
Abstract
We report clinical findings and molecular cytogenetic analyses for two patients with translocations [t(14;17)(p12;p12) and t(15;17)(p12;p13.2)], in which the chromosome 17 breakpoints map at a large low-copy repeat (LCR) and a breakage-prone TRE-2 (USP6) oncogene, respectively. In family 1, a 6-year-old girl and her 5-year-old brother were diagnosed with mental retardation, short stature, dysmorphic features, and Charcot-Marie-Tooth disease type 1A (CMT1A). G-banding chromosome analysis showed a der(14)t(14;17)(p12;p12) in both siblings, inherited from their father, a carrier of the balanced translocation. Chromosome microarray and FISH analyses revealed that the PMP22 gene was duplicated. The chromosome 17 breakpoint was mapped within an ∼383 kb LCR17pA that is known to also be the site of several breakpoints of different chromosome aberrations including the evolutionary translocation t(4;19) in Gorilla gorilla. In family two, a patient with developmental delay, subtle dysmorphic features, ventricular enlargement with decreased periventricular white matter, mild findings of bilateral perisylvian polymicrogyria and a very small anterior commissure, a cryptic duplication including the Miller–Dieker syndrome region was identified by chromosome microarray analysis. The chromosome 17 breakpoint was mapped by FISH at the TRE-2 oncogene. Both partner chromosome breakpoints were mapped on the short arm acrocentric heterochromatin within or distal to the rRNA cluster, distal to the region commonly rearranged in Robertsonian translocations. We propose that TRE-2 together with LCR17pA, located ∼10 Mb apart, also generated the evolutionary gorilla translocation t(4;19). Our results support previous observations that the USP6 oncogene, LCRs, and repetitive DNA sequences play a significant role in the origin of constitutional chromosome aberrations and primate genome evolution.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The majority of recurrent chromosome microdeletion/duplication syndromes result from non-allelic homologous recombination (NAHR) between large (usually >10 kb), highly identical (>95%) low-copy repeat (LCR) structures (Stankiewicz and Lupski 2002; Lupski and Stankiewicz 2006). Recently, the two most frequent recurrent non-Robertsonian constitutional translocations t(11;22)(q23;q11.2) and t(4;8)(p16;p23) have been found to be mediated by the AT-rich cruciform structures in 11q23 and in LCR22-3a in 22q11.2, and by the olfactory receptor-gene cluster LCRs, respectively. However, little is known about the role of genome architecture in the origin of non-recurrent chromosome rearrangements.
Four genomic disorders are caused by constitutional deletion or duplication in proximal 17p, an unstable genomic region that is gene-rich and contains >23% region-specific LCR sequences (Stankiewicz et al. 2003). Charcot-Marie-Tooth type 1A disease (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP) are caused in >99% of cases by copy-number change of a dosage sensitive gene peripheral myelin protein 22 (PMP22) as a result of reciprocal duplication or deletion of a ∼1.4 Mb genomic fragment within 17p12, respectively (Chance et al. 1994; Reiter et al. 1996). This genomic segment is flanked by two ∼24 kb and ∼98.7% identical LCRs, termed proximal and distal CMT1A-REP, which serve as substrates for NAHR (Pentao et al. 1992; Reiter et al. 1997).
The same LCR/NAHR-based mechanism in 17p11.2 results in both an ∼4 Mb deletion del(17)(p11.2p11.2) that is found in 70–80% of patients with Smith–Magenis syndrome (SMS) and its reciprocal duplication in patients with the dup(17)(p11.2p11.2) syndrome (Chen et al. 1997; Potocki et al. 2000; Bi et al. 2003). The rearranged DNA fragment is flanked by proximal (∼256 Kb) and distal (∼176 Kb) SMS-REP LCRs (Chen et al. 1997; Potocki et al. 2000; Park et al. 2002).
We have also identified a novel LCR17p family, with one member, a large ∼383 kb LCR17pA clustering chromosome breakpoints of several different constitutional and evolutionary rearrangements including the translocation t(4;19) in Gorilla gorilla (Stankiewicz et al. 2001a, 2003, 2004, 2005; Shaw et al. 2004a). LCRs in 17p were shown to catalyze non-recurrent chromosomal rearrangements including unusual deletions, translocations, marker chromosomes and complex chromosome rearrangements (Stankiewicz et al. 2001b, 2003; Shaw et al. 2004a; Yatsenko et al. 2005; Lupski and Stankiewicz 2005).
We present the clinical and molecular cytogenetic data of two unique constitutional translocations t(14;17)(p12;p12) and t(15;17)(p12;p13.2), resulting in trisomy 17p12-pter and 17p13.2-pter, respectively. Our data further document that the TRE-2 (USP6) oncogene, LCRs, and repetitive elements in the short arm of acrocentric heterochromatin may all play a significant role in the formation of non-recurrent chromosome translocations.
Material and methods
Patients
We obtained samples from the patients and their family members after acquiring informed consent approved by the Institutional Review Board for Human Subject Research at Baylor College of Medicine and appropriate institutions.
Family 1
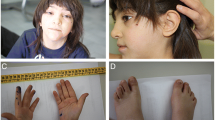
Patient A: The patient is a 6-year-old girl of Mexican–American descent. She is the second child of non-consanguineous parents, both 35 years old. Their first child, a male, was born prematurely at 28 weeks gestation, was ill throughout his life, and died at age 20 months. He reportedly did not have a chromosomal abnormality. The proband was born at 40 weeks gestation by repeat Cesarean section. She has shown developmental delay in fine and gross motor and cognitive areas. She sat at 1 year and walked independently at age 3½ years. At the age of 6 years, she speaks a few intelligible words. On physical examination, her OFC is at 10th centile, weight at 90th centile, and height at 5th centile. She has long straight lashes, large eyes with splotchy melanization of the sclera, down slanting palpebral fissures, protruding ears, micrognathia, and a short neck. She has a transverse smile and mild facial weakness. She has generalized low tone, decreased muscle bulk in bilateral lower limbs, and absent reflexes. She walks on the lateral surface of the left foot with a steppage gait. Gait was improved with ankle–foot orthoses (AFOs), and was even better after reconstructive surgery. An MRI scan of the brain at age 8 months showed callosal hypogenesis, absent anterior commissure, frontal lobe hypoplasia, and paucity of the white matter. MRI of the spine at age 6 years showed mild thoracolumbar scoliosis, and also noted were a left sided superior vena cava and absent right kidney. Nerve conduction velocity testing showed very slow conduction velocities (10–20 m/s) and is consistent with severe demyelination.
The proband’s chromosomal abnormality was diagnosed at age 1 month. Her father is a carrier of a balanced translocation; the mother had a normal karyotype.
Patient B: The 5-year-old brother of the proband has the same unbalanced chromosomal translocation. He was born at 40 weeks by repeat Cesarean section. He sat at age 2 years and walked independently at age 3 years. Presently, he has about five to six words in his vocabulary. His is microcephalic (OFC at 2nd centile), his weight is at 40th centile, and height at 2nd centile. Dysmorphic features were significant for downslanting palpebral fissures, mild ptosis bilaterally, splotchy melanization of sclera, long straight eyelashes, depressed nasal bridge, protruding ears, chronically open mouth, micrognathia, facial weakness, short neck, and tapered fingers. Musculoskeletal exam was remarkable for tapered calves and bilateral moderate to severe varus foot deformities. His eye movements are intact and pupils equally react to light. He has facial weakness, palate is slightly high-arched, and reflexes are absent. He had a steppage gait which improved by both AFOs, and foot reconstruction surgery. Head MRI showed callosal hypogenesis and microcephaly. An MRI scan of the spine is normal.
Family 2
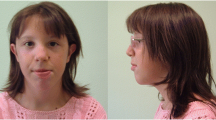
Patient C is a 4-year-old boy referred because of developmental delay. The pregnancy was uneventful except for maternal hypertension. Amniocentesis performed for advanced maternal age revealed a normal male karyotype. He was delivered spontaneously at 38 weeks gestation with a weight of 3.2 kg. The Apgar scores were 9 and 10 at 1 and 5 min, respectively. A heart murmur was discovered in the neonatal period and echocardiography revealed pulmonary valvular stenosis and a ventricular septal defect, which spontaneously closed during the first year of life. He showed global developmental delay. He first walked at 20 months of age. At last evaluation he could form small sentences but pronunciation was not clear. He could draw circles and lines but could not draw a face. He was toilet-trained at 2½ years of age. He did not show any behavioral abnormality. He had strabismus and at age 2½ years successfully underwent surgery. At 4 years of age, his weight was 15.7 kg (25th centile), his height was 101.1 cm (25th centile) and his head circumference was 51.7 cm (50th centile). Physical examination revealed subtle facial dysmorphic features including a broad nasal tip, hypoplastic alae nasi and a small mouth. The hands were characterized by the presence of bilateral clinodactyly of the fifth finger. Deep tendon reflexes were present. Head CT performed at 19 months showed a mild ventricular enlargement involving asymmetrically both lateral ventricles. This enlargement was secondary to a decrease in periventricular white matter in the parietal lobes. The subarachnoid spaces were normal. The frontal horns appeared slightly “squared off” suggesting some degree of ventricular dysmorphism. Brain MRI done at 2½ years of age confirmed the ventricular enlargement and the decrease of the periventricular white matter but showed mild findings of bilateral perisylvian polymicrogyria. The corpus callosum appeared thin but completely formed. The anterior commissure was very small (Fig. 1). The family history was unremarkable.

Brain imaging in patient C. a Axial T2 MR image showing periventricular white matter loss and very small anterior commissure (arrowhead). b Axial T1 MR image showing mild enlargement of lateral ventricles secondary to periventricular white matter loss and subtle signs of bilateral perisylvian polymicrogyria (arrowheads)
FISH analysis
A peripheral blood sample was obtained from the patients and their parents and whole blood lymphocytes were cultured with phytohemagglutinin (PHA) using standard methods. BAC and PAC probes specific for human chromosome region 17p12-p13 were identified from the existing physical maps of this region (Inoue at al. 2001; Bi et al. 2002; NCBI, http://www.ncbi.nlm.nih.gov/; UCSC genome browser, http://www.genome.ucsc.edu) and purchased from the BACPAC Resource Center (Oakland, CA, USA) (Tables 1, 2). For mapping the breakpoints in the short arms of chromosomes 14 and 15, we used an rDNA-specific probe pA (the 28S rRNA gene) (Sylvester et al. 1986) and pU6.2 (the 18S rRNA gene) (Wilson et al. 1978). FISH was performed as previously described (Shaffer et al. 1997).
Array-CGH
A microarray containing 853 BAC and PAC clones designed to cover genomic regions of 75 known genomic disorders, all 41 subtelomeric regions, and 43 pericentromeric regions were used for chromosomal microarray analysis (Cheung et al. 2005; Baylor College of Medicine, Chromosome Microarray Analysis, V.5, http://www.bcm.edu/cma/assets/abnormalities.pdf).
The DNA from BAC and PAC clones was prepared using a standard alkaline lysis method and chemically modified for array printing and the procedures for DNA labeling and hybridization were performed as previously described in detail (Yu et al. 2003). Genomic DNA was isolated from peripheral blood lymphocytes using a PureGene DNA purification kit (Gentra Systems, Mineapolis, MN, USA) following the manufacturer’s protocol. DNA was digested with restriction enzyme DpnII (New England Biolabs, Beverly, MA, USA) and purified using phenol/chloroform extraction.
Genomic DNAs extracted from the patients and from a gender-matched control sample were differentially labeled with Cyanine-3 (Cy3) and Cyanine-5 (Cy5) (Perkin Elmer, Boston, MA, USA) using a Bioprime DNA direct labeling kit (Invitrogen, Carlsbad, CA, USA) and hybridized onto the arrays at 37°C for 24 h. The microarray slides were washed at 45°C twice with 50% formamide/2XSSC for 10 and 15 min and once with 0.2XSSC for 5 min. The resulting fluorescent signals on the slides were scanned into image files using an Axon microarray scanner and ScanArray software (GenePix 4000B from Axon Instruments, Union City, CA, USA). For each sample, two experiments were performed with reversal of the dye labels for the control and test samples, and the data from both dye-reversed hybridizations were integrated to determine inferences for each case.
Microarray image files were quantified using GenePix Pro 4 software. The quantization data were subjected to normalization as described (Shaw et al. 2004b).
DNA sequence analysis
Sequences of the analyzed clones were downloaded from the NCBI and UCSC web sites (http://www.ncbi.nlm.nih.gov/; http://genome.ucsc.edu/). Searches for the LCRs in 17p13.2 were performed using NCBI BLAST against the high-throughput and the non-redundant sequence databases (http://www.ncbi.nlm.nih.gov/blast/) and assembled using the Sequencher software (Gene Codes) and NCBI BLAST 2.
Results
Family 1
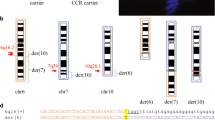
GTG-banded chromosome analysis of patients A and B revealed a derivative chromosome 14, der(14)t(14;17)(p12;p12) (Fig. 2a), resulting in trisomy 17p12-pter, inherited from the father, who is a carrier of the balanced translocation. In chromosome microarray analysis, an increment in log2 ratio (all around or above 0.2) was observed for all of the 15 clones in 17p12-pter, compared to the average log2 of the whole genome normalized as 0 (Fig. 2b). A dramatic transition was observed between clone RP11–726O12 (log2 ratio=0.278) and RP11–692E18 (log2 ratio=0.076), indicating that a breakpoint of the translocation maps in the genomic fragment between these two clones (Table 1) in a large ∼383 kb LCR, LCR17pA, that has been identified previously at this locus (Stankiewicz et al. 2003). Subsequent FISH studies using a PMP22 probe confirmed duplication of this gene and mapped the breakpoint within two overlapping BAC clones CTD-3157E16 and RP11-692E18 (Fig. 2c), at the centromeric edge of LCR17pA. The 14p breakpoint was mapped within or distal to the rDNA sequences on 14p12 (Fig. 2d).

a Partial G-banded karyotype of the patient A in family 1. b Array based comparative genomic hybridization. This profile represents two hybridizations performed simultaneously with dye reversal using reference DNA. In the column marked “raw” for raw data, the mean values of the T/R ratio and error bars in a hybridization are shown in blue and dye reversal is shown in red. The effect of normalization is shown by comparing the middle set of data marked “normalized” with the “raw” data. There are several clones from the distal portion of chromosome 17p that show displacement to the left in blue and to the right in the dye reversal, both indicating a gain of 17p material in the patient versus the reference DNA. In the “combined” column, the sign of one of the two reversed hybridizations is changed and the data are averaged with gains shown to the right and losses to the left. For the combined data, there is a strong indication of a gain detected with 15 clones corresponding to the 17p region from 17p12 to 17pter. The library source of the clone, the location and the T/R ratio are listed in Table 1. c Patient A metaphase chromosomes after FISH with BAC clone CTD-3157E16 specific for LCR17pA showed that this clone flanks the breakpoint on its distal side [der(14) shown by arrow]. d FISH with rDNA probe pU6.2 (green) specific for the p arm of acrocentric chromosomes demonstrated the presence of rDNA on der(14) (white arrow). The clone RP11-820M16 (green) was used as a chromosome 14qter control probe
Family 2
Initial GTG-banding chromosome analysis on patient’s peripheral blood lymphocytes performed in another laboratory showed a normal male karyotype. Subtelomeric FISH analysis showed an additional signal of the telomeric region of chromosome 17p in the distal short arm of one chromosome 15, resulting in duplication of 17pter. The retrospective karyotype analysis at the 600-band resolution revealed the presence of a very small amount of additional material on 15p12 (Fig. 3a). This abnormality was not found in either parent. Subsequent CMA revealed duplication with the most distal seven clones from 17p included on the array, indicating that the breakpoint mapped between clones RP11–115H24 and RP11–960B9 (Table 2; Fig. 3b). FISH analysis narrowed the breakpoint to within the sequence contained in two overlapping clones RP11-333E1 and RP11-420A6 at the TRE-2 (USP6) oncogene (Fig. 3c). By computational analysis, this genomic segment showed a significant (∼95%) sequence homology to a few LCRs on chromosome 17 including short fragments (∼3, 9, and 11 kb) constituting the SMS-REPs (Park et al. 2002) and a cluster of the CCL3 chemokine ligand genes (Gonzalez et al. 2005) interspersed with one of two TRE-2 ancestor genes, TBC1D3. Interestingly, the BLAST analysis revealed that the middle ∼25 kb portion of this LCR has a 95% sequence identity only to a clone 413A16 from the gorilla library CHORI-255, that we previously found to be spanning the evolutionary translocation t(4;19) (Fig. 4) (Stankiewicz et al. 2001a, 2004). In addition, a fossil of a Charlie 3 DNA transposon, previously identified at the evolutionary translocation t(4;19) (Stankiewicz et al. 2004), was found at the edge of the TRE2 (USP6) gene. Similar to family 1, the 15p breakpoint was mapped within or distal to the rDNA sequences on 15p12 (Fig. 3d).

a Partial G-banded karyotype for chromosome 17 in family 2 with arrow indicating the der (17) chromosome. b Array CGH has been performed simultaneously with dye reversal using reference DNA. Seven most distal clones corresponding to 17p13.2-pter showed log2 ratio >0.2, indicating gain of genetic material in the patient versus the reference DNA (also see Table 2 for details). c Metaphase FISH using overlapping BAC clones RP11-333E1 (red) and RP11-420A6 (green) showed the fluorescence signal on der(15) only for RP11-333E1 and narrowed the 17p13.2 breakpoint within these clones. d FISH with rDNA probes (pA and pU6.2—both red) demonstrated the presence of rDNA on der(15) (white arrow). The subtelomere clones RP11-90E5 (green) and RP11-46E14 (red) were used as control probes for chromosomes 15 and 17, respectively

The BLAST analysis of the ∼65 kb LCR harboring TRE-2 revealed DNA sequence homology to nine genomic loci: two TRE-2 ancestor genes: TBC1D3 (17q12), USP32 (17q23.2), all three SMS-REPs in 17p11.2, LCRs on 17q23.3, 17q11.2, chemokine gene cluster at 17q12, and the gorilla clone CHORI-255-413A16 (see Results for details). Note that the genomic structure of the gorilla clone 413A16, that spans the evolutionary translocation t(4;19), resembles perfectly the sequence of the TRE-2 gene, indicating that this gene likely has played an important role in the formation of this translocation t(4;19)
BLAST analysis of TRE2 revealed nine segmental duplications: TBC1D3 (17q12, 93%, RP11-493E8/AC027821), USP32 (17q23.2, 94%, RP11-3K24/AC104763), all three SMS-REPs in 17p11.2 (95%), LCRs on 17q23.3 (94%, RP11-51L5/AC053481), 17q11.2 (NF1-REP, 89%, RP11-271K11/AC005562), chemokine gene cluster at 17q12 (93%, 91J4/AC003976), and the gorilla clone CHORI-255-413A16 (95%). Interestingly, an ∼11 kb portion of TRE-2 has been found to be a core element in several segmental duplications in chromosome 17, including SMS-REPs (Fig. 5) (Park et al. 2002; Zody et al. 2006).

Schematic representation of genomic architecture in proximal 17p. Depiction of proximal chromosome 17p showing the position and orientation of LCRs. The LCR17p structures are depicted in colors to better represent their position and orientation with respect to each other; the colored rectangles and horizontal arrowheads represent the orientation of the LCRs. Black arrows indicate breakpoints of chromosome translocations, interstitial deletions, complex duplication and recurrent isodicentric chromosome 17q (Stankiewicz et al. 2003; Barbouti et al. 2004). The translocation breakpoint in the present case is shown with a red arrow. The evolutionary gorilla translocation breakpoint is indicated by a vertical arrow between BAC clones RP11-640I15 and CTD-3157E15 and LCR17pA/B and LCR17pA/D subunits of the LCR17pA copy (Stankiewicz et al. 2004). The LCRs that are >20 Kb are depicted
Discussion
Patients with trisomy of the short arm of chromosome 17 are affected with a psychomotor delay, pre- and post-natal growth retardation, hypotonia, microcephaly, minor craniofacial anomalies, hypertrichosis, mild skeletal anomalies and congenital heart defects (Jinno et al. 1982; Mascarello et al. 1983; Magenis et al. 1986; Martsolf et al. 1988; Schrander-Stumpel et al. 1990; Spinner et al. 1993; Lurie et al. 1995; Kulharya et al. 1998; Morelli et al. 1999; De Pater et al. 2000).
Partial trisomy of the short arm of chromosome 17 involving the sub-band 17p11.2 due to duplication of the SMS critical region shows mild developmental delay, neurobehavioral abnormalities and minor craniofacial anomalies (Brown et al. 1996; Balarin et al. 1999; Potocki et al. 2000; Schneider et al. 2000). Patients with larger proximal 17p duplication including the PMP22 gene within 17p12 are affected with CMT1A (Chance et al. 1992; Lupski et al. 1992; Upadhyaya et al. 1993; Roa et al. 1996; King et al. 1998; Lupski and Garcia 2001).
Two siblings in family 1 are duplicated for the PMP22 gene and, as anticipated, demonstrate clinical findings consistent with CMT1A peripheral neuropathy (absent reflexes, foot deformities, distal muscle wasting and severely slow nerve conduction velocities) but in addition display a more complex phenotype (microcephaly, mental retardation, craniofacial anomalies, and callosal hypogenesis) likely due to the more extensive genomic duplication.
A common microdeletion of chromosome 17p13.3 involving two dosage sensitive genes LIS1 and 14-3-3 epsilon results in a well known neuronal migration disorder Miller–Dieker lissencephaly syndrome (MDLS) (Dobyns et al. 1992; Reiner et al. 1993; Reiner et al. 1995; Chong et al. 1997; Lo Nigro et al. 1997; Toyo-oka et al. 2003; Cardoso et al. 2003). Isolated duplications of this genomic region have not been described yet. Recently, Ensenauer et al. (2004) and Hwang et al. (2005) reported duplication of the MDLS region due to unbalanced translocations t(5;17)(p15.31;p13.1) and t(17;18)(p13.2;q22.3), respectively. The abnormal phenotypes included developmental delay, growth retardation, microcephaly, flat midface, prominent forehead, down slanting palpebral fissures, hypertelorism, short nose with upturned nares, bitemporal hollowing, low-set ears, micrognathia, short webbed neck, congenital heart defect and clinodactyly of fifth fingers.
Patient C manifests with developmental delay, subtle dysmorphic features including a broad nasal tip, hypoplastic alae nasi and a small mouth and ventricular enlargement with a decrease of the periventricular white matter, mild findings of bilateral perisylvian polymicrogyria and very small anterior commissure and thus defines trisomy 17p13.2-pter. We propose that the neurodevelopmental abnormalities most likely result from increased dosage of LIS1 and 14-3-3 epsilon. Using chromosome microarray analysis, a few patients with isolated submiscroscopic duplications involving the MDLS chromosome region have been identified (S.-W. Cheung, personal communication). Interestingly, the MRI findings also revealed neuronal migration anomalies (Sahoo et al., manuscript in preparation).
Recently, we showed that genomic architecture of proximal 17p also catalyzes non-recurrent chromosomal rearrangements (Stankiewicz et al. 2003). To further investigate the molecular bases of non-recurrent aberrations, we analyzed the breakpoint regions of both translocations.
The chromosome 17 breakpoint of t(15;17)(p12;p13.2) was mapped at the TRE-2 (USP6) gene, a hominoid-specific chimeric oncogene, product of fusion between the TBC1D3 (17q12) and USP32 (17q23.2) ancestral genes (Paulding et al. 2003). TRE-2 encodes a ubiquitin-specific protease and has been found frequently fused with other genes: CDH11, TRAP150, ZNF9, osteomodulin, and COL1A1 in aneurysmal bone cysts (Oliveira et al. 2004a, b, 2005; Althof et al. 2004). TRE-2 has been identified also at the breakpoint of a constitutional translocation t(13;17)(q14;p13) in a patient with Asperger syndrome (Tentler et al. 2003), indicating that it is responsible for genome instability both in tumors and during gametogenesis.
Since SMS-REPs are absent in the mouse genome and have arisen during primate speciation, we speculate that TRE-2 has played an important role in their formation. BLAST analysis revealed that ∼50 kb fragment of TRE-2 has a ∼95% homology exclusively to the gorilla clone spanning the evolutionary translocation t(4;19) breakpoint in LCR17pA (localized ∼10 Mb proximal to TRE-2), indicating that it also participated in the formation of this rearrangement. Moreover, a remnant of a Charlie 3 DNA transposon, identified at the evolutionary translocation t(4;19), was found at the edge of the TRE-2 gene. We have suggested previously that this event might have occurred in a testis of a pre-gorilla individual and was subsequently transmitted, implanted, and accumulated as heterozygous and fixed to the homozygous state due to inbreeding in a small “bottleneck population” (Stankiewicz et al. 2001a). Interestingly, in contrast to the broad expression pattern of its ancestral genes TBC1D3 and USP32, TRE-2 is transcribed exclusively in testes, and has been proposed to play a role in speciation (Paulding et al. 2003).
We have mapped previously the breakpoint of an unbalanced translocation t(10;17)(q26.3;p11.2) just adjacent to the centromeric end of the distal SMS-REP, in the direct vicinity of the evolutionarily unstable portion of the distal SMS-REP, an interstitial ∼39 kb deletion of the genomic segment encompassing TRE-2 pseudogenes (between the KER and CLP) (Park et al. 2002; Stankiewicz et al. 2003). These data demonstrate that TRE-2, or surrounding genomic sequences are a major genome instability factor.
The breakpoint of t(14;17)(p12;p12) has been mapped at the centromeric edge of the LCR17pA (Fig. 5). An ∼383 kb LCR17pA is a breakage-prone genomic region and has been shown to harbor several chromosome breakpoints, including six uncommon but recurrent ∼5 Mb SMS deletions, two unusual sized SMS deletions, marker chromosome, two duplications, complex submicroscopic duplication, and the evolutionary translocation t(4;19) in Gorilla gorilla (Stankiewicz et al. 2001a, b, 2003; Shaw et al. 2004a) (Fig. 5).
Interestingly, in both translocations described herein, the partner chromosome breakpoints were mapped within rDNA in the short arm of acrocentric heterochromatin, suggesting that, together with LCRs, highly repetitive elements may also mediate the formation of chromosome translocations. In conclusion, our results suggest that together with LCRs, the oncogene TRE2 plays an important role not only in primate genome evolution and cancers, but also in the formation of constitutional chromosome translocations.
References
Althof PA, Ohmori K, Zhou M, Bailey JM, Bridge RS, Nelson M, Neff JR, Bridge JA (2004) Cytogenetic and molecular cytogenetic findings in 43 aneurysmal bone cysts: aberrations of 17p mapped to 17p13.2 by fluorescence in situ hybridization. Mod Pathol 17:518–525
Balarin MAS, da Silva Lopes VLG, Varella-Garcia M (1999) A dup(17)(p11.2p11.2) detected by fluorescence in situ hybridization in a boy with Alport syndrome. Am J Med Genet 82:183–186
Barbouti A, Stankiewicz P, Nusbaum C, Cuomo C, Cook A, Birren B, Höglund M, Johansson B, Hagemeijer A, Park S-S, Mitelman F, Lupski JR, Fioretos T (2004) The breakpoint region of the most common isochromosome, i(17q), in human neoplasia is characterized by a 220 kb region containing palindromic low-copy repeats. Am J Hum Genet 74:1–10
Bi W, Yan J, Stankiewicz P, Park S-S, Walz K, Boerkoel CF, Potocki L, Shaffer LG, Devriendt K, Nowaczyk MJM, Inoue K, Lupski JR (2002) Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. Genome Res 12:713–728
Bi W, Park SS, Shaw CJ, Withers MA, Patel PI, Lupski JR (2003) Reciprocal crossovers and a positional preference for strand exchange in recombination events resulting in deletion or duplication of chromosome 17p11.2. Am J Hum Genet 73:1302–1315
Brown A, Phelan MC, Patil S, Crawford E, Rogers RC, Schwartz C (1996) Two patients with duplication of 17p11.2: the reciprocal of the Smith-Magenis syndrome deletion? Am J Med Genet 63:373–377. Erratum in Am J Med Genet 65:254
Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, Martin CL, Allanson J, Pilz DT, Olney AH, Mutchinick OM, Hirotsune S, Wynshaw-Boris A, Dobyns WB, Ledbetter DH (2003) Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet 72:918–930
Chance PF, Bird TD, Matsunami N, Lensch MW, Brothman AR, Feldman GM (1992) Trisomy 17p associated with Charcot-Marie-Tooth neuropathy type 1A phenotype: evidence for gene dosage as a mechanism in CMT1A. Neurology 42:2295–2299
Chance PF, Abbas N, Lensch MW, Pentao L, Roa BB, Patel PI, Lupski JR (1994) Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Mol Genet 3:223–228
Chen K-S, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault AC, Lee CC, Lupski JR (1997) Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet 17:154–163
Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, Lupski JR, Chinault AC, Beaudet AL (2005) Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med 7:422–432
Chong SS, Pack SD, Roschke AV, Tanigami A, Carrozzo R, Smith ACM, Dobyns WB, Ledbetter DH (1997) A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet 6:147–155
De Pater JM, Van Tintelen JP, Stigter R, Brouwers HAA, Scheres JMJC (2000) Precarious acrocentric short arm in prenatal diagnosis: no chromosome 14 polymorphism, but trisomy 17p. Genet Couns 11:241–247
Dobyns WB, Elias ER, Newlin AC, Pagon RA, Ledbetter DH (1992) Causal heterogeneity in isolated lissencephaly. Neurology 42:1375–1388
Ensenauer R, Jalal S, Meyer R, Babovic-Vuksanovic D (2004) Unbalanced cryptic 5p deletion/17p duplication identified by subtelomeric FISH in a family with a boy with chimerism and a balanced t(4;5). Am J Med Genet 125A:86–91
Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O’Connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK (2005) The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307:1434–1440
Hwang KS, Pearson MA, Stankiewicz P, Lennon PA, Cooper ML, Wu J, Ou Z, Cai W-W, Patel A, Cheung SW (2005) Cryptic unbalanced translocation t(17;18)(p13.2;q22.3) identified by subtelomeric FISH and defined by array-based comparative genomic hybridization in a patient with mental retardation and dysmorphic features. Am J Med Genet 137A:88–93
Inoue K, Dewar K, Katsanis N, Reiter LT, Lander ES, Devon KL, Wyman DW, Lupski JR, Birren B (2001) The 1.4-Mb CMT1A duplication/HNPP deletion genomic region reveals unique genome architectural features and provides insights into the recent evolution of new genes. Genome Res 11:1018–1033
Jinno Y, Matsuda I, Kajii T (1982) Trisomy 17p due to a t(5;17)(p15;p11)pat translocation. Ann Genet 25:123–125
King PH, Waldrop R, Lupski JR, Shaffer LG (1998) Charcot-Marie-Tooth phenotype produced by a duplicated PMP22 gene as part of a 17p trisomy-translocation to the X chromosome. Clin Genet 54:413–416
Kulharya AS, Garcia-Heras J, Radtke HB, Norris KS, Keppen LD, Flannery DB (1998) Prenatal diagnosis of a trisomy 17p derived from a de novo non-mosaic satellited marker. Clin Genet 54:421–425
Lo Nigro C, Chong SS, Smith ACM, Dobyns WB, Carrozzo R, Ledbetter DH (1997) Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet 6:157–164
Lupski JR, Garcia CA (1992) Molecular genetics and neuropathology of Charcot-Marie-Tooth disease type 1A. Brain Pathol 2:337–349
Lupski JR, Garcia CA (2001) Charcot-Marie-Tooth peripheral neuropaties and related disorders. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs B, Kinzler KW, Vogelstein B (eds) The metabolic & molecular bases of inherited diseases, vol IV. McGraw-Hill, New York, pp 5759–5788
Lupski JR, Stankiewicz P (2005) Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet 1:e49
Lupski JR, Stankiewicz P (2006) Genomic disorders: the genomic basis of disease. Humana Press, Totowa
Lurie IW, Gurevich DB, Binkert F, Schinzel A (1995) Trisomy 17p11-pter: unbalanced pericentric inversion, inv(17)(p11q25) in two patients, unbalanced translocations t(4;17)(q27;p11) in a newborn and t(4;17)(p16;p11.2) in a fetus. Clin Dysmorphol 4:25–32
Magenis RE, Brown MG, Allen L, Reiss J (1986) De novo partial duplication of 17p [dup(17)(p12→p11.2)]: clinical report. Am J Med Genet 24:415–420
Martsolf JT, Larson L, Jalal SM, Wadahl WA, Miller R, Kukolich M (1988) Complete trisomy 17p a relatively new syndrome. Ann Genet 31:172–174
Mascarello JT, Jones MC, Hoyme HE, Freebury MM (1983) Duplication (17p) in a child with an isodicentric (17p) chromosome. Am J Med Genet 14:67–72
Morelli SH, Deubler DA, Brothman LJ, Carey JC, Brothman AR (1999) Partial trisomy 17p detected by spectral karyotyping. Clin Genet 55:372–375
Oliveira AM, Hsi B-L, Weremowicz S, Rosenberg AE, Dal Cin P, Joseph N, Bridge JA, Perez-Atayde AR, Fletcher JA (2004a) USP6 (Tre2) fusion oncogenes in aneurysmal bone cyst. Cancer Res 64:1920–1923
Oliveira AM, Perez-Atayde AR, Inwards CY, Medeiros F, Derr V, Hsi B-L, Gebhardt MC, Rosenberg AE, Fletcher JA (2004b) USP6 and CDH11 oncogenes identify the neoplastic cell in primary aneurysmal bone cysts and are absent in so-called secondary aneurysmal bone cysts. Am J Pathol 165:1773–1780
Oliveira AM, Perez-Atayde AR, Dal Cin P, Gebhardt MC, Chen C-J, Neff JR, Demetri GD, Rosenberg AE, Bridge JA, Fletcher JA (2005) Aneurysmal bone cyst variant translocations upregulate USP6 transcription by promoter swapping with the ZNF9, COL1A1, TRAP150, and OMD genes. Oncogene 24:3419–3426
Park S-S, Stankiewicz P, Bi W, Shaw C, Lehoczky J, Dewar K, Birren B, Lupski JR (2002) Structure and evolution of the Smith-Magenis syndrome repeat gene clusters, SMS-REPs. Genome Res 12:729–738
Paulding CA, Ruvolo M, Haber DA (2003) The Tre2 (USP6) oncogene is a hominoid- specific gene. Proc Natl Acad Sci USA 100:2507–2511
Pentao L, Wise CA, Chinault AC, Patel PI, Lupski JR (1992) Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat Genet 2:292–300
Potocki L, Chen K-S, Park S-S, Osterholm DE, Withers MA, Kimonis V, Summers AM, Meschino WS, Anyane-Yeboa K, Kashork CD, Shaffer LG, Lupski JR (2000) Molecular mechanism for duplication 17p11.2- the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet 24:84–87
Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH (1993) Isolation of a Miller-Dieker lissencephaly gene containing G protein β-subunit-like repeats. Nature 364:717–721
Reiner O, Albrecht U, Gordon M, Chianese KA, Wong C, Gal-Gerber O, Sapir T, Siracusa LD, Buchberg AM, Caskey CT, Eichele G (1995) Lissencephaly gene (LIS1) expression in the CNS suggests a role in neuronal migration. J Neurosci 15:3730–3738
Reiter LT, Murakami T, Koeuth T, Pentao L, Muzny DM, Gibbs RA, Lupski JR (1996) A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat Genet 12:288–297
Reiter LT, Murakami T, Koeuth T, Gibbs RA, Lupski JR (1997) The human COX10 gene is disrupted during homologous recombination between the 24 kb proximal and distal CMT1A-REPs. Hum Mol Genet 6:1595–1603
Roa BB, Greenberg F, Gunaratne P, Sauer CM, Lubinsky MS, Kozma C, Meck JM, Magenis RE, Shaffer LG, Lupski JR (1996) Duplication of the PMP22 gene in 17p partial trisomy patients with Charcot-Marie-Tooth type-1A neuropathy. Hum Genet 97:642–649
Schneider MC, Hughes CR, Forrester S, Kimonis V (2000) Mild phenotype due to tandem duplication of 17p11.2. Am J Med Genet 94:296–299
Schrander-Stumpel C, Schrander J, Fryns JP, Hamers G (1990) Trisomy 17p due to a t(8;17)(p23;p11.2)pat translocation. Case report and review of the literature. Clin Genet 37:148–152
Shaffer LG, Kennedy GM, Spikes AS, Lupski JR (1997) Diagnosis of CMT1A duplications and HNPP deletions by interphase FISH: implications for testing in the cytogenetics laboratory. Am J Med Genet 69:325–331
Shaw CJ, Withers MA, Lupski JR (2004a) Uncommon Smith-Magenis syndrome deletions can be recurrent by utilizing alternate LCRs as homologous recombination substrates. Am J Hum Genet 75:75–81
Shaw CJ, Shaw CA, Yu W, Stankiewicz P, White LD, Beaudet AL, Lupski JR (2004b) Comparative genomic hybridisation using a proximal 17p BAC/PAC array detects rearrangements responsible for four genomic disorders. J Med Genet 41:113–119
Spinner NB, Biegel JA, Sovinsky L, McDonald-McGinn D, Rehberg K, Parmiter AH, Zackai EH (1993) 46,XX,15p+ documented as dup (17p) by fluorescence in situ hybridization. Am J Med Genet 46:95–97
Stankiewicz P, Lupski JR (2002) Molecular-evolutionary mechanisms for genomic disorders. Curr Opin Genet Dev 12:312–319
Stankiewicz P, Park S-S, Inoue K, Lupski JR (2001a) The evolutionary chromosome translocation 4;19 in Gorilla gorilla is associated with microduplication of the chromosome fragment syntenic to sequences surrounding the human proximal CMT1A-REP. Genome Res 1:1205–1210
Stankiewicz P, Park S-S, Holder SE, Waters CS, Palmer RW, Berend SA, Shaffer LG, Potocki L, Lupski JR (2001b) Trisomy 17p10-p12 resulting from a supernumerary marker chromosome derived from chromosome 17: molecular analysis and delineation of the phenotype. Clin Genet 60:336–344
Stankiewicz P, Shaw CJ, Dapper JD, Wakui K, Shaffer LG, Withers M, Elizondo L, Park S-S, Lupski JR (2003) Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am J Hum Genet 72:1101–1116
Stankiewicz P, Shaw CJ, Withers M, Inoue K, Lupski JR (2004) Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res 14:2209–2220
Stankiewicz P, Vissers LELM, Yatsenko SA, Crawford E, Creswick H, Gutter EM, de Vries BBA, Pfundt R, Marcelis C, Zackowski J, Lupski JR, Veltman JA (2005) Complex chromosome rearrangements in 17p are associated with genomic architecture involving low-copy repeats. In: 55th Annual meeting of the American Society of Human Genetics, 25–29.10, Salt Lake City, Utah, USA. Am J Hum Genet, p 157
Sylvester JE, Whiteman DA, Podolsky R, Pozsgay JM, Respess J, Schmickel RD (1986) The human ribosomal RNA genes: structure and organization of the complete repeating unit. Hum Genet 73:193–198
Tentler D, Johannesson T, Johansson M, Råstam M, Gillberg C, Orsmark C, Carlsson B, Wahlström J, Dahl N (2003) A candidate region for Asperger syndrome defined by two 17p breakpoints. Eur J Hum Genet 11:189–195
Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai L-H, Dobyns W, Ledbetter D, Hirotsune S, Wynshaw-Boris A (2003) 14–3–3ε is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet 34:274–285
Upadhyaya M, Roberts SH, Farnham J, MacMillan JC, Clarke A, Heath JP, Hodges ICG, Harper PS (1993) Charcot-Marie-Tooth disease 1A (CMT1A) associated with a maternal duplication of chromosome 17p11.2→12. Hum Genet 91:392–394
Wilson GN, Hollar BA, Waterson JR, Schmickel RD (1978) Molecular analysis of cloned human 18S ribosomal DNA segments. Proc Natl Acad Sci USA 75:5367–5371
Yatsenko SA, Treadwell-Deering D, Krull K, Glaze D, Horz M, Stankiewicz P, Lupski JR, Potocki L (2005) Trisomy 17p10-p12 due to mosaic supernumerary marker chromosome: delineation of molecular breakpoints and clinical phenotype and comparison to other proximal 17p segmental duplications. Am J Med Genet 138A:175–180
Yu W, Ballif BC, Kashork CD, Heilstedt HA, Howard LA, Cai W-W, White LD, Liu W, Beaudet AL, Bejjani BA, Shaw CA, Shaffer LG (2003) Development of a comparative genomic hybridization microarray and demonstration of its utility with 25 well-characterized 1p36 deletions. Hum Mol Genet 12:2145–2152
Zody MC, Garber M, Adams DJ, Sharpe T, Harrow J, Lupski JR et al (2006) DNA Sequence of human chromosome 17 and comparison with mouse chromosome 11. Nature 44:1045–1050
Acknowledgements
We thank Dr. Arthur L. Beaudet for helpful discussions and suggestions. We are grateful to both families for participation in theses studies. This work was supported in part by grants from the National Institute of Child Health and Human Development (PO1 HD39420 to J.R.L.) and the Mental Retardation Research Center (HD24064).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ou, Z., Jarmuż, M., Sparagana, S.P. et al. Evidence for involvement of TRE-2 (USP6) oncogene, low-copy repeat and acrocentric heterochromatin in two families with chromosomal translocations. Hum Genet 120, 227–237 (2006). https://doi.org/10.1007/s00439-006-0200-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-006-0200-7