
Abstract
Congenital heart disease (CHD) is the most common birth defect in humans and is present in 40% of newborns affected by Down syndrome (DS). The SH3BGR gene maps to the DS-CHD region and is a potential candidate for the pathogenesis of CHD, since it is selectively expressed in cardiac and skeletal muscle. To determine whether overexpression of Sh3bgr in the murine heart may cause abnormal cardiac development, we have generated transgenic mice using a cardiac- and skeletal-muscle-specific promoter to drive the expression of a Sh3bgr transgene. We report here that heart morphogenesis is not affected by overexpression of Sh3bgr.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A 5-Mb region in 21q22.2 encompassing D21S55 to MX1 has been associated with congenital heart disease (CHD) present in 40% of newborns affected by Down syndrome (DS). The complete sequencing of human chromosome 21 has recently shown that this region contains less than 30 genes (Hattori et al. 2000). The functional characterization of these genes is a crucial step towards the identification of genes involved in the pathogenesis of DS-CHD. The SH3BGR gene maps to the DS-CHD critical region and has been previously isolated by cDNA selection in an effort to identify genes that are located in this region and that are expressed in the developing heart (Scartezzini et al. 1997). An expression study in the mouse has revealed that the corresponding murine gene is expressed in the heart from early stages of embryonic development (Egeo et al. 2000).
The mechanisms by which an extra copy of chromosome 21 results in the various clinical aspects of DS are still unknown. It remains to be established whether the defects seen in DS are attributable to the overexpression of specific individual genes or small groups of genes located on the triplicated chromosome (Chrast et al. 2000) or to a general stochastic deregulation of transcription when three copies of the chromosome are present (Saran et al. 2003). The use of both microarray approaches and single gene overexpression will be necessary to dissect the molecular mechanisms that underlie the development of DS phenotype. We have used a transgenic approach to explore the functional role of the Sh3bgr gene in heart development and its possible involvement in DS-CHD.
Materials and methods
Two different constructs, one containing only the mouse Sh3bgr gene and another also containing a lacZ gene placed after an internal ribosomal entry site element, were used for the production of the transgenic lines on an FVB genetic background (Fig. 1A). Gene expression in both constructs was driven by the beta/slow MyHC promoter that, in mouse embryos, is expressed exclusively in heart and skeletal muscle (Knotts et al. 1996). We obtained several transgenic lines (Fig. 1B); however, only two lines (164 and 192) were found to express the transgene (not shown). The Sh3bgr level of expression in the two transgenic lines was analyzed by real-time polymerase chain reaction (PCR).

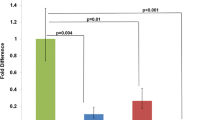
A Schematic representation of the two constructs used to overexpress the Sh3bgr gene. The relative position of the primers used for genotyping is indicated. B Transgenic positive lines are identified by PCR from genomic DNA. The 1-kb PCR product corresponds to the α-5 integrin gene, amplified as a control. C Level of expression of Sh3bgr gene determined in E17 transgenic hearts by quantitative real-time PCR. Values are expressed as the fold-increase over wild-type (wt) littermates and are normalized for β-actin (n=5–8 for each group; *P<0.05 versus wt). D Expression pattern of Sh3bgr transgenes. Part a E10 embryo, line 164. Whole mount LacZ staining; β-gal positive cells are present exclusively in heart and skeletal muscle. Parts b, c E14 embryo, line 962. Sections processed for in situ hybridisation with 35S-labelled cRNA probes specific for the HGHpolyA gene, counterstained with eosin and viewed with darkfield (Part b) or brightfield (Part c) optics. Note that both septa (asterisks) and atrioventricular valves (arrows) are normal. Parts d–f E16 embryo, line 164. Serial sections of the heart processed for β-gal staining (Part d) or in situ hybridisation with probes specific for lacZ gene viewed with darkfield (Part e) or brightfield (Part f) optics. Note that both septa (asterisks) and atrioventricular valves (arrows) are normal
Results and discussion
As shown in Fig. 1C, a moderate but significant overexpression of Sh3bgr gene (1.6 for line 164 and 2.1 for line 962) was observed at day 17 of embryonic development (E17) in the hearts of transgenic embryos compared with wild-type littermates.
In human trisomy 21, the most frequent and specific heart abnormalities are endocardial cushion defects (atrioventricular canal defects and atrioventricular septal defects). In the mouse, endocardial cushion development starts at E10 and septation is almost completed by E14. The transgenes were expressed in cardiac muscle at E10, as determined by reverse transcription/PCR in both lines (not shown) and by whole mount lacZ staining for line 164 (Fig. 1D, part a) and was also observed at subsequent developmental stages, as determined by lacZ staining or by in situ hybridization with specific radioactive probes (Fig. 1D, parts b–f). Heart morphology was not altered in transgenic hearts at E14 and E16. In particular, the four chambers were correctly separated by valves and septa (Figs. 1D, parts c, f); the outflow tract, which is also frequently altered in the murine model for DS (Ts16), was normally developed in both lines (data not shown). The transgene was expressed at birth and in the adult; heart morphology was also normal at these stages in both transgenic lines (not shown).
In conclusion, we have produced two transgenic lines that moderately overexpress the Sh3bgr gene with an expression profile that resembles endogenous Sh3bgr gene expression throughout cardiac development. These lines therefore provide an appropriate model to study the contribution of SH3BGR gene to the heart phenotype of DS. We have not observed any gross difference in heart morphology between the transgenic animals and their wild-type littermates and, hence, Sh3bgr gene overexpression per se is not accompanied by congenital heart defects in the mouse, at least not in those with an FVB genetic background. Our results do not rule out the possibility that modifier genes are involved in this phenotype; indeed, only a proportion of the patients with Down syndrome develop heart malformations. Furthermore, overexpression of the Sh3bgr gene together with other genes of the critical region may be necessary to induce the heart phenotype.
References
Chrast R, Scott HS, Madani R, Huber L, Wolfer DP, Prinz M, Aguzzi A, Lipp HP, Antonarakis SE (2000) Mice trisomic for a bacterial artificial chromosome with the single-minded 2 gene (Sim2) show phenotypes similar to some of those present in the partial trisomy 16 mouse models of Down syndrome. Hum Mol Genet 9:1853–1864
Egeo A, Di Lisi R, Sandri C, Mazzocco M, Lapide M, Schiaffino S, Scartezzini P (2000) Developmental expression of the SH3BGR gene, mapping to the Down syndrome heart critical region. Mech Dev 90:313–316
Hattori M, Fujiyama A, Taylor T.D, Watanabe H, Yada T, Park H.-S, Toyoda A, et al (2000) The DNA sequence of human chromosome 21. Nature 405:311–319
Knotts S, Sanchez A, Rindt H, Robbins J (1996) Developmental modulation of a beta myosin heavy chain promoter-driven transgene. Dev Dyn 206:182–192
Saran NG, Pletcher MT, Natale JE, Cheng Y, Reeves RH (2003) Global disruption of the cerebellar transcriptome in a Down syndrome mouse model. Hum Mol Genet 12:2013–2019
Scartezzini P, Egeo A, Colella S, Fumagalli P, Arrigo P, Nizetic D, Taramelli R, Rasore-Quartino A (1997) Cloning a new human gene from chromosome 21q22.3 encoding a glutamic acid-rich protein expressed in heart and skeletal muscle. Hum Genet 99:387–392
Acknowledgements
We thank Dr. J. Robbins for kindly providing the beta/slow MyHC construct, Fiorella Altruda for the production of the transgenic mice and Lisa Agatea for technical assistance. This study was supported by a grant from EC (Biomed Contract BMH4/CT98/404).
Author information
Authors and Affiliations
Corresponding author
Additional information
C.S. and R.D.L. contributed equally to this work
Rights and permissions
About this article
Cite this article
Sandri, C., Di Lisi, R., Picard, A. et al. Heart morphogenesis is not affected by overexpression of the Sh3bgr gene mapping to the Down syndrome heart critical region. Hum Genet 114, 517–519 (2004). https://doi.org/10.1007/s00439-004-1088-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-004-1088-8