world (Kidd et al. 2006). While others have similarly discussed the utility of autosomal SNPs in forensics and some have presented preliminary panels (Syvanen et al. 1993; Gill et al. 2004; Inagaki et al. 2004; Amorim and Pereira 2005; Vallone et al. 2005; Petkovski et al. 2005; Lee et al. 2005; Dixon et al. 2005; Li et al. 2006; Sanchez et al. 2006), as far as we know, no one previously has thought to propose and seek especially optimal SNP markers with globally low Fst and high heterozygosity to complement the multi-allelic polymorphisms in the CODIS set (Budowle et al. 1998). There are two commonly recognized problems with SNPs replacing STRPs (short tandem repeat polymorphisms) in forensics. One is the inability to reliably detect mixtures, which are a significant occurrence in case work. The other is the inertia created by the large existing databases of standard panels of STRP markers, such as CODIS in the United States. A third problem, we consider even more significant than those two is the population genetics of SNPs. As illustrated in our previous paper (Kidd et al. 2006) a major problem with SNPs is that the frequency of an allele can range from zero to one among different populations, causing a very large dependence of the match probability on the population frequencies used for the calculation, a dependence potentially many fold larger than for CODIS markers. With modern cosmopolitan populations one must know the allele frequencies for all forensically relevant populations to avoid successful courtroom challenges. Thus, we strongly disagree with authors, e.g., Li et al. (2006), who imply that the population genetics of SNPs is not a problem. For a diallelic SNP chosen at random the allele frequency in one population in one region of the world does not predict the frequency in another region of the world with sufficient accuracy and the level of predictability (e.g., correlation of allele frequency profiles between populations) declines fairly rapidly for populations in adjacent geographical regions. Our focus is to identify SNPs for which this problem does not exist.
A SNP with high heterozygosity and essentially identical allele frequencies in all populations would be ideal for several purposes because the probability of unrelated individuals having the same genotype would be nearly constant irrespective of population. In forensics a panel of such markers would not only be easy to defend in court but could constitute a universally applicable forensic panel. While national pride may tend to favor a country-specific forensic panel, a panel with near maximum heterozygosity of all markers in all parts of the world is ideal in cosmopolitan regions and nearly ideal for any one specific population. Moreover, from an economic perspective a single “kit” that can be sold around the world can be produced more cheaply than multiple different “kits” each with limited sales. Gill et al. (2005, 2006) have discussed the value of a common European STRP panel; the same logic extends to a globally applicable, i.e., “universal”, SNP panel. The same characteristics also make such a panel useful in parentage testing. Sample identification and tracking in large biomedical and epidemiologic studies of diverse ethnic origins could also use these SNPs for an efficient initial molecular labeling as the samples are being acquired.
One component of our screening procedure for finding appropriate SNPs was quantifying allele frequency variation among populations using Fst. While many other measures exist, we chose to use Wright’s (1951) Fst because of its population genetics significance including its relationship to the substructure correction factor θ used in forensics [NRC Committee on DNA Technology in Forensic Science, 1996 (NRC Committee 1996)]. Very low Fst assures low allele frequency variation thereby minimizing differences in match probabilities among populations. The 40-population samples we have employed provide a sampling of human genetic variation from all the major continental regions of the world and this set intentionally includes some relatively isolated, inbred samples that help to test the robustness of the marker set since such groups, when included, will tend to increase the magnitude of Fst values. The other screening criterion, high heterozygosity, maximizes the information at each SNP. Thus, the combination of high heterozygosity and low Fst increases the efficiency of a panel for forensic and sample tracking applications, that is, it will take fewer SNPs to produce lower probabilities of identity between two unrelated individuals than if random SNPs are used. Here we report the updated results of our efforts: a panel of 40 SNPs that yields an average match probability in most populations we have studied of less than 10−16. These become candidate SNPs to be considered, among others, by the forensic community for inclusion in a universal identification panel. The screening process has also been illuminating from a population genetics perspective.
Methods
Strategy and criteria
Our screening strategy and criteria were described in detail in our initial report (Kidd et al. 2006). Briefly, we selected markers from a list provided by Applied Biosystems (AB) of a subset of the TaqMan assays in the Assays-on-Demand catalog. AB provided allele identities for those SNPs. We then selected SNPs that had high heterozygosity and minimal allele frequency variation from among the 90,483 SNPs tested by AB in four populations. Because allele frequencies of Japanese and Chinese are very similar and generally different from allele frequencies of the other two populations (European Americans and African Americans), allele frequencies and heterozygosities of Japanese and Chinese were averaged and these averages were used along with those values for African Americans and European Americans in screening the AB database. The 436 SNPs we selected were typed in our laboratory on a total of 371 individuals from seven populations in order to independently sample genetic variation from all major geographical regions [See Supplemental Table 1; Table 1 in Kidd et al. (2006) for the full list of populations and links to the descriptions of the populations and samples in ALFRED, the ALlele FREquency Database (http://www//alfred.med.yale.edu)]. All subjects gave informed consent for genetic marker testing under a human subjects protocol at Yale University as well as other protocols required in various countries of origin. The markers from the seven-population screen that had a Fst ≤ 0.02 and average heterozygosity >0.4 were then tested on an additional 33 populations. Thus, markers making it through the second screen will have been typed on ∼2,070 individuals from 40 populations. These 40-population samples represent one of the best available samples of worldwide human genetic variation. By geographic region the total numbers of individuals tested are: Africa (including African Americans) (459), Southwest Asia (211), Europe (558), Northwest Asia (90), East Asia (345), Northeast Asia/Siberia (51), Pacific Islands (60), North America (105), and South America (191). Our final panel consists of markers with a 40-population Fst below 0.06 and average heterozygosity >0.4. Such markers correspond to the least varying 1.24% of markers studied in our lab for other purposes (Kidd et al. 2004; unpublished data).
Marker typing
Marker typing was done with TaqMan assays ordered from the Assays-on-Demand catalog of AB. We chose these assays so that we could evaluate individual SNPs for appropriateness without having to develop assays. The manufacturer’s protocol was followed using 3 μl reactions in 384-well plates. PCR was done on either an AB9600 or MJ Tetrad. Reactions were read in an AB 7900HT and interpreted using sequence detection system (SDS) 2.1 software. All scans were visually checked for genotype clustering by the software. Assays which failed to give distinct genotype clusters or failed the Hardy–Weinberg test were discarded. All individual DNA samples that failed to give a result, that is, did not fall within an otherwise callable genotype cluster, were repeated once only to provide the final data set.
Analytic methods
Allele frequencies for each marker were estimated by gene counting within each population sample assuming each marker to be a two-allele, co-dominant system. Agreement with Hardy–Weinberg ratios was tested for each marker in each population using a simple Chi-square test comparing the expected and observed number of individuals occurring for each possible genotype. The statistical independence of the markers was assessed by calculating linkage disequilibrium (LD) as r 2 (Devlin and Risch 1995) for all of the 780 unique, pairwise combinations of the final 40 markers within each of the 40 populations. The LD values were then examined in various ways for evidence of meaningful associations among the markers.
The match probability that two unrelated individuals will have the same multi-locus genotype was calculated as described previously (Kidd et al. 2006). The frequency of the most common 40-locus genotype was calculated assuming Hardy–Weinberg ratios and the independence of the loci.
Results
The yield from screening
We screened the 90,483 SNPs that have allele frequencies for four populations (European American, African American, Chinese, and Japanese) and identified 436 markers that we have typed on the seven-population screen. Four failed to show acceptable clusters or failed Hardy–Weinberg ratios in multiple populations and were discarded as unacceptable/unreliable. 73 SNPs or 17% of the remaining 432 had an Fst of 0.02 or less on the seven populations and we typed these on all 40 populations.
The Fst distribution on the seven-population screen is shown in Fig. 1. This is a very “wide” distribution considering that all of these markers had a three-population Fst of 0.01 or less. However, the majority of these Fst values (median = 0.054) are below the mean and median of a distribution of markers unselected for Fst. Our published Fst distribution of 369 similarly unselected SNPs on 38 populations had a mean Fst of 0.138 and a standard deviation of 0.068 (Kidd et al. 2004); a recent update (unpublished) of this distribution has 813 SNPs on 40 populations with a mean of 0.139 and a standard deviation of 0.070.

The Fst distribution for the 432 SNPs successfully followed up on 7 population samples. The seven populations are those flagged with an asterisk in Fig. 3
Figure 2 compares the Fst values for these 73 markers on 7 and 40 populations. Due to the contraction in the range of values studied at this low end of the global, multi-population Fst distribution no significant correlation exists. Having started our screening process with SNPs giving essentially identical allele frequencies in populations representing three regions of the world, we end with a relatively small fraction (∼10%) of SNPs still showing little allele frequency variation when tested on a broader sample of populations from around the world. However, over 50% of those 73 SNPs with low Fst and high heterozygosity on our seven-population screen still met our 40-population criteria.

Scatterplot of the 73 markers tested on all 40 populations by the Fst values for the seven populations in the initial screen and for all 40 populations (All 40 populations are listed in Fig. 3 and those in the seven-population screen are flagged with an asterisk). The 40 SNPs included in the panel are plotted as diamonds, the 33 SNPs with final Fst above 0.06 are plotted as triangles. The Pearson correlation coefficient is 0.02, P = 0.44, NS. Note that nine SNPs have Fst (based on 40 populations) values between 0.06 and 0.07 and that another three SNPs have Fst values of 0.070
The heterozygosities calculated for the initial three populations (>0.45) remain high for the 40 populations (>0.43 for 40 best SNPs and >0.37 for 73 SNPs). The 40 SNPs that met the criterion of an Fst of 0.06 or less for all 40 populations (Fig. 2) are listed in Table 1.
Missing typings were not concentrated in any population sample or SNP. For the seven-population screening (371 individuals) of 432 SNPs, 95.9% of the 160,272 typings succeeded and 4.1% failed. For the individual populations, missing/failed typings ranged from 1.6% in the Cambodians to 5.8% in the Maya. For 2,053 individuals in 40-population samples, 98.8% of the 82,120 possible typings for the 40 best SNPs succeeded and 1.2% failed. An average of 39.51 SNPs were typed per individual; 97.86% of the individuals had typings completed for 36–40 of the SNPs. For individual populations the rate of missing typings ranged from 0.3 (Chagga, Komi Zyrian) to 2.6% (Ethiopians, Nasioi) and had a simple average of 1.2% (1.1% median). For the 40 SNPs individually the rate of missing typings ranged from about 0.1 to 3.9%. So far as we can tell, it was the random occurrences of these few missing typings that resulted in the relatively low (∼76%) frequency of individuals with complete typing results for all 40 SNPs (Table 3).
Independence of the 40 best SNPs
As shown in Table 1, because the ascertainment did not consider chromosomes per se, the 40 best SNPs are distributed across only16 different autosomes with 11 chromosomes having more than 1 SNP. In order to assess the population independence of variation for the 40 markers, all pairwise LD values (r 2) were computed in each of the 40-population samples. The pattern of results across the 780 × 40 = 31,200 LD values clearly supports the conclusion that each SNP contributes essentially independent variation for each of the 40-population samples tested. The vast majority of the r 2 values are close to zero (e.g., the median is 0.010 and the average is 0.029) and these are not statistically different from equilibrium given our sample sizes and the numbers of tests done. The distribution of nominal significance levels is approximately what can be expected by chance with an average across populations of 11.1% of the 780 comparisons in a population nominally significant at the 0.01 level, 3.7% nominally significant at the 0.001 level, and 1.3% nominally significant at the 0.0001 level. An ultraconservative Bonferroni correction assigns the equivalent 1% significance level to 0.0000128 (=0.01/780). In all of these comparisons two populations are noticeable outliers: the Karitiana and Ticuna. Both are known to contain significant numbers of close relatives. While the exact relationships among these samples are not known, the entire Karitiana population is equivalent to a single extended family so a sample of unrelated individuals is an impossibility (Kidd et al. 1993). Inclusion of biological relatives in a sample does not bias gene frequency estimates (Cotterman 1954) but does bias LD measures upward. Not surprisingly, other small populations such as the Rondonian Surui and Samaritans also consistently have among the highest percentages of nominally significant comparisons at all levels of significance.
There is also a positive bias in LD estimates that increases as sample size decreases (Teare et al. 2002). This bias is demonstrated in our results by our examination of the largest LD values ranging from 0.25 to 0.54 to see if they might contain evidence of weak levels of association. There are only 99 LD values in this range, the most extreme one-third of 1% of the 31,200 calculated. Of these 99 largest LD values 88 involve SNPs paired across different chromosomes. There are several reasons for believing these represent chance. We noted above that 780 comparisons were done for each population so that these large LD values that involve different chromosomes likely represent the chance occurrences that can arise when carrying out a large number of comparisons. This seems especially so in conjunction with the bias in LD values for small samples since most of the 99 most extreme LD values involve samples of less than 40 individuals (Fig. 3 ). Because there is no plausible biological explanation for expecting SNP alleles on different chromosomes or those far apart on the same chromosome to be associated only in a few small samples but not in the majority of samples except by chance, we provisionally conclude that all of these large LD values are chance deviations. Larger samples from these populations will be necessary to confirm this but they are not currently available.

The distribution of r 2 values above 0.25 by population. The 40 populations are sorted by sample size (number of individuals in parentheses next to population name) to show clearly that most of these large values occur in the smallest samples as expected from the known bias due to small sample size. The seven populations used in the initial screen are indicated with an asterisk
The 11 of the largest 99 LD values that involve markers located on the same chromosome are also likely due to chance. Table 2 summarizes the LD results for these SNP pairs on the same chromosome that have LD values >0.25. All of the marker pairs in Table 2 have median LD values of 0.03 or less and mean values of 0.06 or less across the 40 populations. Most of these LD values for these pairs of markers are not significantly different from zero in the majority of population samples. These 11 SNP pairs involve distances of at least 2.8 Mb, most from 22 to 108 Mb. All of these distances are at least 10 times larger than the 200 or so kilobases that is the maximum extent of LD usually seen (Peltonen et al. 1999; Varilo and peltonen 2004). As is evident from these very low mean and median values, these maximum LD values are likely global outliers and probably represent chance in light of the many comparisons. Moreover, most of the populations involved are those with the smaller sample sizes and hence the values are biased upward. We expect that independent re-samplings of these populations would not show these associations and provisionally conclude that these 11 SNP pairs in Table 2 are statistically independent. In addition, small inbred populations necessarily contain related individuals and can be expected to show extended LD—the R. Surui (Calafell et al. 1999) and Karitiana (Kidd et al. 1993) account for three of the four smallest intervals in Table 2. The two instances where the number of populations total 39 arise because the Nasioi sample is fixed for an allele at site RSPO3 (row 35 in Table 1). Out of 31,200 LD calculations, only 39 could not be computed due to fixation of an allele for RSPO3 in the Nasioi.
Statistics for the 40-SNP panel
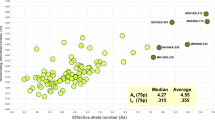
The frequencies of the most probable 40-locus genotype (assuming Hardy–Weinberg ratios) for each population are given in Fig. 4 (by the line connecting the diamond shaped points). Most values are less than 10−12 and the largest value is less than 10−9. The larger values in the small isolated populations are relevant in that they should provide a reasonable upper bound to the match probability in any population.

The frequencies of the most frequent genotype for 40 SNPs in each population are represented by the diamond shaped points. The average match probability for the best 40 markers for each of 40 population samples is represented by the filled-circles. Populations are ordered by geographic region roughly by increasing distance from Africa. The five outliers are highlighted by the population names. These are all small populations that are relatively isolated reproductively
Figure 4 also presents the average match probability by population as shown by the values represented by filled circles. This value is the weighted average of the match probabilities of the 340 possible genotypes, assuming exact H–W ratios within each population. Most populations have values less than 10−16 but the values range across approximately four orders of magnitude, from less than 10−12 to less than 10−16. We note only five populations have values about or larger than 10−15 and in none of those populations are there more than 104 individuals. The probability of discrimination, i.e., the probability that two individuals are different, for each population is one minus the values shown in this figure. Thus, in all populations, the probability of discrimination is greater than 0.999999999999.
Discussion
In terms of the diversity of the populations on which data have been collected this study represents the largest single study to date to find SNPs with globally low Fst and high heterozygosity. The final panel of 40 SNPs has a narrow range for the average match probability across almost all populations. This validates the low Fst, high heterozygosity strategy for identifying SNPs that are appropriate for use in human identification. While Fst depends on the specific set of populations studied, it is clear that a global set of DNA samples can be used to screen for markers with globally low Fst values. A maximum global Fst of 0.06 functions well as a criterion even when small isolated populations are included. Similarly, because we also selected for high heterozygosity, the globally low Fst reflects not just similar allele frequency but also uniformly high heterozygosity. The actual cause of the low Fst in the SNPs we screen is most likely that they are drawn from the lower tail of the distribution of Fst for random neutral SNPs. The fact that 39 of the 40 best SNPs are located in intronic, intergenic, or untranslated regions reinforces this idea; one SNP is located in an exon of the SSTR4 gene and the polymorphism produces a nonsynonomous, missense change. We are not aware of any phenotypic consequences either of this polymorphism or of any polymorphism in linkage disequilibrium with any of the 40 SNPs. However, the possibility of such cannot be excluded.
The data from our step-wise screening also demonstrate an important fact relevant to extrapolating to a global level the allele frequency variation found in a smaller set of population samples. The Fst range for the 90,483 AB markers screened in the three populations we used for our original selection of candidate markers was 5.6 × 10−8 to 0.93, with mean = 0.087 and median = 0.063. Only 14,638 SNPs in this large pool had heterozygosities >0.45 in all three populations and this marker subset had Fst values ranging from 5.6 × 10−8 to 0.1. We selected 436 SNPs to follow-up because they all had an Fst ≤ 0.01 on the initial three populations and the highest heterozygosities out of 2,723 SNPs with Fst ≤ 0.01. Nonetheless, on our seven-population screen we obtained a wide range of Fst values for the successful 432 SNPs extending from 0.003 to 0.232 (mean = 0.054, median = 0.046) (Fig. 1). On the 813 essentially random markers we have tested on these seven populations the Fst range is even larger (range 0.020 to 0.534, mean = 0.139, SD = 0.070), but Fst for these potentially low Fst markers spans half of that range. The same imprecision in extrapolation occurs with our selection of markers with a seven-population Fst ≤ 0.02 for typing on all 40 populations, as can be seen in Fig. 2. There is no correlation between the variation of Fst among SNPs in 40 populations and that in seven populations for this lower tail of the seven-population distribution. When markers are selected in a nearly random manner, there is a high correlation between the Fst seen on these seven populations and on all 40 populations (Kidd et al. 2006), but the present results show the impossibility of accurately predicting or extrapolating to the relative Fst of a larger set of populations from values on a subset, even if that subset includes a set of populations from the four major continents.
We conclude that the 40 SNPs in our “final” panel are statistically independent at the population level. The median (0.01) and mean (0.03) LD values are close to zero and the computed LD values that are nominally significantly different from zero are approximately what would be expected by chance and primarily involve markers on different chromosomes and/or the smallest populations. About 99.68% of all LD values are ≤0.25. The relatively small number of LD values greater than 0.25 (i.e., 99 values or <0.3%) occurred almost entirely between unlinked markers (88 involve SNPs paired from different chromosomes and 2 are >50MB apart on the same chromosome) and predominantly involved different SNP pairs (89 of 99 SNP pairs).
General implications
Two especially interesting aspects of our screening results are (1) the large variation among SNPs in Fst value when additional populations were tested (Figs. 1, 2), (2) yet the relatively high yield of markers having both low Fst values and high heterozygosity when a large number of population samples was studied. The first has implications for the search for balancing selection based solely on data for a small number of populations, such as is true for the HapMap data (The International HapMap Consortium 2003, 2005). The HapMap data are a very valuable resource but cannot be considered to represent the extent of global allele frequency variation very accurately. The second finding also has implications for the search for balancing selection in that there must be a very large number of such SNPs with low Fst and high heterozygosity. It is improbable that most would be maintained by balancing selection. In our screening study of 90,483 AB SNPs we found that 0.0442% or about 4.4 per 10,000 SNPs screened met our criteria for the combination of low Fst and high heterozygosity. Among our other research projects (enriched for SNPs and InDels varying around the world) 11 out of 887 markers screened (1.24%) could be identifed that met the same criteria for low Fst and high heterozygosity. Thus, it may be challenging to unequivocally demonstrate balancing selection in humans against a background of such SNPs.
Discrimination among individuals
Our panel of 40 SNPs resulted in unique genotypes for every one of the individuals with complete typings for all 40 SNPs. The distribution (Table 3) of the number of SNP genotypes matching for the more than 1.22 million pairwise comparisons of 1,568 individuals shows that no individuals match at all the markers. We obtained the nearly symmetric distribution around 15 (out of 40) matches expected by chance and no comparisons with more than 34 matches out of the 40. Thus, even with an occasional typing error generating an incorrect genotype and hence a false match or mismatch, the panel is robust. The expected number of real mismatches between unrelated samples is large enough to be certain of non-identity. A single mismatch between two 40-SNP profiles has a high probability of being an error and should be replicated. One would suspect biological relatedness or errors masking true identity if only a few mismatches occur. This also makes the marker set appropriate for tagging and tracking DNA samples in large biomedical, association, and epidemiological studies.
Toward a universal panel
This preliminary panel of 40 SNPs has excellent characteristics for individual identification, already yielding match probabilities that come close to the theoretical average match probability of just under 10−17 for 40 “perfect” SNPs, i.e., all with heterozygosity equal to 0.5. The yield of 40 acceptable SNPs from an initial set of 436 selected SNPs is encouraging. While our use of Fst < 0.06 is arbitrary, it has proven to be very good at identifying markers with very similar allele frequencies in most populations. As more populations are typed, especially smaller and/or more isolated populations, some of these 40 SNPs may have much less uniformly high heterozygosities. Certainly, their rank order may change and some of the SNPs with Fst just larger than 0.06 may end up better than those with Fst just smaller than 0.06. Therefore, in order to obtain a universally applicable panel of SNPs it will be necessary to have an even larger panel of candidates from which to eventually select a final panel. That panel of candidates must also be sufficiently large that allowance is made for the inability of some markers to be included in multiplexed reactions. Other sources of potentially acceptable SNPs exist. Thousands of additional candidates for screening are available from the HapMap. Other researchers have identified SNPs with high heterozygosity in several diverse populations (e.g., Shriver et al. 2005; Sanchez et al. 2006) corresponding roughly to our seven-population screen. Our 40-population data from other projects can also yield suitable candidates. Thus, the forensic community should have no problem extending the panel of candidates to >>45 SNPs and even reducing the variation among populations provided many candidate markers can be tested on sufficiently large and diverse sets of populations. At the levels of heterozygosity we are achieving, a panel of 45 SNPs would give match probabilities less than 10−18 for most populations, easily in the range achieved with the CODIS markers. Were we to incorporate markers with 0.06 < Fst < 0.07 into the preliminary panel, the variation in average match probability among populations we have studied would increase somewhat, but match probabilities would decrease for all populations.
Our panel should be considered in conjunction with markers in other panels to attempt to reach a consensus among the global research and forensic communities. Among SNP panels that have been proposed for use in individual identification (e.g., Inagaki et al. 2004; Lee et al. 2005; Sanchez et al. 2006), ours is the first to screen simultaneously for high heterozygosity and low Fst in a large global sample of populations. Others have tested only one or a few populations and/or have not imposed a specific criterion of low Fst to evaluate the uniformity of the high heterozygosity. (Note, uniformly high heterozygosity means that the Fst will be low but a low Fst does not mean a high heterozygosity, just a relatively uniform heterozygosity). When allele frequencies have been available for multiple populations, most previously published markers fail our criteria. The SNPforID study (Sanchez et al. 2006) has the next largest set of populations and loci but all but 13 of their 52 markers have either an Fst > 0.06 on their panel of eleven populations (calculations not shown; data from SNPforID web site, http://www.snpforid.org) or an average heterozygosity lower than our criterion of 0.4. (Among those 13 markers, only 5 are unlinked to each other and are unlinked to any of our best SNPs). Their panel of population samples is less diverse geographically than ours, but these 13 SNPs provide another source of markers potentially meeting our criteria when tested on more populations.
Independence in populations versus unlinked in families
Other groups (e.g., Sanchez et al. 2006; Lee et al. 2005) have screened for unlinked SNPs so that the panel would also be appropriate for paternity testing and for forensic work that involved relatives. While all 40 SNPs in our panel are statistically independent at the population level (the objective of our study), several of them are close enough molecularly to show linkage in families. If a universally applicable panel of SNPs is ever adopted by the international forensic community, it would be ideal for all markers in the panel to be both independent at the population level and unlinked.
The syntenic SNPs among the best 40 in our study were examined to determine which would likely show genetic linkage among close biological relatives. The 25 syntenic SNP pairs are separated on average by 37.5 MB but cluster into two very distinct groups—six pairs that are 75 to 172 MB apart and 19 pairs that are all <34 MB apart (median separation ∼15 MB). The six pairs >75 MB apart should be essentially unlinked. An estimate of the genetic map distance between each of the 19 SNP pairs that are <34 MB was obtained via the NCBI MapViewer (http://www.ncbi.nlm.nih.gov/mapview). The nucleotide positions for each pair were entered and the map distance was gauged by averaging the Genethon, deCode, and Marshfield estimates of map distance. A scatterplot (data not shown) of physical distance in MB by map distance in centi-Morgans (cM) for the 19 closest SNP pairs displays a relationship not too different from the genome-wide expectation of roughly one cM per MB although most of the 19 points are above the >1 cM/MB line, such that the median ratio is 1.28 cM/MB and the range is 0.8–2.7 cM/MB. If we eliminate 15 SNPs because of linkage, retaining only the SNP with the best combination of low Fst and high heterozygosity from each set of linked SNPs, the 25 remaining SNPs are both unlinked and independent (Table 1, column 3). However, it is premature to discard any of these syntenic candidate SNPs for at least two reasons. The rank order of the 40 SNPs will likely change as additional populations is tested for these markers. Also, additional appropriate markers identified in the future may be unlinked to some of these syntenic loci but not others.
Some forensic considerations
The values in Figs. 4 and 5 are calculated for ideal populations with no allowance for substructure. As noted by the NRC Committee (1996), the correction factor θ is equivalent to Fst for markers having Hardy–Weinberg ratios, as is the case for all our markers within each population. We assume that any correction factor for substructure within a large ethnically more homogeneous population will be small and not greatly alter the match probabilities for the large populations in Fig. 4 (filled-circles). We note that the relationships of measures of within population substructure to the global Fst are not simple (Balding 2003). However, the similarity of allele frequencies globally greatly reduces the likelihood of substantial allele frequency differences among subgroups within an ethnically heterogeneous population. Moreover, by selecting for a globally low Fst we should also be reducing the likelihood of relevant substructure within each population. For these 40 loci the average “global” (40-population) Fst is 0.047. In an actual forensic application ignoring ethnicity one could use the global average allele frequencies (appropriately weighted from population-specific data available for these 40 SNPs in ALFRED) and the average global Fst as the value of θ used in standard forensic calculations (NRC Committee 1996) to account for global substructure.
Candidate SNPs being considered for forensic applications need to be tested by several laboratories before being introduced into actual casework, both to demonstrate robustness of the methodology and to provide additional population data. Especially, for a potentially universally applicable panel many additional populations will need to be tested and independent samples of those we have studied should be tested. Except for very small endogamous (tribal) populations it seems unlikely that very different allele frequencies will result for the 40 SNPs we have identified since we know from many years of data being accumulated on populations that allele frequencies tend to be similar in geographically close populations (Cavalli-Sforza et al. 1994; Rosenberg et al. 2002; Tishkoff and Kidd 2004). The 40 populations studied here cover most major regions of the world; the regions not covered are flanked by those that have been studied. However, as additional data accumulate on these markers and similar data become available for other markers, the rank order of markers for a universal panel may well change. Also, we would expect the Fst values to increase as more small, isolated populations are studied for these markers. Even so, the frequencies of the most common genotype and the average probabilities of identity are not likely to greatly exceed the ranges seen for the 40 populations that we have studied since we have deliberately included some isolated populations from various parts of the world as test of the robustness/generality of the results. Also important would be independent samples to show that the few large associations among markers are indeed the chance events they seem to be. That may be impossible for the very isolated populations such as the Nasioi because of the cost of a specific expedition as well as the problems of obtaining cooperation of a new group of individuals.
We used TaqMan for the screening procedures because we were screening markers individually and did not have to develop or optimize the assays. While TaqMan low density arrays allow samples to be co-loaded, TaqMan is not capable of being multiplexed for the entire analysis through to the reading of the plate. It is not our intention to advocate any typing protocol nor, at this stage, to invest effort in developing multiplexing for these markers. Because dozens of SNPs can be routinely multiplexed, that is not an issue with modern “chip” methods such as those of Illumina or Affymetrix. Some typing methods might require a different multiplexing procedure and one would need to be developed. One important caveat is that any new typing method must be evaluated to demonstrate that there are not common nearby variants that would interfere with typing the target SNP (e.g., Osier et al. 2002). However, the SNPs we are identifying are in the public domain and any individual or corporation wishing to can work on developing methods for implementing this panel in a forensic or research setting. We do not advocate such effort for a forensic application of this panel. For a research application these SNPs are an efficient small panel but we do note that large numbers of “random” SNPs should also provide uniqueness irrespective of ethnicity. For a forensic application many more candidate SNPs need to be developed and all such need to be tested on more populations. In identifying those candidate SNPs we recommend researchers use screening criteria similar to those we have used because, though arbitrary, they have been demonstrated to yield SNPs with the desirable population genetic characteristics. When larger numbers of appropriate SNPs are available, the best set can be selected both in terms of their population genetics and the ability to develop an appropriate assay for forensic applications.
References
Amorim A, Pereira L (2005) Pros and cons in the use of SNPs in forensic kinship investigation: a comparative analysis with STRs. Forensic Sci Int 150:17–21
Balding DJ (2003) Likelihood-based inference for genetic correlation coefficients. Theor Popul Biol 63:221–230
Budowle B, Moretti TR, Niezgoda SJ, Brown BL (1998) CODIS and PCR-based short tandem repeat loci: law enforcement tools. Second European symposium on human identification, Promega Corporation, Madison, pp 73–88
Calafell F, Shuster A, Speed WC, Kidd JR, Black FL, Kidd KK (1999) Genealogy reconstruction from short tandem repeat genotypes in an Amazonian population. Am J Phys Anthropol 108:137–146
Cavalli-Sforza LL, Menozzi P, Piazza A (1994) The history and geography of human genes. Princeton University Press, Princeton
Cotterman CW (1954) “Estimation of gene frequencies in nonexperimental populations. In: Kempthorne O, Bancroft TA, Gowen JW, Lush JL (eds) Statistics and mathematics in biology. Iowa State College Press, Ames, pp 449–465
Devlin B, Risch N (1995) A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29:311–322
Dixon LA, Murray CM, Archer EJ, Dobbins AE, Koumi P, Gill P (2005) Validation of a 21-locus autosomal SNP multiplex for forensic identification purposes. Forensic Sci Int 154:62–77
Gill P, Werrett DJ, Budowle B, Guerrieri R (2004) An assessment of whether SNPs will replace STRs in national DNA databases—joint considerations of the DNA working group of the European network of forensic science institutes (ENFSI) and the scientific working group on DNA analysis methods (SWGDAM). Sci Justice 44:51–53
Gill P, Fereday L, Morling N, Schneider PM (2005) The evolution of DNA databases—recommendations for new European STR loci. Forensic Sci Int 156:242–244
Gill P, Fereday L, Morling N, Schneider PM (2006) Letter to the Editor: new multiplexes for Europe—amendments and clarification of strategic development. Forensic Sci Int 163:155–157
Inagaki S, Yamamoto Y, Doi Y, Takata T, Ishikawa T, Imabayashi K, Yoshitome K, Miyaishi S, Ishizu H. (2004) A new 39-plex analysis method for SNPs including 15 blood group loci. Forensic Sci Int. 144:45–57
International HapMap Consortium (2003) The international HapMap project. Nature 406:789–796
International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320
Kidd KK, Pakstis AJ, Speed WC, Kidd JR (2004) Understanding human DNA sequence variation. J Hered 95:406–420
Kidd KK, Pakstis AJ, Speed W, Grigorenko E, Kajuna SLB, Karoma N, Kungulilo S, Kim J-J, Lu A, Odunsi R.-B, Okonofua F, Parnas J, Schulz L, Zhukova O, Kidd JR (2006) Developing a SNP panel for forensic identification of individuals. Forensic Sci Int: 164:20–32
Kidd JR, Pakstis AJ, Kidd KK (1993) Global levels of DNA variation. Proceedings of the fourth international symposium on human identification 1993 (Promega), pp 21–30
Lee HY, Park MJ, Yoo J-E, Chung U, Han G-R, Shin K-J (2005) Selection of 24 highly informative SNP markers for human identification and paternity analysis in Koreans. Forensic Sci Int 148:107–112
Li L, Li C-T, Li R-Y, Liu Y, Lin Y, Que T-Z, Sun M-Q, Li Y (2006) SNP genotyping by multiplex amplification and microarrays assay for forensic application. Forensic Sci Int 162:74–79
National Research Council Committee on DNA Technology in Forensic Science (1996) The evaluation of forensic DNA evidence/Committee on DNA forensic science: an update. National Academy Press, Washington D.C.
Osier MV, Pakstis AJ, Goldman D, Edenberg HJ, Kidd JR, Kidd KK (2002) A proline-theronine substitution in codon 351 of ADH1C is common in native Americans. Alcohol Clin Exp Res 26:1759–1763
Peltonen L, Jalanko A, Varilo T (1999) Molecular genetics of the Finnish disease heritage. Hum Mol Genet 8:1913–1923
Petkovski E, Keyser-Tracqui C, Hienne R, Ludes B (2005) SNPs and MALDI-TOF MS: tools for DNA typing in forensic paternity testing and anthropology. J Forensic Sci 50:535–541
Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW (2002) Genetic structure of human populations. Science 298:2381–2385
Sanchez JJ, Phillips C, Borsting C, Balogh K, Bogus M, Fondevila M, Harrison CD, Musgrave-Brown E, Salas A, Syndercombe-Court D, Schneider PM, Carracedo A, Morling N (2006) A multiplex assay with 52 single nucleotide polymorphisms for human identification. Electrophoresis 27:1713–1724
Shriver MD, Mei R, Parra EJ, Sonpar V, Halder I, Tishkoff SA, Schurr TG, Zhadanov SI, Osipova LP, Brutsaert TD, Friedlaender J, Jorde LB, Watkins WS, Bamshad MJ, Guiterrez G, Loi H, Matsuzaki H, Kittles RA, Argyropoulos G, Fernandez JR, Akey JM, Jones KW (2005) Large-scale SNP analysis reveals clustered and continuous patterns of human genetic variation. Hum Genomics 2:81–89
Syvanen AC, Sajantila A, Lukka M (1993) Identification of individuals by analysis of biallelic DNA markers, using PCR and solid-phase minisequencing. Am J Hum Genet 52:46–59
Teare MD, Dunning AM, Durocher F, Rennart G, Easton DF (2002) Sampling distribution of summary linkage disequilibrium measures. Ann Hum Genet 66:223–233
Tishkoff SA, Kidd KK (2004) Implications of biogeography of human populations for race and medicine. Nat Genet 36(suppl):s21-s27
Vallone PM, Decker AE, Butler JM (2005) Allele frequencies for 70 autosomal SNP loci with US Caucasian, African–American, and Hispanic samples. Forensic Sci Int 149:279–286
Varilo T, Peltonen L (2004) Isolates and their potential use in complex gene mapping efforts. Curr Opin Genet Dev 14:316–323
Wright S (1951) The genetical structure of populations. Ann Eugenics 15:323–354
Acknowledgments
This work was funded primarily by NIJ Grant 2004-DN-BX-K025 to KKK awarded by the National Institute of Justice, Office of Justice Programs, US Department of Justice. Points of view in this document are those of the authors and do not necessarily represent the official position or policies of the US Department of Justice. We thank Applied Biosystems for making their allele frequency database available to us. We also want to acknowledge and thank the following people who helped assemble the samples from the diverse populations: F. L. Black, B. Bonne-Tamir, L. L. Cavalli-Sforza, K. Dumars, J. Friedlaender, L. Giuffra, E. L. Grigorenko, S. L. B. Kajuna, N. J. Karoma, K. Kendler, J-J. Kim, W. Knowler, S. Kungulilo, R-B. Lu, A. Odunsi, F. Okonofua, F. Oronsaye, J. Parnas, L. Peltonen, L. O. Schulz, D. Upson, K. Weiss, and O. V. Zhukova. In addition, some of the cell lines were obtained from the National Laboratory for the Genetics of Israeli Populations at Tel Aviv University, Israel, and the African American samples were obtained from the Coriell Institute for Medical Research, Camden, NJ. Special thanks are due to the many hundreds of individuals who volunteered to give blood samples for studies of gene frequency variation.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Pakstis, A.J., Speed, W.C., Kidd, J.R. et al. Candidate SNPs for a universal individual identification panel. Hum Genet 121, 305–317 (2007). https://doi.org/10.1007/s00439-007-0342-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-007-0342-2