
Abstract
Congenital cataracts are an important cause of blindness worldwide. In a family of Chinese descent, a dominant congenital nuclear cataract locus was mapped to chromosome 17q11.1-12. The maximum LOD score, 2.49, at recombination fraction 0, was obtained for marker D17S1294. The results of both linkage and haplotype analyses defined a disease-gene to an 11.78-cM region harboring the gene coding for βA1/A3 crystallin (CRYBA1/A3). Mutation analysis of the CRYBA1/A3 gene identified a 3-bp deletion in exon 4, which cosegregated with the disease risk in this family and was not observed in 100 normal chromosomes. This mutation resulted in the deletion of a highly conserved glycine at codon 91 (ΔG91) and could be associated with an incorrect folding of βA1/A3 crystallin. It highlights the physiological importance of crystallin and supports the role of CRYBA1/A3 in human cataracts formation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Congenital cataracts are phenotypically and genetically heterogeneous, with an estimated prevalence of 1–6 per 10,000 live births (Lambert and Drack 1996). Approximately one-third of all cases are familial, and autosomal dominant cataract (ADC) appears to be the most common transmission mode (Ionides et al. 1999). Despite congenital cataracts being a leading cause of blindness worldwide, the mechanisms of lens opacification remain poorly understood (Arnold 1998).
So far, causative mutations in ADC have been identified in 12 distinct genes, including seven genes coding for crystallins: CRYAA on chromosome 21q (Litt et al. 1998), CRYAB on 11q (Berry et al. 2001), CRYBA1/A3 on 17q (Kannabiran et al. 1998; Bateman et al. 2000), CRYBB1 on 22q (Mackay et al. 2002), CRYBB2 on 22q (Litt et al. 1997; Gill et al. 2000; Vanita et al. 2001), CRYGC on 2q (Heon et al. 1999; Ren et al. 2000), and CRYGD on 2q (Stephan et al. 1999; Nandrot et al. 2003); as well as two coding for gap junctional channel proteins, GJA3 on 13q (Mackay et al. 1999; Rees et al. 2000), and GJA8 on 1q (Shiels et al. 1998; Berry et al. 1999; Polyakov et al. 2001), one for heat-shock transcription factor 4, HSF4 on 16q (Bu et al. 2002), one for major intrinsic protein, MIP on 12q (Berry et al. 2000), and one for beaded-filament structural protein-2, BFSP2 on 3q (Conley et al. 2000; Jakobs et al. 2000). Only two genes are associated with human autosomal recessive cataracts: one coding for crystallin gene, CRYAA on 21q (Pras et al. 2000), the other for intrinsic membrane protein 19, MP19 on 19q (Pras et al. 2002). Of interest, the CRYAA gene causes both autosomal dominant and autosomal recessive congenital cataracts (Litt et al. 1998; Pras et al. 2000).
Nuclear cataract, which is one of most familial types of severe congenital cataracts, was first reported by Brown (1924). The opacity was located at the center of the lens, which can have a remarkable effect on visual acuity. So far, dominantly inherited nuclear cataracts have been linked to 2p12 (MIM 607304) (Khaliq et al. 2002) and CRYAA on 21q (MIM 123580) (Litt et al. 1998).
In this report, we linked the dominantly inherited nuclear cataracts affecting a Chinese family to 17q11.1-12, and identified a deletion mutation in the CRYBA1/A3 gene that is responsible for the cataracts in this pedigree. This is the first report of nuclear cataracts caused by this gene.
Materials and methods
Clinical evaluation and DNA specimens
We studied a large family that is affected with congenital nuclear cataracts (Fig. 1). This family resides in a relatively isolated region of China. Fourteen members of the family participated the study; all participants gave informed consent, which was approved by the Institution Review Board of the Harbin Medical University, China. The dilated slit-lamp examinations were carried out. Among 14 participants, 11 individuals were identified to be affected with nuclear cataract, the remaining three were normal. The cataract was bilateral in all cases and consisted of a well-defined and dense opacity that was located at the embryonic nucleus of the lens and was 1.5–3 mm in diameter. The opacity was present at birth and developed during infancy but did not progress with age. There was no family history of other ocular or systemic abnormalities.

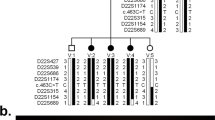
Pedigree and haplotype of the autosomal dominant congenital cataract. Open white and solid black symbols denote unaffected and affected individuals, respectively. The affected haplotype is indicated by a black vertical bar. The sequence of markers is from centromere to telomere. Uninformative makers are indicated by a vertical line in the haplotype bar
Blood samples were collected in EDTA and leukocyte genomic DNA was extracted.
Genotyping and linkage analysis
The initial strategy consisted of screening 12 known candidate genes related to ADC formation and one locus related to congenital nuclear cataract (Khaliq et al. 2002). The primer sequences were taken from the Genome Database (http://www.gdb.org). Microsatellites used in this study included: D1S252, D1S305, D1S2721, D2S157, D2S325, D2S2333, D3S1290, D3S1744, D11S1986, D11S898, D12S90, D12S1676, D13S175, D13S1236, D16S421, D16S3043, D17S1294, D21S212, CRYBB2, D22S1174, TOP1P2, D22S315. Fifty nanograms of template DNA was used in a 25-μl reaction volume, with 2.5 mM of dNTPs, 10 pmol of fluorescently labeled forward primers and unlabeled reverse primers, 1.0 U Taq DNA polymerase, and 10×PCR buffer [100 mM Tris-HCl, 80 mM (NH4)2SO4, 100 mM KCl, and 25 mM MgCl2, (pH 9.0); Sangon, Shanghai, China]. After an initial denaturation of 5 min at 95 °C, 31 cycles were performed at 94 °C for 30 s, 53–58 °C for 30 s, and 72 °C for 1 min, followed by an extension at 72 °C for 10 min and a final hold at 4 °C. PCR products were mixed with loading buffer equally (v/v), as well as with 0.5 μl of internal lane standard (ROX-400; Perkin-Elmer Applied Biosystems). PCR products were denatured at 98 °C for 2 min, and were electrophoresed on 4% denaturing polyacrylamide gels on an automatic DNA sequencer (ABI-PRISM 377; Perkin-Elmer Applied Biosystems). Data were collected by GENESCAN 2.1 software (Perkin-Elmer Applied Biosystems ) and individual genotypes were analyzed with GENOTYPER version 2.0 (Perkin-Elmer Applied Biosystems).
Two-point linkage analysis was performed with LINKAGE program package version 5.1 (freely available at http://linkage.rockfeller.edu). Autosomal dominant inheritance, with a full penetrance and a disease-gene frequency of 0.0001 was considered. The marker allele frequencies were assumed to be uniformly distributed.
Mutation analysis
A strong candidate gene, the βA1/A3 crystallin gene (CRYBA1/A3) (GenBank accession number NM_005208), is comprised of six exons. To screen the coding regions of CRYBA1/A3, gene-specific PCR primers were designed flanking each exon and intron-exon junction (Table 1). All primers were derived from relevant articles, except exon 2 (Kannabiran et al. 1998; Bateman et al. 2000).
The purified PCR products (using an Agarose Gel DNA Purification kit; TAKARA, Dalian, China) were sequenced from both directions using an automated fluorescence sequencer (ABI PRISM 377; Perkin-Elmer Applied Biosystems). One affected (III:2) and an unaffected family member (IV:5) were compared. After identifing a mutation in exon 4, all family members and 50 unrelated normal individuals were screened.
Results
Autosomal dominant inheritance of the cataract was supported by the presence of affected individuals in each of the four generations, and male-to-male transmission (Fig. 1).
After excluding candidate genes which lack segregation between the markers and the disease, a maximum LOD score of 2.49 without recombination (θ=0), was obtained with D17S1294 on chromosome 17q11.1-12. Five additional markers flanking D17S1294 were analysed in this pedigree (Table 2). The order and genetic distances of the markers were derived from the Marshfield database (http://research. marshfieldclinic.org).
The results of both linkage and haplotype analyses (Fig. 1) defined a disease-gene to an 11.78-cM region bounded by D17S1288 and D17S933.
Direct sequencing of amplified exons, a 3-bp (GGA) deletion was detected at nucleotide position 278–280 (GenBank accession number NM_005208) of exon 4 (Fig. 2B).

Sequence analysis of CRYBA1/A3 at exon 4. A Sequence chromatograms of the wild-type allele, showing the translation of glycine (GGA) at codon 91 (underlined). B Sequence chromatograms of mutant allele showing a 3-bp deletion (underlined). C A multiple alignment of protein sequence of CRYBA1/A3 with the corresponding segments in different species, and five other β-crystallins in humans. The sequences were selected using BLASTP (http://www.ncbi.nlm.nih.gov/BLAST). The arrow indicates the ΔG91 mutated position
This deletion is predicted to cause an in-frame deletion of a glycine residue at position 91 (ΔG91) from the mature gene product (Entrez-protein accession number P_05813) and produced an aberrant protein consisting of 214 residues. No other exonic sequence variants were observed in one affected (III:2) and an unaffected family member (IV:5). The six base changes reported by Lampi et al. (1997) were confirmed. In addition, this mutation was detected in all affected members in this family, but neither in normal relatives nor in 50 unrelated individuals, excluding the possibility that it is a rare polymorphism.
Discussion
In mammals, three major groups of lens proteins can be distinguished: the α-, β-, and γ-crystallins. The β- and γ-crystallins are also considered to be superfamilies. Crystallography has shown that both the β- and γ-crystallins consist of two similar domains (the N-terminal domain and C-terminal domain) separated by a short connecting peptide. Each domain folds into two similar “Greek key” motifs, with distinctive β-sheet folding. Each Greek key motif consists of four consecutive anti-parallel β-strands, known as a, b, c and d strands (Fig. 3) (Blundell et al. 1981; Slingsby and Clout. 1999). The four motifs form four β-sheets: two (the β1- and β3-sheets) lie on the outside of the molecule, and two (the β2- and β4-sheets) are in partial contact (domain association) (Fu and Liang 2003).

Greek key motif topology. Strands are represented by vertical arrows. The lengths of β-strand are reflected by arrow lengths. The horizontal arrow indicates the site of the ΔG91 mutation in the N-terminal domain. The hydrogen bonds between strands are represented by dashed lines
The β-crystallin family consists of seven proteins, βB1-, βB2-, βB3-, and βA4-crystallin on chromosome 22q11.2-13.1 (Hulsebos et al. 1991, van Rens et al. 1992), βA1/A3-crystallin on 17q11.1-12 (Sparkes et al. 1986) and βA2-crystallin on 2q33-35 (Hulsebos et al. 1995). Different β-crystallin proteins can interact with each other to form oligomers of different sizes range from dimers to octamers (Werten et al. 1996) and can also interact with other lens proteins. The protein-protein interactions are predicted to be key in maintaining the transparency of lens (Bax et al. 1990; Russell and Chambers 1990). It is predicted that the transparency of the lens depends on the tertiary structure of the crystallins. A disruption of stabilization or oligomerization by any means would result in lens opacification (Delaye and Tardieu 1983).
By in situ hybridization, Xu et al. (1988)had localized a gene that encodes crystallin, βA1 (MIM 123610) to chromosome 17q11.1-q12. The chromosomal localization, as well as the important role of the protein in maintaining lens transparency and development, and its association with cataract formation, suggested that CRYBA1/A3 was an excellent candidate gene for this pedigree.
The βA1/A3-crystallin gene encodes both the βA3- and βA1-crystallins. The latter is 17 aa shorter than βA3-crystallin and they are generated by use of an alternative translation initiation site (Quax-Jeuken et al. 1984). The first two exons of the βA1-crystallin gene encode the N-terminal extension, and exons 3–6 encode the four Greek key motifs (Hogg et al. 1986).
Kannabiran et al. (1998) reported a splice mutation (G→A) in CRYBA1/A3 in an Indian family affected with autosomal dominant zonular cataracts with sutural opacities (CCZS). Bateman et al. (2000) also reported a splice junction mutation (G→C) occurred in an identical locus.
Alignment of protein sequences of CRYBA1/A3 in five different species with five other β-crystallins of human species revealed that the glycine at this position is highly conserved (Fig. 2C). The deletion of this highly conserved glycine identified in our family is located at the c2 strand of the second Greek key motif, which is a key region for the tertiary structure of the βA1/A3-crystallin.The deletion could destroy the interstrand hydrogen bonds between c2 and d1 strands (Fig. 3), disrupting the proper folding of whole N-terminal domain, which, in turn, could destroy the intramolecular interactions between domains (Fu and Liang 2003), causing the disruption of the highly symmetrical structure of βA1/A3-crystallin. The change of tertiary structure ultimately may contribute to decrease the stability and/or solubility of this protein. If there is an aberrant aggregation of mutant protein, it may cause light scattering and finally lead to lens opacification.
The β-crystallins tend to abundantly expressed at an early developmental stage in elongating fiber cells, so they are found primarily in the lens nucleus (Aarts et al. 1989), which is consistent with the location of the opacity in the embryonic nucleus in this pedigree.
In conclusion, we have identified, in βA1/A3-crystallin gene (CRYBA1/A3), a novel deletion mutation (ΔG91) causing nuclear cataract. This mutation supports the previously proposed role of CRYBA1/A3 in the lens opacification process and may help delineate the functional domains of CRYBA1/A3. This is the first report of the autosomal dominant nuclear cataract caused by CRYBA1/A3, which should contribute to a better understanding of the relationships of genotype-phenotype.
References
Aarts HJ, Lubsen NH, Schoenmakers JG (1989) Crystallin gene expression during rat lens development. Eur J Biochem 183:31–36
Arnold J (1998) Global cataract blindness: the unmet challenge. Br J Ophthalmol 82:593–594
Bateman JB, Geyer DD, Flodman P, Johannes M, Sikela J, Walter N, Moreira AT, Clancy K, Spence MA (2000) A new betaA1-crystallin splice junction mutation in autosomal dominant cataract. Invest Ophthalmol Vis Sci 41:3278–3285
Bax B, Lapatto R, Nalini V, Driessen H, Lindley PF, Mahadevan D, Blundell TL, Slingsby C (1990) X-ray analysis of beta B2-crystallin and evolution of oligomeric lens proteins. Nature 347:776–780
Berry V, Mackay D, Khaliq S, Francis PJ, Hameed A, Anwar K, Mehdi SQ, Newbold RJ, Ionides A, Shiels A, Moore T, Bhattacharya SS (1999) Connexin 50 mutation in a family with congenital “zonular nuclear” pulverulent cataract of Pakistani origin. Hum Genet 105:168–170
Berry V, Francis P, Kaushal S, Moore A, Bhattacharya S (2000) Missense mutations in MIP underlie autosomal dominant ‘polymorphic’ and lamellar cataracts linked to 12q. Nat Genet 25:15–17
Berry V, Francis P, Reddy MA, Collyer D, Vithana E, MacKay I, Dawson G, Carey AH, Moore A, Bhattacharya SS, Quinlan RA (2001) Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am J Hum Genet 69:1141–1145
Blundell T, Lindley P, Miller L, Moss D, Slingsby C, Tickle I, Turnell B, Wistow G (1981) The molecular structure and stability of the eye lens: X-ray analysis of gamma-crystallin II. Nature 289:771–777
Brown AL (1924) Hereditary cataract. Am J Ophthalmol 7:36–38
Bu L, Jin Y, Shi Y, Chu R, Ban A, Eiberg H, Andres L, Jiang H, Zheng G, Qian M, Cui B, Xia Y, Liu J, Hu L, Zhao G, Hayden MR, Kong X (2002) Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat Genet 31:276–278
Conley YP, Erturk D, Keverline A, Mah TS, Keravala A, Barnes LR, Bruchis A, Hess JF, FitzGerald PG, Weeks DE, Ferrell RE, Gorin MB (2000) A juvenile-onset, progressive cataract locus on chromosome 3q21-q22 is associated with a missense mutation in the beaded filament structural protein-2. Am J Hum Genet 66:1426–1431
Delaye M, Tardieu A (1983) Short-range order of crystallin proteins accounts for eye lens transparency. Nature 302:415–417
Fu L, Liang JJ (2003) Alteration of protein-protein interactions of congenital cataract crystallin mutants. Invest Ophthalmol Vis Sci 44:1155–1159
Gill D, Klose R, Munier FL, McFadden M, Priston M, Billingsley G, Ducrey N, Schorderet DF, Heon E (2000) Genetic heterogeneity of the Coppock-like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci 41:159–165
Heon E, Priston M, Schorderet DF, Billingsley GD, Girard PO, Lubsen N, Munier FL (1999) The gamma-crystallins and human cataracts: a puzzle made clearer. Am J Hum Genet 65:1261–1267
Hogg D, Tsui LC, Gorin M, Breitman ML (1986) Characterization of the human beta-crystallin gene Hu beta A3/A1 reveals ancestral relationships among the beta gamma-crystallin superfamily. J Biol Chem 261:12420–12427
Hulsebos TJ, Bijlsma EK, Geurts van Kessel AH, Brakenhoff RH, Westerveld A (1991) Direct assignment of the human beta B2 and beta B3 crystallin genes to 22q11.2-q12: markers for neurofibromatosis 2. Cytogenet Cell Genet 56:171–175
Hulsebos TJ, Cerosaletti KM, Fournier RE, Sinke RJ, Rocchi M, Marzella R, Jenkins NA, Gilbert DJ, Copeland NG (1995) Identification of the human beta A2 crystallin gene (CRYBA2): localization of the gene on human chromosome 2 and of the homologous gene on mouse chromosome 1. Genomics 28:543–548
Ionides A, Francis P, Berry V, Mackay D, Bhattacharya S, Shiels A, Moore A (1999) Clinical and genetic heterogeneity in autosomal dominant cataract. Br J Ophthalmol 83:802–808
Jakobs PM, Hess JF, FitzGerald PG, Kramer P, Weleber RG, Litt M (2000) Autosomal-dominant congenital cataract associated with a deletion mutation in the human beaded filament protein gene BFSP2. Am J Hum Genet 66:1432–1436
Kannabiran C, Rogan PK, Olmos L, Basti S, Rao GN, Kaiser-Kupfer M, Hejtmancik JF (1998) Autosomal dominant zonular cataract with sutural opacities is associated with a splice mutation in the betaA3/A1-crystallin gene. Mol Vis 4:21
Khaliq S, Hameed A, Ismail M, Anwar K, Mehdi SQ (2002) A novel locus for autosomal dominant nuclear cataract mapped to chromosome 2p12 in a Pakistani family. Invest Ophthalmol Vis Sci 43:2083–2087
Lambert SL, Drack AV (1996) Infantile cataracts. Surv Ophthalmol 40:427–458
Lampi KJ, Ma Z, Shih M, Shearer TR, Smith JB, Smith DL, David LL (1997) Sequence analysis of βA3, βB3, and βA4 crystallins completes the identification of the major proteins in young man lens. J Biol Chem 272:2268–227
Litt M, Carrero-Valenzuela R, LaMorticella DM, Schultz DW, Mitchell TN, Kramer P, Maumenee IH (1997) Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Hum Mol Genet 6:665–668
Litt M, Kramer P, LaMorticella DM, Murphey W, Lovrien EW, Weleber RG (1998) Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum Mol Genet 7:471–474
Mackay D, Ionides A, Kibar Z, Rouleau G, Berry V, Moore A, Shiels A, Bhattacharya S (1999) Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet 64:1357–1364
Mackay DS, Boskovska OB, Knopf HL, Lampi KJ, Shiels A (2002) A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q. Am J Hum Genet 71:1216–1221
Nandrot E, Slingsby C, Basak A, Cherif-Chefchaouni M, Benazzouz B, Hajaji Y, Boutayeb S, Gribouval O, Arbogast L, Berraho A, Abitbol M, Hilal L (2003) Gamma-D crystallin gene (CRYGD) mutation causes autosomal dominant congenital cerulean cataracts. J Med Genet 40:262–267
Polyakov AV, Shagina IA, Khlebnikova OV, Evgrafov OV (2001) Mutation in the connexin 50 gene (GJA8) in a Russian family with zonular pulverulent cataract. Clin Genet 60:476–478
Pras E, Frydman M, Levy-Nissenbaum E, Bakhan T, Raz J, Assia EI, Goldman B, Pras E (2000) A nonsense mutation (W9X) in CRYAA causes autosomal recessive cataract in an inbred Jewish Persian family. Invest Ophthalmol Vis Sci 41:3511–3515
Pras E, Levy-Nissenbaum E, Bakhan T, Lahat H, Assia E, Geffen-Carmi N, Frydman M, Goldman B, Pras E (2002) A missense mutation in the LIM2 gene is associated with autosomal recessive presenile cataract in an inbred Iraqi Jewish family. Am J Hum Genet 70:1363–1367
Quax-Jeuken Y, Janssen C, Quax W, van den Heuvel R, Bloemendal H (1984) Bovine beta-crystallin complementary DNA clones. Alternating proline/alanine sequence of beta B1 subunit originates from a repetitive DNA sequence. J Mol Biol 80:457–472
Rees MI, Watts P, Fenton I, Clarke A, Snell RG, Owen MJ, Gray J (2000) Further evidence of autosomal dominant congenital zonular pulverulent cataracts linked to 13q11 (CZP3) and a novel mutation in connexin 46 (GJA3). Hum Genet 106:206–209
Ren Z, Li A, Shastry BS, Padma T, Ayyagari R, Scott MH, Parks MM, Kaiser-Kupfer MI, Hejtmancik JF (2000) A 5-base insertion in the gammaC-crystallin gene is associated with autosomal dominant variable zonular pulverulent cataract. Hum Genet 106:531–7
Russell P, Chambers C (1990) Interaction of an altered beta-crystallin with other proteins in the Philly mouse lens. Exp Eye Res 50:683–687
Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S (1998) A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am J Hum Genet 62:526–532
Slingsby C, Clout NJ (1999) Structure of the crystallins. Eye 13:395–402
Sparkes RS, Mohandas T, Heinzmann C, Gorin MB, Zollman S, Horwitz J (1986) Assignment of a human beta-crystallin gene to 17cen-q23. Hum Genet 4:133–136
Stephan DA, Gillanders E, Vanderveen D, Freas-Lutz D, Wistow G, Baxevanis AD, Robbins CM, VanAuken A, Quesenberry MI, Bailey-Wilson J, Juo SH, Trent JM, Smith L, Brownstein MJ (1999) Progressive juvenile-onset punctate cataracts caused by mutation of the gammaD-crystallin gene. Proc Natl Acad Sci USA 96:1008–1012
van Rens GL, de Jong WW, Bloemendal H (1992) A superfamily in the mammalian eye lens: the beta/gamma-crystallins. Mol Biol Rep 16:1–10
Vanita, Sarhadi V, Reis A, Jung M, Singh D, Sperling K, Singh JR, Burger J (2001) A unique form of autosomal dominant cataract explained by gene conversion between beta-crystallin B2 and its pseudogene. J Med Genet 38:392–396
Werten PJ, Carver JA, Jaenicke R, de Jong WW (1996) The elusive role of the N-terminal extension of beta A3- and beta A1-crystallin. Protein Eng 9:1021–1028
Xu W, Gorman PA, Rider SH, Hedge PJ, Moore G, Prichard C, Sheer D, Solomon E (1988) Construction of a genetic map of human chromosome 17 by use of chromosome-mediated gene transfer. Proc Nat Acad Sci USA 85:8563–8567
Acknowledgements
The authors are grateful to the families for their enthusiastic participation. We also acknowledge the financial support of the Natural Scientific Fund of Heilongjiang Provincial Scientific and Technical Bureau (Grant D0218).
Author information
Authors and Affiliations
Corresponding author
Additional information
Y. Qi and H. Jia contributed equally to this work
Rights and permissions
About this article
Cite this article
Qi, Y., Jia, H., Huang, S. et al. A deletion mutation in the βA1/A3 crystallin gene (CRYBA1/A3) is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Hum Genet 114, 192–197 (2004). https://doi.org/10.1007/s00439-003-1049-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-003-1049-7