
Abstract
With the recent advances in genomic research, it has become apparent that a substantial part of human malformation and mental retardation is caused by imbalances in genomic content. Thus, there is an increasing need for versatile methods allowing a detailed mapping and cloning of the actual rearrangements. We have combined the flexibility of real-time quantitative PCR with the knowledge of human genome sequence to perform a copy number scanning in three patients known to harbour a deletion in the 7p14p15 locus. In two of the patients the actual breakpoints were cloned and sequenced, whereas the breakpoint of the third patient was mapped to a region previously predicted to be prone for rearrangements. One patient also harboured an inversion in connection with the deletion that disrupted the HDAC9 gene. All three patients showed clinical characteristics reminiscent of the hand-foot-genital syndrome and were deleted for the entire HOXA cluster. Two patients were also deleted for DFNA5, a gene implicated in dominant nonsyndromic hearing impairment, but neither patient showed signs of reduced hearing capabilities. The described copy number scanning approach is largely independent of the genomic locus and may be a valuable tool for characterising a large spectrum of deletions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the fast expanding services of genetic screening, it has become clear that a substantial fraction of human mutations results from the gain or loss of genetic material rather than single base-pair substitutions. Based on the recent update of the Human Gene Mutation Database, deletions represent 22% of reported mutations (Stenson et al. 2003). The majority of these are very small deletions that can be detected by traditional mutation scans. However, almost 7% of the reported mutations are gross deletions, which would escape detection by a conventional sequencing approach. The figure is likely to be an underestimate illustrated by the observation that alternative mutation detection strategies can improve the detection of mutations dramatically (Nystrom-Lahti et al. 1995; Petrij-Bosch et al. 1997).
Larger genomic deletions have traditionally been identified and characterised by various banding techniques or by Southern blotting, but the resolution of these techniques is usually low and it often requires a substantial amount of effort to resolve the extent of individual deletions. Fluorescent in situ hybridisation (FISH) has been the method of choice for deletion analysis, but is, however, limited for detailed deletion mapping by the high cost, the scarcity of commercial probes, and by the fact that probes usually are in the range of 100–150 kb. Recently, some of these problems have been circumvented by the refinement of probes (Knoll and Rogan 2003) and by the use of primed in situ labelling (PRINS) (Tharapel and Kadandale 2002), but whether these improvements will facilitate the high-resolution mapping of individual deletions remains to be seen. PRINS does, however, emerge as an attractive method for the identification of translocation breakpoints (Nimmakayalu et al. 2003). Loss of heterozygosity mapping has also been employed for characterising deletions, but the technique usually does not aid a high-resolution mapping, and optimally requires samples from the patient’s relatives, who are not always available for analysis. A more technical demanding strategy for deletion mapping is conversion to haploidy by fusion of lymphocytes and rodent cell to create hybrids with individual human chromosomes (Yan et al. 2000).
During the last decade, a variety of PCR-based quantitative and semi-quantitative techniques have been described for deletion analysis (Armour et al. 2002). Most recently, the MAPH and MLPA techniques have been developed and successfully employed in copy number measurements (Armour et al. 2000; Schouten et al. 2002). Both assays are well suited for analysing a limited number of loci in large sample sizes, however, the workup is very labour intensive and the techniques are therefore not favourable for characterising individual deletions. Other commonly used approaches are competitive PCR, which relies on a coamplification of two targets, one of which is of a known concentration (Prediger 2001), and differential PCR, which is a semi-quantitative method determining the relative concentrations of two amplicons (Rowland et al. 2001). These techniques also require substantial fine-tuning and are becoming obsolete after the development of real-time PCR, which is now widely used for quantification of transcripts. Although real-time PCR is frequently used in determining the copy number of specific genomic loci, there is a surprising lack of reports applying real-time quantitative PCR as a universal application for mapping and characterising imbalances in genomic content (Armour et al. 2002; Ginzinger 2002).
We have previously shown that 12% of mentally retarded and dysmorphic children with normal conventional karyotypes have chromosomal aberrations that can be identified by high-resolution comparative genomic hybridisation (HR-CGH) (Kirchhoff et al. 2000, 2004). To validate and characterise the deletions found by routine HR-CGH screening, we have combined the flexibility of real-time quantitative PCR with the knowledge of the human reference sequence to design numerous very short test PCRs allowing a real-time quantitative scanning of the genomic region of interest. By this scanning approach, we have located and cloned the exact breakpoints of a recurrent 7p14p15 deletion spanning the entire HOXA cluster. Thus, this strategy allows us to go from the HR-CGH resolution of approximately 3–4 Mb down to the actual base pair, and as the approach is largely independent of the genomic locus, it can be applied on virtually any deletion of interest.
Materials and methods
Human subjects
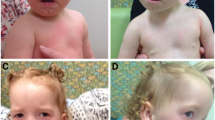
Case 1 was referred to our cytogenetic facility for HR-CGH on the basis of idiopathic mental retardation combined with dysmorphic features and normal conventional karyotypes. The patient (Fig. 1A) was the second child of unrelated, healthy parents. The pregnancy was uneventful and a girl was born at term with a weight of 2,700 g and a length of 48 cm. Psychomotor development was retarded: she sat alone at 8–10 months old, walked at 22 months and speech was delayed. At age 5 years, she was dysmorphic with up-slanting palpebral fissures, a broad nose with flat root and anteverted nostrils, broad lips, short extremities with extension defect in the right knee, and short fingers with tapering of terminal phalanges, clinodactyly of the third right finger, short and broad first toes. After receipt of informed consent, genomic DNA was extracted from peripheral blood by standard methods and HR-CGH (Kirchhoff et al. 1998) subsequently identified a de novo deletion of the entire 7p15 band (Fig. 2A) (Kirchhoff et al. 2001), which was assumed to be the underlying cause of the clinical phenotype.

A Case 1, B Case 2. Note that apart from the clinodactyly, the two patients did not share obvious major dysmorphic similarities

High resolution comparative genomic hybridisation results of chromosome 7 in cases 1 and 2. Vertical lines represent ratios of 0.5, 0.75, 1, 1.25 and 1.5. Both deletions were estimated to comprise approximately 8–10 Mb. The red bars indicate the deletions. Black lines represent the 99.5% dynamic standard reference intervals, while grey lines represent the 99.5% confidence interval of the samples (Kirchhoff et al. 2001, 2004)
Case 2 was referred to our cytogenetic facility for HR-CGH on the same basis as case 1. The patient was the first child of unrelated, healthy parents (Fig. 1B). The pregnancy was uneventful and a boy was born at term with a weight of 2,680 g and a length of 48 cm. Psychomotor development was delayed: he sat alone at 7 months old, walked at 18 months and speech was delayed. At 4 years of age, he was dysmorphic with short, slightly malformed ears, small down-slanting palpebral fissures, hypermetropia (+6), a broad neck, short extremities, short fingers with tapering of terminal phalanges, clinodactyly of fifth fingers, short toes, and pes planus. HR-CGH subsequently identified a de novo deletion of the 7p14p15 band (Fig. 2B) (Kirchhoff et al. 2004), which was assumed to be the underlying cause of the clinical phenotype.
Case 3 has been described elsewhere (Devriendt et al. 1999). Briefly, conventional karyotype revealed a de novo deletion of the 7p14 band, and loss of heterozygosity mapping demonstrated that the deletion encompassed the two microsatellites D7S529 and D7S2496 located at 25,128,129 and 29,309,086, respectively. The patient exhibited clinical characteristics consistent with hand-foot-genital syndrome.
Real-time copy number scanning
To confirm and map the three microdeletions, we undertook a real-time quantitative approach (Fig. 3).

Genomic DNA from two normal individuals and the three patients were initially diluted to give a nearly identical CT value to the GAPDH reference PCR. If the test PCR was located outside the deletion all samples should essentially have the same CT value, whereas if the test PCR was located in the deleted area, the CT value was shifted by approximately one cycle compared with the normal samples. The upper panel shows the reference real-time PCR in a normal and a patient sample, whereas the lower panel depicts an example of a test PCR located within the deletion (each performed in duplex)
A reference PCR was developed according to the PrimerExpress/SDS7000 guidelines that allowed a robust and highly reproducible amplification of a 51-bp genomic fragment of the GAPDH gene located on chromosome 12 (ctccccacacacatgcactta; ttgccaagttgcctgtcctt). In the presence of SYBR Green this reference PCR could be monitored on an SDS7000 real-time PCR machine. Next, high quality DNA was isolated from the three patients and from two normal reference cases, and the five samples were individually diluted (approximately 5 ng/μl) to obtain nearly exact cycle thresholds (CT) values for the reference GAPDH PCR. By utilizing the human reference sequence (http://www.genome.ucsc.edu) numerous test PCR reactions of 50–55 bp, likewise designed according to the PrimerExpress/SDS7000 guidelines (tm between 58 and 60°C, C+G content between 20 and 80%, primer length between 9 and 24 bp, and amplicon length between 50 and 58 bp), were created inside and outside the region presumed to be involved in the two deletions. All primers were designed to run under similar conditions. Care was taken not to locate the different test PCRs in repeated sequences using the BLAT feature of the Human Genome Browser (http://genome.ucsc.edu/cgi-bin/hgBlat). All PCRs were performed in duplex and followed by a melting curve analysis to ensure the generation of specific amplicons. Optimal threshold levels were set for each individual run. The CT value of the case test PCR reactions were related relative to the CT value of the GAPDH reference PCR and the five cases using the comparative CT method (described in User Bulletin 2 from Applied Biosystems). If the test PCR fragment was located outside the deleted sequence a relation of 1:1 (±20%) was seen, whereas if the test PCR was located inside the deleted sequence a relation of 0.5:1 (±20%) was usually observed (Table 1). If the relation was between 0.65 and 0.75, the test PCR was repeated. Primer sequences are available on request.
PCR and sequencing
Test and reference PCR reactions were performed on an SDS7000 using the SYBR Green PCR Master Mix according to the manufacture’s instructions (Applied Biosystems). PCRs spanning the mapped breakpoints were performed using the Expand Long Template PCR system (Roche). Sequencing the breakpoint-spanning PCR products was performed on an ABI310 automated sequencer, using BigDyeTerminator V1.1 by standard methods.
Sequence location
All sequence locations refer to the July 2003 assembly of the human reference sequence at the UCSC database (http://genome.ucsc.edu/).
Results
Deletion mapping
Three patients were included in the deletion mapping approach. Cases 1 and 2 were identified by routine HR-CGH screening of idiopathic mentally retarded children with a dysmorphic appearance (Fig. 1), whereas the third case was identified by conventional karyotyping. Case 1 exhibited an HR-CGH signal consistent with a deletion of the entire 7p15 band, whereas the deletion of case 2 was assigned to 7p14p15 (Fig. 2). Both deletions were interpreted to comprise approximately 8–10 Mb (Kirchhoff et al. 2001, 2004). The deletion in case 3 was assigned to 7p14 and microsatellite and FISH analysis demonstrated that the deletion encompassed D7S529 and D7S2496 (Devriendt et al. 1999). Based on these initial findings, we undertook a more detailed mapping of the three deletions (Fig. 3). Preliminary real-time quantitative copy number scanning of the 7p14p15 chromosomal region indicated that the deletions in cases 1 and 2 both encompassed approximately 9 Mb with roughly a 6-Mb overlap (Fig. 4a). This finding corresponded nicely with the HR-CGH data. The deletion in case 3 spanned approximately 12 Mb and was located within the region defined by cases 1 and 2 (Fig. 4). Both the distal and proximal breakpoints of cases 1 and 2 were subsequently mapped to a resolution of 3–7 kb.

A Schematic presentation of the extent and location of the case deletions. B Modified screen picture of the UCSC Human Genome Browser (http://genome.ucsc.edu) showing a 155-kb segment (chromosome 7: 34,832,356–34,988,039) with ‘Ensembl Genes’, ‘Simple Repeat’ and the ‘Self Chain’ feature (showing alignments of the human genome with itself) in the full display mode (minor Self Chain alignments have been omitted). The positions of the aligned sequences are indicated by chromosome band, strand (+/−), and first base (in thousands). The identified Ensembl gene is a 317-bp segment potentially encoding a putative calcium-binding protein but is likely to represent a pseudogene. The proximal breakpoint of case 3 is mapped to be located within the 77.8-kb segment indicated by the vertical arrows. Note that the segment contains high homology to sequences just distal to the telomeric breakpoints of cases 1 and 3, and to sequences within the mapped deletions. The homology between the displayed segment and the sequence of 7p15.1 has been proposed to be a potential site for duplication-mediated rearrangements (Hillier et al. 2003)
The distal breakpoint of case 3 was mapped to be located within a 10.3-kb segment (chromosome 7: 22,815,150–22,825,525) harbouring a LINE and an LTR element, which made it impossible to map the breakpoint further. The proximal breakpoint was located to a 77.8-kb segment (chromosome 7: 34,855,706–34,933,550) that apart from various short repetitive elements, also showed very high homology to different regions of chromosomes 7 and 12 (Fig. 4B). Database analysis using the Chained Blastz feature of the UCSC Genome Browser (Schwartz et al. 2003), identified various degrees of homology to sequences located in the presumed deleted region (7p14.3 and 7p15.1), and to a segment in close vicinity of the distal breakpoint (7p15.3). Moreover, a large segment of 7p14.2 spanning more than 150 kb showed near 100% identity to a sequence located on the long arm of chromosome 7 (7q22.1) and to a region of chromosome 12 (12q14.2). This segmental duplication of 7p14.2 complicated a further mapping of the proximal breakpoint.
Breakpoint cloning
Having narrowed down the proximal and distal breakpoints in cases 1 and 2, primers were designed for long-range PCR, which would allow the actual breakpoint to be PCR amplified. The breakpoint-spanning PCR resulted in a nice specific band in case 1. Subsequent sequencing of the junction fragment demonstrated that both breakpoints of case 1 were located outside known genes and that the deletion exhibited a 6-bp insertion in the breakpoint (Fig. 5). According to the human reference sequence, the deletion encompasses a total of 71 genes, of which 29 were predicted on a theoretical basis.

A Sequence of the deletion junction of case 1. B Sequence of the distal and C proximal deletion junction of case 2. The numbers identify the base on each side of the specific deletion junction according to the July 2003 assembly of the Human reference sequence. Arrows indicate strand direction
The breakpoint-spanning PCR of case 2 did, after a number of attempts, result in a cryptic PCR product several kilobases longer that expected. The PCR product could not be sequenced with the proximal primer. However, sequencing with distal the primer revealed a fusion to the HDAC9 gene (Zhou et al. 2001) located 7.8 Mb distal to the presumed breakpoint. Moreover, according to the human reference sequence, the strand shifted from the + strand to the − strand at the fusion, indicating the presence of an inversion. The inversion could indeed be confirmed by designing a reverse primer in the HDAC9 gene that together with the proximal breakpoint primer amplified a specific fragment in case 2. Subsequent sequencing revealed an 8-bp deletion in the inversion breakpoint (Fig. 5). Thus, besides a 9,028,219-bp deletion encompassing 58 predicted genes (of which 26 are hypothetical), case 2 also harboured a 7,849,544-bp inversion disrupting the HDAC9 gene (Fig. 4). At the deletion junction the inverted fragment disrupted the SNX10 gene, which is predicted to encode a protein of the sorting nexin family involved in intracellular trafficking.
Several attempts were made to clone the breakpoint of case 3, but due to the high degree of sequence homology at the mapped breakpoint regions a deletion-specific PCR product could not be generated.
Discussion
During the last couple of years, real-time quantitative PCR (Higuchi et al. 1993) has become a widespread technique for mRNA expression studies, copy number measurements of genomic, viral, and mitochondrial DNA, as well as for allelic discrimination assays, and lately for confirmation of microarray data (Ginzinger 2002). In the majority of these assays, the quantitative signal is detected by sequence-specific fluorogenic probes which provide high specificity on behalf of additional costs and PCR optimisation, and the detection method is therefore not suitable for mapping a broad spectrum of deletions as described here. Alternatively, the quantitative signal can be monitored by the intercalating dye SYBR green I, which binds double-stranded DNA in a sequence-independent manner. In this assay the specificity and efficiency of the PCR is only determined by the primers, template quality, and reaction conditions. In the copy number scanning described here, all amplifications were carried out under identical conditions, on the same DNA dilutions, leaving primers as the only variable.
In order to compare the reference and test PCR by the comparative CT method, the two reactions should have identical efficiencies (Pfaffl 2001). Typically, a five- to ten-fold dilution series is used to generate standard curves from which the efficiency is determined. To avoid generating standard curves for every test PCR, we carefully titrated our template DNA to give CT values that were practically identical to those of the GAPDH reference PCR. Thus, any difference in the PCR efficiency between the reference and test PCR would affect all samples equally, and therefore eliminate the need for assaying the efficiency. Imbalances in genomic copy number could theoretically be detected solely by evaluating the test PCR across the five samples. However, the GAPDH PCR was always included to provide extra security and for monitoring pipetting accuracy. The vast majority of test PCRs in the reference individuals exhibited a CT value close to the CT value of the GAPDH PCR indicating comparative efficiencies. The mean ΔCT value for 25 representative test PCRs of the normal sample was −0.025±0.84 consistent with comparative efficiencies. This is likely related to the fact that all primers were designed according to the same criteria creating very small amplicons of only 51–55 bp, that are expected to have an efficiency close to 100%. Occasionally, a difference of up to two cycles in the CT value between the GAPDH and a test PCR was observed, however, this did not influence the expected 1:1 relation between the normal samples after the comparative CT calculation. A summary of 64 test PCRs located either within (32) or outside (32) the deletions clearly demonstrates that the quantitative relationship between the test and reference PCRs is easily designable in the two areas (Table 1).
The mapping strategy outlined here requires successive designing and testing of primers, and for each deletion approximately 20–25 primer pairs were used. Depending on the pre-knowledge of the deletion, 10–15 primer pairs would allow a deletion to be mapped to a 100-kb resolution. Alternatively, a single breakpoint can be mapped and a different technique, such as Genome Walker (Siebert et al. 1995) or traditional Southern blot, can be used to clone the junction fragment. Although more labourious, such an approach should be able to detect cryptic sequence rearrangements, such as inversions and insertions at the breakpoint fusion, which otherwise are hard to detect by a breakpoint-spanning PCR.
The short arm of chromosome 7 appears to be a relatively potent site for chromosomal aberrations and more that 100 cytogenetic abnormalities have so far been assigned to 7p. A substantial part of these represents 7p duplications, which are believed to arise from malsegregation of balanced translocation carriers (Kozma et al. 2000). Cai et al. (1999) summarise 47 of such cases and recognise 18 where the breakpoint is located in the 7p14.2p15.3 interval deleted in our three patients. Indeed, duplication of 7p15p22 can be recognised clinically as a distinctive phenotype (Cox et al. 2002; Reish et al. 1996), and an interesting case of trisomy 7p has been described where a 7p13p21.2 segment was inserted into 8p23 (Megarbane et al. 2001). The area deleted in our three patients has also been implicated in several other deletions. Chotai et al. (1994) review 32 patients of which 11 harbour, an interstitial deletion where at least one of the presumed breakpoints appears to be located within the 7p14.2p15.3 segment. The underlying mechanism for the instability of this particular region is unknown, but the recent determination of the entire chromosome 7 sequence has revealed an unusually high degree of intrachromosomal segmental duplications (Hillier et al. 2003). By scanning the chromosome 7 sequence for regions flanked by highly homologous segmental duplications, 11 regions were identified susceptible to duplication mediated rearrangements (Hillier et al. 2003). One of these regions (29,334,859–34,878,685) ends exactly at the proximal breakpoint identified in case 3 and it is also in close vicinity to the proximal breakpoint of case 2 (Fig. 4). Thus, it is highly likely that the deletions in our patients are mediated through intrachromosomal segmental duplicated sequences. In this respect, it is interesting to note that the sequence harbouring the proximal breakpoint in case 3, is also present in the 7p15.3 band just telomeric to the distal breakpoint of cases 1 and 3, and at 7p15.1 which is absent in all three cases (Fig. 4). The existence of virtually the same 100-kb sequence at both the short and the long arm of chromosome 7 (7p14.2 and 7q21.1) as well as on 12q14.2 points to the possibility of quite complex rearrangements. However, we are not aware of any specific rearrangements between these areas.
The three deletions described here are quite large and comprise, all in all, 80 putative genes, of which about half are uncharacterised (according to the human genome browser). It is, therefore, quite difficult to evaluate the clinical phenotypes with respect to individual genes. The three cases do, however, show characteristics reminiscent of the dominant hand-foot-genital syndrome (HFGS) (Fig. 1), which is caused by mutations in or haploinsufficiency of the HOXA13 gene (Devriendt et al. 1999; Frisen et al. 2003; Mortlock and Innis 1997; Utsch et al. 2002). All three patients are deleted for the entire HOXA cluster, which is one of the four homeobox gene clusters encoding transcription factors essential for correct limb patterning and growth (Goodman 2002). Thus, cases 1 and 2 represent two new examples of HOXA13 haploinsufficiency resulting in the HFGS. Interestingly, the deletions of cases 1 and 3 involve the DFNA5 gene, which has been implicated in dominant nonsyndromic hearing impairment. Only three DFNA5 mutations have been described, which all lead to skipping of exon 8 whereby the reading frame is interrupted (Bischoff et al. 2004; Van Laer et al. 1998; Yu et al. 2003). As neither case 1 nor case 3 has obvious hearing impairments, it appears that the three splice mutations act by a dominant negative mechanism rather than creating a state of DFNA5 haploinsufficiency. The 7.8-Mb inverted segment of case 2 disrupts two protein-coding sequences, the HDAC9 and SNX10 genes. The first gene belongs to a family of histone deacetylases, which are involved in transcriptional regulation by modifying the core histones (Zhou et al. 2001), whereas the latter gene, which is uncharacterised, might be involved in intracellular trafficking. Recently, a Portuguese family has been described where a balanced translocation t(1;7)(q41;p21) co-segregated with a congenital eye defect known as Peters’ anomaly, and subsequent identification of the translocation breakpoints demonstrated a disruption of the HDAC9 gene (David et al. 2003). The identified inversion breakpoint of case 2 is located 192 kb proximal to this translocation breakpoint, but as case 2 does not show any obvious eye defects, the HDAC9 gene is not likely to be involved in the pathogenesis of Peters’ anomaly. However, the existence of a translocation breakpoint in such close vicinity of the inversion breakpoint supports the possible existence of rearrangement-prone sequence motifs. This is further substantiated by the recognition of a microdeletion syndrome at 7p21.1 (Johnson et al. 1998). As for the deletions characterised here, the exact underlying mechanism for the instability is not known.
In conclusion, we described a strategy for mapping deletions where numerous short test PCRs were designed according to the human reference sequence and used to scan a genomic area for the relative copy number. By consecutive testing and designing new test PCRs we cloned two different deletion breakpoints, proving the concept of the mapping strategy. As the approach is largely independent of the genomic locus and easy to handle, it may prove helpful for characterising a broad spectrum of deletions.
References
Armour JA, Sismani C, Patsalis PC, Cross G (2000) Measurement of locus copy number by hybridisation with amplifiable probes. Nucleic Acids Res 28:605–609
Armour JA, Barton DE, Cockburn DJ, Taylor GR (2002) The detection of large deletions or duplications in genomic DNA. Hum Mutat 20:325–337
Bischoff AM, Luijendijk MW, Huygen PL, van Duijnhoven G, De Leenheer EM, Oudesluijs GG, Van Laer L, Cremers FP, Cremers CW, Kremer H (2004) A novel mutation identified in the DFNA5 gene in a Dutch family: a clinical and genetic evaluation. Audiol Neurootol 9:34–46
Cai T, Yu P, Tagle DA, Xia J (1999) Duplication of 7p21.2→pter due to maternal 7p;21q translocation: implications for critical segment assignment in the 7p duplication syndrome. Am J Med Genet 86:305–311
Chotai KA, Brueton LA, van Herwerden L, Garrett C, Hinkel GK, Schinzel A, Mueller RF, Speleman F, Winter RM (1994) Six cases of 7p deletion: clinical, cytogenetic, and molecular studies. Am J Med Genet 51:270–276
Cox H, Stewart H, Hall L, Donnai D (2002) Phenotypic spectrum of interstitial 7p duplication in mosaic and non-mosaic forms. Am J Med Genet 109:306–310
David D, Cardoso J, Marques BB, Marques R, Silva ED, Santos H, Boavida MG (2003) Molecular characterization of a familial translocation implicates disruption of HDAC9 and possible position effect on TGFbeta2 in the pathogenesis of Peters’ anomaly. Genomics 81:489–503
Devriendt K, Jaeken J, Matthijs G, Van Esch H, Debeer P, Gewillig M, Fryns JP (1999) Haploinsufficiency of the HOXA gene cluster, in a patient with hand-foot-genital syndrome, velopharyngeal insufficiency, and persistent patent ductus Botalli. Am J Hum Genet 65:249–251
Frisen L, Lagerstedt K, Tapper-Persson M, Kockum I, Nordenskjold A (2003) A novel duplication in the HOXA13 gene in a family with atypical hand-foot-genital syndrome. J Med Genet 40:e49
Ginzinger DG (2002) Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol 30:503–512
Goodman FR (2002) Limb malformations and the human HOX genes. Am J Med Genet 112:256–265
Higuchi R, Fockler C, Dollinger G, Watson R (1993) Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N Y) 11:1026–1030
Hillier LW, Fulton RS, Fulton LA, Graves TA, Pepin KH, Wagner-McPherson C, Layman D, Maas J, Jaeger S, Walker R, Wylie K, Sekhon M, Becker MC, O’Laughlin MD, Schaller ME, Fewell GA, Delehaunty KD, Miner TL, Nash WE, Cordes M, Du H, Sun H, Edwards J, Bradshaw-Cordum H, Ali J, Andrews S, Isak A, Vanbrunt A, Nguyen C, Du F, Lamar B, Courtney L, Kalicki J, Ozersky P, Bielicki L, Scott K, Holmes A, Harkins R, Harris A, Strong CM, Hou S, Tomlinson C, Dauphin-Kohlberg S, Kozlowicz-Reilly A, Leonard S, Rohlfing T, Rock SM, Tin-Wollam AM, Abbott A, Minx P, Maupin R, Strowmatt C, Latreille P, Miller N, Johnson D, Murray J, Woessner JP, Wendl MC, Yang SP, Schultz BR, Wallis JW, Spieth J, Bieri TA, Nelson JO, Berkowicz N, Wohldmann PE, Cook LL, Hickenbotham MT, Eldred J, Williams D, Bedell JA, Mardis ER, Clifton SW, Chissoe SL, Marra MA, Raymond C, Haugen E, Gillett W, Zhou Y, James R, Phelps K, Iadanoto S, Bubb K, Simms E, Levy R, Clendenning J, Kaul R, Kent WJ, Furey TS, Baertsch RA, Brent MR, Keibler E, Flicek P, Bork P, Suyama M, Bailey JA, Portnoy ME, Torrents D, Chinwalla AT, Gish WR, et al (2003) The DNA sequence of human chromosome 7. Nature 424:157–164
Johnson D, Horsley SW, Moloney DM, Oldridge M, Twigg SR, Walsh S, Barrow M, Njolstad PR, Kunz J, Ashworth GJ, Wall SA, Kearney L, Wilkie AO (1998) A comprehensive screen for TWIST mutations in patients with craniosynostosis identifies a new microdeletion syndrome of chromosome band 7p21.1. Am J Hum Genet 63:1282–1293
Kirchhoff M, Gerdes T, Rose H, Maahr J, Ottesen AM, Lundsteen C (1998) Detection of chromosomal gains and losses in comparative genomic hybridization analysis based on standard reference intervals. Cytometry 31:163–173
Kirchhoff M, Rose H, Maahr J, Gerdes T, Bugge M, Tommerup N, Tumer Z, Lespinasse J, Jensen PK, Wirth J, Lundsteen C (2000) High resolution comparative genomic hybridisation analysis reveals imbalances in dyschromosomal patients with normal or apparently balanced conventional karyotypes. Eur J Hum Genet 8:661–668
Kirchhoff M, Rose H, Lundsteen C (2001) High resolution comparative genomic hybridisation in clinical cytogenetics. J Med Genet 38:740–744
Kirchhoff M, Pedersen S, Kjeldsen E, Rose H, Dunø M, Kølvraa S, Lundsteen C (2004) A prospective study comparing HR-CGH and subtelomeric FISH for investigation of mentally retarded and dysmorphic individuals and an update of a study using only HR-CGH. Am J Med Genet 127A:111–117
Knoll JH, Rogan PK (2003) Sequence-based, in situ detection of chromosomal abnormalities at high resolution. Am J Med Genet 121A:245–257
Kozma C, Haddad BR, Meck JM (2000) Trisomy 7p resulting from 7p15;9p24 translocation: report of a new case and review of associated medical complications. Am J Med Genet 91:286–290
Megarbane A, Le Lorc HM, Elghezal H, Joly G, Gosset P, Souraty N, Samaras L, Prieur M, Vekemans M, Turleau C, Romana SP (2001) Pure partial 7p trisomy including the TWIST, HOXA, and GLI3 genes. J Med Genet 38:178–182
Mortlock DP, Innis JW (1997) Mutation of HOXA13 in hand-foot-genital syndrome. Nat Genet 15:179–180
Nimmakayalu MA, Gotter AL, Shaikh TH, Emanuel BS (2003) A novel sequence-based approach to localize translocation breakpoints identifies the molecular basis of a t(4;22). Hum Mol Genet 12:2817–2825
Nystrom-Lahti M, Kristo P, Nicolaides NC, Chang SY, Aaltonen LA, Moisio AL, Jarvinen HJ, Mecklin JP, Kinzler KW, Vogelstein B et al (1995) Founding mutations and Alu-mediated recombination in hereditary colon cancer. Nat Med 1:1203–1206
Petrij-Bosch A, Peelen T, van Vliet M, van Eijk R, Olmer R, Drusedau M, Hogervorst FB, Hageman S, Arts PJ, Ligtenberg MJ, Meijers-Heijboer H, Klijn JG, Vasen HF, Cornelisse CJ, van ’t Veer LJ, Bakker E, van Ommen GJ, Devilee P (1997) BRCA1 genomic deletions are major founder mutations in Dutch breast cancer patients. Nat Genet 17:341–345
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Prediger EA (2001) Quantitating mRNAs with relative and competitive RT-PCR. Methods Mol Biol 160:49–63
Reish O, Berry SA, Dewald G, King RA (1996) Duplication of 7p: further delineation of the phenotype and restriction of the critical region to the distal part of the short arm. Am J Med Genet 61:21–25
Rowland JS, Barton DE, Taylor GR (2001) A comparison of methods for gene dosage analysis in HMSN type 1. J Med Genet 38:90–95
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G (2002) Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30:e57
Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W (2003) Human-mouse alignments with BLASTZ. Genome Res 13:103–107
Siebert PD, Chenchik A, Kellogg DE, Lukyanov KA, Lukyanov SA (1995) An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res 23:1087–1088
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN (2003) Human gene mutation database (HGMD): 2003 update. Hum Mutat 21:577–581
Tharapel SA, Kadandale JS (2002) Primed in situ labeling (PRINS) for evaluation of gene deletions in cancer. Am J Med Genet 107:123–126
Utsch B, Becker K, Brock D, Lentze MJ, Bidlingmaier F, Ludwig M (2002) A novel stable polyalanine [poly(A)] expansion in the HOXA13 gene associated with hand-foot-genital syndrome: proper function of poly(A)-harbouring transcription factors depends on a critical repeat length? Hum Genet 110:488–494
Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, Van de Heyning P, McGuirt WT, Smith RJ, Willems PJ, Legan PK, Richardson GP, Van Camp G (1998) Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet 20:194–197
Yan H, Papadopoulos N, Marra G, Perrera C, Jiricny J, Boland CR, Lynch HT, Chadwick RB, de la Chapelle A, Berg K, Eshleman JR, Yuan W, Markowitz S, Laken SJ, Lengauer C, Kinzler KW, Vogelstein B (2000) Conversion of diploidy to haploidy. Nature 403:723–724
Yu C, Meng X, Zhang S, Zhao G, Hu L, Kong X (2003) A 3-nucleotide deletion in the polypyrimidine tract of intron 7 of the DFNA5 gene causes nonsyndromic hearing impairment in a Chinese family. Genomics 82:575–579
Zhou X, Marks PA, Rifkind RA, Richon VM (2001) Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci USA 98:10572–10577
Acknowledgements
We would like to thank the three patients and their families for their cooperation. In memory of the late Claes Lundsteen (1944–2003), who initiated the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dunø, M., Hove, H., Kirchhoff, M. et al. Mapping genomic deletions down to the base: a quantitative copy number scanning approach used to characterise and clone the breakpoints of a recurrent 7p14.2p15.3 deletion. Hum Genet 115, 459–467 (2004). https://doi.org/10.1007/s00439-004-1174-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-004-1174-y