
Abstract
Transient neonatal diabetes mellitus (TNDM) is associated with overexpression of an imprinted locus on chromosome 6q24; this locus contains a differentially methylated region (DMR) consisting of an imprinted CpG island that normally allows expression only from the paternal allele of genes under its control. Three types of abnormality involving 6q24 are known to cause TNDM: paternal uniparental disomy of chromosome 6 (pUPD6), an isolated methylation defect of the imprinted CpG island at chromosome 6q24 and a duplication of 6q24 of paternal origin. A fourth group of patients has no identifiable anomaly of 6q24. Bisulphite sequencing of the DMR has facilitated the development of a diagnostic test for TNDM based on ratiometric methylation-specific polymerase chain reaction. We have applied this method to 45 cases of TNDM, including 12 with pUPD6, 11 with an isolated methylation mutation at 6q24, 16 with a duplication of 6q24 and six of unknown aetiology, together with 29 normal controls. All were correctly assigned. The method is therefore capable of detecting all known genetic causes of TNDM at 6q24, although pUPD6 and methylation mutation cases are not distinguished from one another. In addition, we have carried out bisulphite sequencing of the DMR to compare its methylation status between six TNDM patients with a known methylation mutation, six patients with no identifiable 6q24 mutation and six normal controls. Whereas methylation mutation patients showed a near-total absence of DNA methylation at the TNDM locus, the patients with no identified molecular anomaly showed no marked methylation variation from controls.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transient neonatal diabetes mellitus (TNDM; OMIM 601410) affects approximately 1:400,000 infants. Growth-retarded neonates present with persistent hyperglycaemia (Temple and Shield 2002; Temple et al. 2000). Insulin therapy is required for an average of 3 months, after which time the diabetes resolves. However, the majority of TNDM patients develop type 2 diabetes in adolescence or early adulthood.
Three genetic causes of TNDM have been identified that together account for 49 of the 55 cases in the Wessex cohort: paternal uniparental disomy of chromosome 6 (17 probands; Temple et al. 1995; unpublished), a duplication of chromosome 6q24 of paternal origin (21 probands; Temple et al. 1996; unpublished) and an isolated methylation defect of an imprinted CpG island at chromosome 6q24 (11 probands; Gardner et al. 2000; unpublished). Six additional patients with a clinical diagnosis of TNDM have no detectable abnormality of chromosome 6q24. These observations indicate that more than 85% of cases of TNDM are caused by the overexpression of a paternally expressed imprinted gene on chromosome 6q24 (Kamiya et al. 2000; Arima et al. 2001; Mackay et al. 2002).
The imprinted TNDM CpG island is methylated only on the maternally inherited allele. It contains the promoter and transcriptional start sites for two imprinted transcripts: HYMAI (OMIM 606546), an untranslated transcript of unknown function (Arima et al. 2000, 2001), and ZAC (OMIM 603044), the major TNDM candidate gene (Spengler et al. 1997). ZAC is widely expressed in human tissues (Varrault et al. 1998; Abdollahi et al. 1997; Kas et al. 1998) and shows in vitro activity as a transcriptional activator, co-activator and co-repressor and as a tumour suppressor protein (Huang and Stallcup 2000, Huang et al. 2001; Bilanges et al. 2001; Hoffmann et al. 2003). Its expression in vitro is inhibited by promoter methylation (Bilanges et al. 2001; Varrault et al. 2001; Abdollahi et al. 2003).
Current protocols for the molecular diagnosis of TNDM entail: (1) polymerase chain reaction (PCR) of genomic DNA digested with methylation-sensitive restriction enzymes to determine the presence or absence of a maternal methylated allele; (2) if no methylated allele is detected, microsatellite analysis of chromosome 6 to determine UPD6; and (3) if a methylated allele is detected, ratiometric PCR to detect the duplication of 6q24 (Gardner et al. 2000). We have undertaken extensive bisulphite sequencing of the ZAC promoter CpG island in order to develop a single-tube diagnostic test for TNDM and to look for subtle methylation changes in TNDM patients in whom no mutation of 6q24 had been found.
Materials and methods
Patients
Patients were referred for molecular diagnosis of TNDM from or via the Wessex Clinical Genetics Service. Diagnosis had previously been performed by means of microsatellite analysis, ratiometric PCR of unique sequences in and around the ZAC locus and PCR of the TNDM CpG island following restriction digest by methylation-sensitive enzymes (Gardner et al. 2000). In six cases, TNDM was diagnosed from the clinical phenotype of neonatal diabetes with intra-uterine growth retardation and subsequent remission but without a positive outcome from any 6q24 molecular test. These patients were clinically indistinguishable from those with known mutations and five of them have been previously described (Temple et al. 2000, nos. 24 and 26–29). They were also negative for mutations in the KCNJ11 gene, which has been shown to cause neonatal diabetes (Gloyn et al. 2004).
Bisulphite sequencing
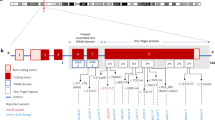
The TNDM CpG island spans nucleotides 52443–53359 of genomic sequence accession no. AL109755 and includes 101 CpG dinucleotides and the transcriptional start sites of ZAC and HYMAI (Fig. 1). Bisulphite-treated genomic DNA was prepared from TNDM patients and controls and primers were designed to amplify both methylated and unmethylated alleles of a sequence encompassing 53 CpG dinucleotides at the centre of the CpG island. The amplification products were cloned and at least 18 clones were sequenced from each individual. The normal methylation status of the region was determined from six controls. Six methylation-defective TNDM patients and six patients with TNDM of unknown aetiology were also sequenced.

Diagram of the TNDM CpG island. Numbering refers to genomic sequence accession no. AL109755. The thickened line represents the extent of the CpG island. The sequence subjected to bisulphite sequencing is indicated above the genomic sequence. The region used in methylation-specific PCR is indicated below the genomic sequence. The orientation of the common primer is marked, as are the relative positions of the C and T primers (specific for methylated and unmethylated allele, respectively). Curved arrows indicate the two transcriptional start sites of ZAC.
Genomic DNA (2 μg) was bisulphite-treated essentially by the method of Zeschnigk et al. (1997), except that free bisulphite was removed by using a desalting column (Wizard DNA clean-up system, Promega). DNA was resuspended in 20 μl TE buffer (10 mM TRIS-HCl pH 7.4, 1 mM EDTA). Templates for sequencing were produced in two rounds of PCR by using hemi-nested primers and conditions as detailed in Table 1. PCR was performed with HotStar Taq and buffer (Qiagen) with 1.5 mM MgCl2, 0.2 mM dNTP and 1 μM primers. First round PCRs contained 50 ng bisulphite-treated genomic DNA in a 20-μl reaction volume and second-round PCRs contained 1 μl of the first-round product in a 20-μl reaction volume.
PCR products were cloned and transformed by using the TOPO PCR cloning kit (Invitrogen). Minipreps were grown and harvested with the Montage miniprep system (Millipore), sequenced with BigDye version 1.1 (Applied Biosystems) and analysed on a Genetic Analyser 3100 or 3730 (Applied Biosystems).
Methylation-specific PCR
By using the information on methylation status provided by bisulphite sequencing, methylation-specific duplex PCR targeting CpG sequences between 52808 and 53005 (AL109755) was designed after the method of Zeschnigk et al. (1997) in order to estimate the relative proportions of methylated and unmethylated alleles in patients and controls. A set of three primers was designed in the region demonstrated by bisulphite sequencing to show differential methylation in normal individuals. One primer was non-selective for methylated or unmethylated DNA and was fluorescently labelled. Two adjacent reverse primers were designed: one specific for the methylated and one for the unmethylated allele. A reaction containing all three primers should generate maternal and paternal products of 175 bases and 187 bases, respectively, in a ratio reflecting that of the source DNA. Use of a restricted number of PCR cycles and a known quantity of input DNA kept amplification in the linear range and ratiometry was possible.
Methylation-specific PCR (MS-PCR) was performed in a blinded manner on 29 control samples, 12 pUPD6 TNDM cases, 11 methylation defect TNDM cases, 16 cases with paternal duplication of 6q24, six samples with maternal duplication of 6q24 and six TNDM patients with no identified chromosome 6 anomaly (it was not possible to study the whole Wessex cohort by MS-PCR becuase of the shortage of material).
PCR was carried out in a reaction volume of 10 μl in a Tetrad thermal cycler (MJ Research) under the following conditions: 50–100 ng DNA, 200 μM each dNTP, 0.5 μM each primer, 2 mM MgCl2 and HotStar Taq and buffer (Qiagen) according to the manufacturer’s specification. Primer sequences and PCR conditions are listed in Table 1. DNA fragments were resolved on a Genetic Analyser 3100 (Applied Biosystems). Fragments were analysed by using GeneScan and Genotyper software (Applied Biosystems) and the peak height ratio T/C or [methylated]/[unmethylated] ratio was calculated by using peak heights taken from electropherograms; tests giving very strong (>7000) or very weak (>100) peak heights were discarded, since inaccuracy in peak height calculation would be liable to lead to incorrect ratiometry and, in these cases, the PCR products were reanalysed or the amplifications repeated with adjusted template concentration. Normalised T/C ratios were calculated for each experiment with respect to the ratios of control PCRs.
Results
Bisulphite sequencing
Bisulphite sequencing was performed to assess the methylation of 53 CpG dinucleotides in the central portion of the TNDM imprinted CG island; the results are represented in Fig. 2. Figure 2a shows the sequencing data from three individuals, one control individual (A), one with TNDM of unknown aetiology (B), and a third with TNDM caused by methylation defect (C). The cloned sequences are shown in each case.

Summary of bisulphite sequencing data. a Bisulphite sequencing of the TNDM CpG island. The three blocks represent sequence data from a control individual (A), a TNDM patient without identified 6q24 anomaly (B), and a TNDM patient with isolated methylation defect (C). Numbers over the data blocks represent the 53 CpG dinucleotides sequenced (shaded squares methylated CpG, white squares unmethylated CpG). Each row of squares represents a single cloned sequence, except where a figure appears to the right of the row, in which case it indicates the number of times that the sequence was observed. Similar data were obtained from a further five controls, five methylation mutation cases and five cases with no known mutation. The clones with complete demethylation are assumed to represent the paternal allele, which amplified less efficiently than the methylated maternal allele and is therefore represented by a smaller proportion of clones. b Sporadic demethylation in TNDM CpG island in TNDM cases and controls. The x-axis denotes CpG position, as in a. The y-axis represents the quotient of [demethylations observed/total sequences counted] for each CpG. Methylation-absent sequences were omitted from the calculation. The TNDM group represents six individuals with classic clinical TNDM but no 6q24 anomaly (total 155 sequences), whereas the control group represents six control individuals (total 156 sequences).
At the high cycle number required for cloning and sequencing, the PCR products of A and B are mainly derived from CG-rich templates, indicating that the PCR is selective for the CG-rich DNA derived from the methylated allele.
In cases A and B, some products, which were assumed to be derived from the paternal allele, showed demethylation of every CpG dinucleotide but several sequences showed sporadic demethylation at various CpG positions. There was no CpG position at which demethylation was seen in a high proportion of sequences in B, as might have been expected if isolated demethylation of any group of CpG dinucleotides was sufficient to cause TNDM. By contrast, the sequences derived from C had absent or near-absent methylation at all CpG positions.
In all, the sequences from the six methylation-defective TNDM patients revealed the effective absence of methylation across a total of 166 sequences. Data obtained from six TNDM cases of unknown aetiology totalled 172 sequences, of which 17 were of near-absent methylation (paternal allele); the data from nine controls totalled 177 sequences, of which 21 were of near-absent methylation (paternal allele). In order to compare all data from these two sample sets, the methylated allele sequences were pooled into the two groups and the individual incidences of sporadic demethylation were summed across each CpG position and presented graphically (Fig. 2b). The incidence of sporadic demethylation was compared between the two groups; no marked differences were observed.
Ratiometric MS-PCR
The sequencing information was used to design primers for MS-PCR, which, in normal individuals, were predicted to reside within highly methylated (CpG 10–13) or completely unmethylated (CpG 13–18) sequences (Fig. 2a). MS-PCR generated two products in a ratio reflecting that of methylated and unmethylated alleles of the genomic DNA. Figure 3 shows examples of product quantitation of MS-PCR.

Electropherogram of amplification products of MS-PCR. The x-axis scale represents the calculated product size (in bp), whereas the y-axis indicates peak height, as do the numbers under each peak. The ratio (C/T) was calculated as the peak height ratio of methylated versus unmethylated amplification products.
To permit inter-assay comparisons, the methylated:unmethylated peak height ratio was calculated for normal controls and test data were normalised to this ratio. To estimate inter-assay variability, DNA from three individuals (maternal duplication, paternal duplication and control) were subjected to five separate PCRs on different occasions by using DNA derived from two bisulphite reactions performed on different occasions. With this method, the peak height ratios from paternal and maternal duplication and normal control approximated reliably to 0.5, 2.0 and 1.0, respectively (Table 2).
MS-PCR was performed in a blinded manner on a total of 81 samples, comprising 29 controls, 17 paternal duplications, 6 maternal duplications, 11 with a methylation defect, 12 with paternal UPD6 and 6 TNDM cases of unknown aetiology. The results of these determinations are presented in Table 3. The peak height ratios from paternal UPD6 cases and methylation-defective cases were zero, reflecting the absence of a methylated DNA product. In the TNDM patients of unknown aetiology, the peak height ratio approached 1.0, viz. no methylation changes were detected. In paternal and maternal duplication, peak height ratios approximated to 0.5 and 2.0, respectively. In all cases, genotype was correctly predicted, with one interesting exception. One additional sample was also tested that was derived from a fibroblast cell line from a patient with paternal duplication; the ratio for this patient, 1.02 (not included in Table 3), approximated that of normal controls.
On the assumption that a similar methylation pattern prevailed throughout the CpG island, primers were designed for MS-PCR of its proximal and distal portions (52480–52637 and 53051–53247, respectively) and the patient samples blind-tested as described above. Essentially identical patterns were observed for all three MS-PCRs (data not shown).
Discussion
By using this method, bisulphite treatment and a single PCR produces a genetic diagnosis of TNDM, with further testing being necessary only to distinguish UPD6 and the methylation anomaly. This represents a considerable improvement over current methods, which require at least two rounds of genotyping and include restriction enzyme digestion. Such methods have also been developed for diagnosis of UPD11, UPD14 and UPD7 (Fisher et al. 2002; Kosaki et al. 1997, 2000; Murphy et al. 2003, Moore et al. 2003). We note the additional benefit gained by quantifying amplification products: this enables diagnosis not only of paternal UPD6 and methylation mutation cases, but also of duplications of 6q24. Moreover, this quantified method clearly distinguishes between duplication of maternal and paternal origin. Identification of maternal duplication of 6q24 is not of diagnostic value to the proband, since TNDM is not caused by maternal duplication but is of value to the proband’s family, since it indicates the origin of the inherited duplication and thereby directs the referring clinician to the branch of the extended family that requires further testing.
Some caveats should be noted. First, if no maternal methylated allele is seen in methylation-sensitive PCR, microsatellite analysis is required to distinguish pUPD6 from the 6q24 methylation defect, although in either case, a genetic diagnosis of TNDM is indicated. Second, cytogenetic analysis is of value in duplication cases, since some cytogenetically visible duplications cause mild mental retardation in addition to TNDM (Zneimer et al. 1998). Third, approximately 10% of patients have a clinical diagnosis of TNDM but no identified genetic cause and this method, like other methods, does not yield a diagnosis in these cases. Fourth, the DNA sample derived from a cell line gave abnormal results, suggesting that such samples should not be used in this assay.
This study was undertaken partly to search for subtle, hitherto unrecognised changes in the methylation of the TNDM CpG island that might be causing disease in patients with no known 6q24 mutation. Six such patients were analysed, all of whom had a clinical course of disease indistinguishable from patients in which 6q24 mutations had been identified. We performed bisulphite sequencing of 53 CpG dinucleotides from the centre of the CpG island in DNA from controls and TNDM patients. We found sporadic demethylation of isolated CpG dinucleotides in every individual studied but these demethylations did not appear either more marked or distinctly localised in the TNDM patients of unknown aetiology. In this, they contrast with methylation-defective TNDM samples, which show essentially complete demethylation of all CpG sequences.
The patients of unknown aetiology could have been mosaic for completely demethylated maternal alleles. With regard to the bisulphite sequencing protocol, a comparison of the relative number of unmethylated alleles in the control group and in the TNDM cases of unknown aetiology is not possible because of the bias of PCR amplification towards methylated sequences and the large number of PCR cycles required. However, MS-PCR analysis shows no evidence of an increase in umethylated sequences in DNA from blood, the methylation ratios of the cases of unknown aetiology being in the same range as those of the controls (Table 2). We assume therefore that the completely demethylated alleles seen upon bisulphite sequencing represent the paternal allele.
Nevertheless, some TNDM patients might still have had mosaic or tissue-specific methylation changes that were not detected in the blood samples available for study. Alternatively, they may have had mutations in different genes. Mutations in KCNJ11 have been described in cases of neonatal diabetes but none have been found in the six patients of unknown aetiology described above. It should also be noted that extensive molecular study of these patients has revealed no point mutations or subtle deletions/duplications of the TNDM locus (D. Mackay, unpublished).
In summary, we have developed a new rapid method for the molecular diagnosis of TNDM. However, we have been unable to identify any significant changes in methylation profile in those TNDM cases that do not have any of the three known TNDM mutations.
References
Abdollahi A, Godwin AK, Miller PD, Getts LA, Schultz DC, Taguchi T, Testa JR, Hamilton TC (1997) Identification of a gene containing zinc-finger motifs based on lost expression in malignantly-transformed rat ovarian surface epithelial cells. Cancer Res 57:2029–2034
Abdollahi A, Pisarcik D, Roberts D, Weinstein J, Cairns P, Hamilton TC (2003) LOT1 (PLAGL1/ZAC1), the candidate tumor suppressor gene at chromosome 6q24–25, is epigenetically regulated in cancer. J Biol Chem 278:6041–6049
Arima T, Drewell RA, Oshimura M, Wake N, Surani MA (2000) A novel imprinted gene, HYMAI, is located within an imprinted domain on chromosome 6 containing ZAC. Genomics 67:248–255
Arima T, Drewell RA, Arney KL, Inoue J, Makita Y, Hata A, Oshimura M, Wake N, Surani MA (2001) A conserved imprinting control domain at the HYMAI/ZAC domain is implicated in transient neonatal diabetes mellitus. Hum Mol Genet 10:1475–1483
Bilanges B, Varrault A, Mazumdar A, Pantaloni C, Hoffmann A, Bockaert J, Spengler D, Journot L (2001) Alternative splicing of the imprinted candidate tumor suppressor gene ZAC regulates its antiproliferative and DNA binding activities. Oncogene 20:1246–1253
Fisher AM, Thomas NS, Cockwell A, Stecko O, Kerr B, Temple IK, Clayton P (2002) Duplications of chromosome 11p15 of maternal origin result in a phenotype that includes growth retardation. Hum Genet 111:290–296
Gardner RJ, Mackay DJ, Mungall AJ, Polychronakos C, Siebert R, Shield JP, Temple IK, Robinson DO (2000) An imprinted locus associated with transient neonatal diabetes mellitus. Hum Mol Genet 9:589–596
Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining J, Slingerland AS, Howard N, Srinivasan S, Silva JMCL, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay DJG, Shield JPH, Sumnik Z, Rhijn A van, Wales JKH, Clark P, Gorman S, Aisenberg J, Ellard S, Njølstad PR, Ashcroft FM, Hattersley AT (2004) Activating mutations in the ATP-sensitive potassium channel subunit Kir6.2 gene are associated with permanent neonatal diabetes. N Engl J Med 350:1838–1849
Hoffmann A, Ciani E, Boeckardt J, Holsboer F, Journot L, Spengler D (2003) Transcriptional activities of the zinc finger protein Zac are differentially controlled by DNA binding. Mol Cell Biol 23:988–1003
Huang S-M, Stallcup MR (2000) Mouse Zac1, a transcriptional coactivator and repressor for nuclear receptors. Mol Cell Biol 20:988–1003
Huang S-M, Schönthal AH, Stallcup MR (2001) Enhancement of p53-dependent gene activation by the transcriptional coactivator Zac1. Oncogene 20:2134–2143
Kamiya M, Judson H, Okazaki Y, Kusakabe M, Muramatsu M, Takada S, Takagi N, Arima T, Wake N, Kamimura K, et al (2000) The cell cycle control gene ZAC/PLAGL1 is imprinted—a strong candidate gene for transient neonatal diabetes. Hum Mol Genet 9:453–460
Kas K, Voz ML, Hensen K, Meyen E, Van de Ven WJ (1998) Transcriptional activation capacity of the novel PLAG family of zinc finger proteins. J Biol Chem 273:23026–23032
Kosaki K, McGinniss MJ, Veraksa AN, McGinnis WJ, Jones KL (1997) Prader-Willi and Angelman syndromes: diagnosis with a bisulphite-treated methylation-specific PCR method. Am J Med Genet 73:308–313
Kosaki K, Kosaki R, Robinson WP, Craigen WJ, Shaffer LG, Sato S, Matsuo N (2000) Diagnosis of maternal uniparental disomy of chromosome 7 with a methylation specific PCR assay. J Med Genet 37:E19
Mackay DJ, Coupe A-M, Shield JP, Storr JN, Temple IK, Robinson DO (2002) Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum Genet 110:139–144
Moore MW, Dietz LG, Tirtoahardjo B, Cotter PD (2003) A multiplex methylation PCR assay for identification of uniparental disomy of chromosome 7. Hum Mut 21:645–648
Murphy SK, Wylie AA, Coveler KJ, Cotter PD, Papenhausen PR, Sutton VR, Schaffer LG, Jirtle RL (2003) Epigenetic detection of human chromosome 14 uniparental disomy. Hum Mut 22:92–97
Spengler D, Villalba M, Hoffman A, Pantaloni C, Houssami S, Bockaert J, Journot L (1997) Regulation of apoptosis and cell cycle arrest by Zac1, a novel zinc finger protein expressed in the pituitary gland and the brain. EMBO J 16:2814–2825
Temple IK, Shield JP (2002) Transient neonatal diabetes, a disorder of imprinting. J Med Genet 39:872–875
Temple IK, James RS, Crolla JA, Sitch FL, Jacobs PA, Howell WM, Betts P, Baum JD, Shield JP (1995) An imprinted gene(s) for diabetes? Nat Genet 9:110–112
Temple IK, Gardner RJ, Robinson DO, Kibirige MS, Ferguson AW, Baum JD, Barber JC, James RS, Shield JP (1996) Further evidence for an imprinted gene for neonatal diabetes localised to chromosome 6q22-q23. Hum Mol Genet 5:1117–1121
Temple IK, Gardner RJ, Mackay DJ, Barber JC, Robinson DO, Shield JP (2000) Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes 49:1359–1366
Varrault A, Ciani E, Apiou F, Bilanges B, Hoffmann A, Pantaloni C, Bockaert J, Spengler D, Journot L (1998) hZAC encodes a zinc finger protein with antiproliferative properties and maps to a chromosomal region frequently lost in cancer. Proc Natl Acad Sci USA 95:8835–8840
Varrault A, Bilanges B, Mackay DJ, Basyuk E, Ahr B, Fernandez C, Robinson DO, Bockaert J, Journot L (2001) Characterisation of the methylation-sensitive promoter of the imprinted ZAC gene supports its role in transient neonatal diabetes mellitus. J Biol Chem 276:18653–18656
Zeschnigk M, Lich C, Buiting K, Doerfler W, Horsthemke B (1997) A single-tube PCR test for the diagnosis of Angelman and Prader-Willi syndrome based on allelic methylation differences at the SNRPN locus. Eur J Hum Genet 5:94–98
Zneimer SM, Ziel B, Bachman R (1998) Partial trisomy of chromosome 6q: an interstitial duplication of the long arm. Am J Hum Genet 80:133–135
Acknowledgements
The authors thank all patients and clinicians who sent samples for analysis. This study was funded by Diabetes UK (D.J.G.M.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mackay, D.J.G., Temple, I.K., Shield, J.P.H. et al. Bisulphite sequencing of the transient neonatal diabetes mellitus DMR facilitates a novel diagnostic test but reveals no methylation anomalies in patients of unknown aetiology. Hum Genet 116, 255–261 (2005). https://doi.org/10.1007/s00439-004-1236-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-004-1236-1