
Abstract
The highly heterogeneous epithelial mucins show considerable inter-individual variability attributable to allelic variations in their tandem repeat (TR) peptide domains. Most mucins are known to show variations in repeat number but variation in the sequence of the individual TRs is not as well characterised. Here, we have studied variation in the immunodominant PDTR motif in the TR domain of the membrane-associated "cancer" mucin MUC1 by using the Minisatellite Variant Repeat-Polymerase chain reaction (MVR-PCR) technique. We have fully or partially mapped two nucleotide changes that encode two amino-acid changes, PDTR to PESR, across the arrays of 149 alleles. A total of 103 different maps was obtained when these changes alone were considered and additional variations were also observed. Most maps showed blocks of PDTR repeats interspersed with PESR repeats, although these were possibly more irregular in the longer alleles that also tended to have more PESR repeats. This variability has potential functional consequences and possible implications for some individuals with respect to the efficacy of immune targetting and immune therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In mammals, internal epithelial tracts are protected from sheer and abrasion and from invasion by pathogens by a highly heterogeneous mucous layer. The major proteins of this mucus are highly glycosylated glycoproteins known as mucins and some 16 MUC genes have been identified to date (http://www.gene.ucl.ac.uk/nomenclature/). Some of these glycoproteins are true secreted mucins and some are, at least in part, membrane-associated. However, all share the common feature of containing a large serine-rich and threonine-rich domain that usually contains tandemly repeated DNA and protein sequences (Fowler et al. 2001). In most cases, this tandem repeat (TR) region exhibits length polymorphism attributable to variation in the number of TRs (VNTR), which is directly reflected in the length of the protein. Several studies suggest that variation in TR length is associated with susceptibility to inflammatory disease of the epithelia (Kirkbride et al. 2001; Kyo et al. 1999; Vinall et al. 2000a, 2002).
Another less well-characterised source of mucin variability is the inter-repeat differences in nucleotide and amino acid sequence. Such differences have been reported in the initial cloning of MUC2, MUC3, MUC4, MUC5AC, MUC5B, MUC6 and MUC7 and, in the case of MUC2, there is evidence that these repeat sequence variations are genetically variable (Toribara et al. 1991). In this investigation, we have examined the TR region of MUC1, the gene for a membrane-associated mucin, well known for many years because of its aberrant expression in tumours (Taylor-Papadimitriou et al. 1999). The protein was originally detected by the many monoclonal antibodies that recognise the TR domain of this protein (Price et al. 1998).
MUC1 shows extensive TR polymorphism with alleles ranging in repeat number from about 20 to 125 and encoding polypeptides that range in size (M r ), before glycosylation, from about 90,000 to more than 450,000. The distribution of allele length is bimodal with modes at approximately 40 and 80 repeat units (as calculated from HinfI fragment sizes). Because of this bimodality, the alleles can be readily subdivided into two classes, short (S) and long (L). In addition to the VNTR variation, two polymorphisms have been identified that flank the tandem repeat region: a single-nucleotide polymorphism (SNP) in exon 2 (g.3506G→A, numbering according to the MUC1 genomic sequence (g) Genbank no. M61170) and a CA microsatellite polymorphism in intron 6 (g.6003(CA)11–14, Genbank no. M61170). Previous studies have shown a high level of linkage disequilibrium between these two flanking markers and TR length (Pratt et al. 1996). The common haplotypes are 3506A/VNTRS/CA12 or 13 and 3506G/VNTRL/CA11.
In early studies, the TRs of the MUC1 gene were thought to be identical across the array, with the exception of three poorly conserved repeats at the 5' side of the array and two at the 3' side (Gendler et al. 1990). However, some published cDNA sequences (Siddiqui et al. 1988), unpublished evidence from a cDNA clone (Pum24P; Yonezawa et al. 1991), a genomic clone isolated in our own laboratory (PUMGRep; Pratt et al. 1996), and much more recent protein work by Müller and colleagues (1999) have shown several nucleotide and amino acid substitutions in the TR region of the MUC1 gene.
In this investigation, we have examined the allelic differences in nucleotide sequence variations in the TRs of the MUC1 gene that alter the consensus motif PDTR to PESR. These particular changes were chosen because the PDTR sequence is in the most immunogenic part of the MUC1 TR units and overlaps the epitopes to which many MUC1 monoclonal antibodies bind (Price et al. 1998) and which are targets for immunotherapy. The two amino-acid substitutions are caused by two nucleotide changes that we have detected by using the Minisatellite Variant Repeat-Polymerase chain reaction technique (MVR-PCR; Jeffreys et al. 1991) with repeat specific primers that cover both the changes.
While this work was in progress, a similar study was reported by Englemann and colleagues (2001). Unlike these authors, we have separated the alleles of MUC1, succeeded in reading across the entire TR array of shorter alleles and haplotyped them with respect to the flanking markers.
Material and methods
Population tested
Samples tested were obtained, with informed consent, from 94 individuals (51 female, 43 male), 86 of whom were UK residents of European extraction, with eight from elsewhere, (age range: 21–82 years), and comprised healthy laboratory volunteers and patients and controls from our ongoing ethically approved MUC polymorphism and disease association studies (Vinall et al. 2000a, 2002). The samples were selected on the basis of the quality of the DNA and, in some cases, because of allele length homozygosity. They were unselected with respect to disease status.
DNA preparation
Blood DNA was prepared as previously described by using the Puregene DNA extraction kit (Flowgen, Leicestershire, UK; Vinall et al. 2000b).
Allele length polymorphism
Southern blot analysis of genomic DNA digested with HinfI (New England Biolabs, Beverly, Mass.) and probing with the MUC1 TR cDNA probe PUM24P were used to determine HinfI allele lengths as previously described (Vinall et al. 2000b). The number of repeats in the conserved TR array was calculated by subtracting the sum of the two flanking sequences located between the HinfI sites and the beginning and end of the conserved array, namely a total of 1.446 kb. This value obtained was then divided by 60, the length of each repeat unit and was accurate to ±1 repeat.
PCR technique
All oligonucleotide primers (Table 1) were purchased from PE Applied Biosystems (Warrington). A Techne Genius "Phoenix" PCR machine (Helena Biosciences, Cambridge) with a heated lid was used for the reactions.
Isolating single alleles
Single alleles were isolated by PCR across the TR region (Jeffreys et al. 1990); 100–150 ng genomic DNA was used as template for the PCR, each reaction taking place in a 7-μl volume and containing 5% glycerol (v/v), 45 mM TRIS (added at pH 8.8), 2.7 mM TRIS (added unbuffered), 11 mM ammonium sulphate, 4.5 mM MgCl2, 6.7 mM 2-mercaptoethanol, 4.4 μM EDTA pH 8.0, 1 mM dNTPs, 113 μg/ml bovine serum albumin, and oligonucleotide primers, Exon2S and MUC1E2AS (at a concentration 0.25 μM and 0.25 U, respectively; Dynazyme EXT, Finnzyme, GRI Research, Braintree, UK). Reactions were subjected to an initial denaturation of 96°C for 1 min 30 s for 1 cycle and then cycled at 96°C for 40 s, 60°C for 30 s and 68°C for 3 min for 22 cycles.
The entire 7-μl PCR product was subjected to electrophoresis in a 14-cm 1% agarose gel in TBE (1×TBE solution =0.088 M TRIS, 0.088 M boric acid, 0.002 M EDTA pH 8.2–8.4) at 2.1 V/cm for 20 h. A 2-μg aliquot of Kb ladder (Gibco BRL, Rockville, USA) was used as a size marker. After electrophoresis, the gels were stained with 0.055 μg/ml ethidium bromide in TBE. The bands were visualised at 400–500 nm by using a Dark Reader Transilluminator (Clare Chemical Research, Denver, USA) to prevent UV damage. The positions of the bands, which were not visible to the naked eye, were deduced in relation to the position of the molecular weight markers. Gel slices were cut out, placed in an Eppendorf tube, crushed with a pipette tip after addition of 50 μl sonicated herring sperm DNA (5 μg/ml), frozen at −70°C, thawed at room temperature (3×) to release the DNA, and then centrifuged at 11,290 g for 2 min.
PCR for testing gel slices
To check the concentration of DNA extracted, a test PCR was conducted on the gel slice extract by using primers Exon2S and Exon2AS (Table 1).
MVR-PCR of the PDTR to PESR nucleotide changes
MVR-PCR was performed in a 7-μl volume by using 0.7 μl (~40 pg) single-allele PCR product DNA or 0.3 μl (~100 ng) genomic DNA. Each reaction contained 5% glycerol (v/v), 41 mM TRIS (added buffered at pH 8.8), 2.7 mM TRIS (added unbuffered), 10 mM ammonium sulphate, 4 mM MgCl2, 6 mM 2-mercaptoethanol, 4 μM EDTA pH 8.0, 0.25 U Taq DNA polymerase in storage buffer A (Promega, Southampton), oligonucleotide primers and 0.007 U Pfu DNA polymerase (Promega). TAG and external flanking primers (MUC1E2AS or Exon2S) were at a concentration of 0.25 μM, with repeat specific primers at 5 nM or 7 nM as indicated. For the forward MVR, one reaction contained primers Exon2S, TAG and the repeat-specific GTAG (at 5 nM) and the other reaction contained primers Exon2S, TAG and CTAG (at 5 nM). For the reverse reaction, one PCR contained primers MUC1E2AS, TAG and CTAGS (at 7 nM) and the other reaction contained primers MUC1E2AS, TAG and GTAGS (at 7 nM). It should be noted that these primers have several mismatches with the poorly conserved TRs that flank the main array and that are seen in each of the published MUC1 sequences, so that the maps begin in the conserved array.
All MVR-PCRs were subjected to an initial denaturation at 96°C for 1 min 30 s; reactions were then cycled at 96°C for 40 s, 66°C for 30 s and 70°C for 2 min 30 s for 22 cycles.
The entire 7-μl PCR product was electrophoresed thorough a 22-cm 2% TBE agarose gel for 24 h at 2.5 V/cm for the forward maps. The reverse MVR-PCR products were electrophoresed for the first 6 h at 2.5 V/cm and then at 1.8 V/cm for the remaining 18 h. An aliquot of 2 μg Kb ladder and 0.8 μg Raoul Marker (Quantum Appligene, Harefield, Middlesex) were run as size standards.
Gels were then subjected to standard Southern blotting (Vinall et al. 2000b).
Haplotypes
The Exon 2 marker (g.3506G→A, Genbank no. M61170) was determined by PCR with the primers Exon2S and GAS (Table 1), followed by digestion with the restriction enzyme AlwNI and electrophoretic separation of the products. Haplotypes were determined in doubly heterozygous individuals by using the long PCR protocol described above, but with the allele-specific sense primers ExonA2S and ExonG2S (Table 1). Bands were detected by Southern blot analysis and sized by comparison with the bands of the Raoul molecular weight marker.
Results
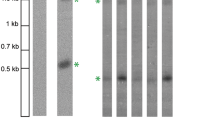
Figure 1 shows the allele length variability of MUC1 together with the calculated number of conserved TRs. In particular, the positions of the bands corresponding to the modal sizes are indicated. Figure 2 shows the consensus sequence of an MUC1 TR unit with the nucleotide changes under investigation in this study being marked above.

Southern blot analysis of MUC1 showing allelic variability and calculated repeat numbers corresponding to modal allele lengths. Lanes R Raoul molecular weight markers, lane C control genomic DNA sample, lane S mix of two control genomic DNA samples of known allele length run in every experiment as additional size standards

MUC1 consensus repeat sequence showing the nucleotide changes that convert PDTR to PESR and the nucleotide change responsible for the null repeats. The start of the repeating unit is selected here to allow clear depiction of the position of the two TR-specific primers used for MVR (arrows)
In the initial experiments, genomic DNA samples were used to construct diploid maps. Figure 3 shows a map from an individual homozygous for both allele length and MVR map. The two left-hand lanes represent a forward MVR map and the two right-hand lanes represent a reverse map. The presence of bands in lane G indicates the "consensus" sequence, which encodes PDTR, and bands in lane C indicate the alternate sequence (PESR). The map shows 37 repeat units. In the forward map, repeats 3, 4 and 12 amplify with neither of the repeat-specific primers and are designated as "null" repeats. Sequencing of a genomic clone (PumGRep) shows that there is a synonymous guanine to cytosine transversion present in some G (PDTR) repeats located underneath the CTAG and GTAG primers, at 10 nucleotides from their 3' end (Fig. 2). In long runs, the GTAGS primer used in the reverse MVR map bind to these forward map null repeats (data not shown), supporting the notion that they are all consensus repeats with respect to the PDTR sequence.

Complete homozygous MVR map. The forward and reverse reactions are shown together with the fully interpreted map. G Consensus PESR repeat, C PESR repeat
The MVR patterns obtained were reproducible and shown to be a characteristic of the studied individual. The presence of bands at the same position in both tracks indicated map heterozygosity (data not shown). Diploid maps of parents and five children of family 104 from the Centre d'Étude du Polymorphisme Humain were tested; the presence or absence of bands and the deduced allelic maps were consistent with Mendelian inheritance.
More powerful information was obtained by constructing single-allele maps, by using size-separated alleles as the source of MVR template. Figure 4 shows examples and illustrates the large amount of inter-allelic variability of the TR region with respect to the "PDTR" consensus repeats and "PESR" alternative repeats for both the forward and reverse MVR maps. All individuals studied had one or two copies of the "null" repeat at positions 3 and 4 counted from the 5' end of the array and zero, one or two "null" repeats at positions 11 and 12.

Examples of single-allele MVR reactions. A Four forward MVR maps from differently sized alleles. B Four reverse MVR maps from differently sized alleles. Arrows (below) Lanes containing the consensus PDTR repeats. The adjacent lanes to the right represent the alternative PESR repeats of the same allele. Numbers (right) Number of repeats from the 5' end (A) or 3' end (B) of the TR array, grey spots positions at which the null repeats occur
In total, 119 complete and 30 partial MUC1 alleles were mapped for the PDTR/PESR substitutions. For the purpose of these comparisons, the "null" repeats were considered as consensus repeats. A total of 103 different maps was obtained, examples of which are shown in Fig. 5. Some maps were found several times but it is interesting to note that the large group of 26 identical 37 repeat alleles showed four different patterns with respect to the 5' null repeats (data not shown). The diploid map shown in Fig. 3 is from one of these individuals, who is also homozygous for the null repeat substitutions. The HinfI size of this allele is 3.7 kb which falls within the modal size range for short alleles (3.5–4.0 kb, 34–42 repeat units). Several of the other short allele maps differ by only one or two repeats from this more frequent map.

MVR PDTR/PESR maps shown diagrammatically. All eight maps that have been found more than once are included, together with a selection of eight of the unique alleles. Left Number of alleles found with the particular map (No), the repeat number (Rep), the S/L classification and haplotype (G/A) with respect to the exon 2 g.3506G→A SNP. All of the maps shown were found in individuals of northern European origin, although the map of 36 repeats represented five times was only found once in a European, the four other alleles being found in two homozygous individuals of Japanese origin, the Japanese being a population in which short alleles are very frequent (Ando et al. 1998). Dark grey squares PDTR repeats, white squares PESR repeats, pale grey squares unmapped regions
Inspection of the short allele MVR maps showed that most carried blocks of five to nine PDTR repeats interspersed with two or three PESR repeats. The short alleles could be grouped into classes, which differed with respect to the number of blocks. Although most of the maps of the longer alleles were incomplete, they clearly also had a block structure; however, the blocks tended to contain fewer PDTR repeats and there seemed to be more alternative PESR repeats. In almost all cases, the long alleles had a cluster of three (rather than two) alternative repeats at repeat numbers 9, 10 and 11 from the 5' end. In addition, they generally had ten consensus repeats at the 3' end of the array compared with the nine consensus repeats seen in the short alleles.
Figure 5 also shows the allelic status for the Exon 2 g.3506G→A SNP for each of the MVR alleles. It is noteworthy that some of the S G alleles show the three alternative repeats at position 9, 10 and 11 from the 5' end, as is more usually found in the long alleles. Others show ten consensus repeats at the 3' end of the TR array, which is also more commonly found in the long alleles.
Discussion
In this study, we have shown, by selecting only two nucleotide changes in the MUC1 repeat unit, that there is considerable sequence diversity. Some TR arrays carry more than 40% of these alternate repeats, which encode PESR rather than PDTR. The high frequency of PESR repeats present in some alleles has implications as to antibody interactions in the context of both antigen detection and cancer immune therapy, since the PDTR motif overlaps the most immunogenic part of the TR domain and the epitopes recognised by most of the monoclonal antibodies used in diagnostic assays and immune targetting (Price et al. 1998). Experiments are needed to determine the quantitative effect of PESR substitutions on the binding of MUC1 TR mAbs to normal and cancer MUC1 mucin, since this might influence the sensitivity and specificity of diagnostic assays for tumour detection. The sequence variations may also affect innate or induced immune responses to aberrantly expressed MUC1.
The nucleotide changes mapped here represent only a fraction of the true allelic variability of this gene. We, like Engelmann and colleagues (2001), have seen, amongst others, changes in the amino acid at position 17 of the sequence shown in Fig. 2 but, so far, these changes have only been mapped in a few alleles. Many of these changes will clearly have an impact on TR domain peptide structure and glycosylation (Irimura et al. 1999) and this may lead to differences in micro-organism interaction, such as the binding of Helicobacter pylori to Leb carbohydrate structure (Boren et al. 1993). The T of PDTR has been shown to be glycosylated (Hanisch et al. 2001) and the glycosylation of the PESR variants is probably somewhat different. All this diversity provides enormous flexibility for a molecule that lies at the interface between the organism and the environment and that plays a role in defence (Irimura et al. 1999; Kardon et al. 1999) and signalling (Zrihan-Licht et al. 1994).
Previous studies have shown a higher frequency of short MUC1 alleles in patients with gastric cancer and also in patients with H. pylori gastritis (Carvalho et al. 1997; Silva et al. 2001; Vinall et al. 2002). MUC1 is also aberrantly expressed in H. pylori gastritis (Vinall et al. 2002), showing high intra-cellular expression but loss of detection of the TR domain on the apical surface. One hypothesis to explain these findings is that H. pylori interacts with MUC1 (to a different extent in different alleles) and that this directly or indirectly affects H. pylori colonisation and progression to gastric cancer. It will thus be important to determine whether particular kinds of short alleles are more frequent or underrepresented in patients with gastritis.
It is interesting to speculate as to the evolutionary origins of the variations in the repeat array. Examination of the pattern of blocks of repeats gives the impression that the 5' end is more conserved than the 3' end, suggesting polarity of the mutational events as has been observed for other MVR maps (May et al. 1996). The longer alleles probably arose from the shorter ones by a series of duplications, together with gene conversion events that led to the spread of the mutation that resulted in the PDTR to PESR change. Examination of the TR array of five chimpanzees by MVR PCR showed that the repeat number was much smaller, varying from 9 to 18, and no PESR repeats were detected.
References
Ando I, Kukita A, Soma G, Hino H (1998) A large number of tandem repeats in the polymorphic epithelial mucin gene is associated with severe acne. J Dermatol 25:150–152
Boren T, Falk P, Roth KA, Larson G, Normark S (1993) Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 262:1892–1895
Carvalho F, Seruca R, David L, Amorim A, Seixas M, Bennett E, Clausen H, Sobrinho-Simoes M (1997) MUC1 gene polymorphism and gastric cancer—an epidemiological study. Glycoconj J 14:107–111
Engelmann K, Baldus SE, Hanisch FG (2001) Identification and topology of variant sequences within individual repeat domains of the human epithelial tumor mucin MUC1. J Biol Chem 276:27764–27769
Fowler J, Vinall L, Swallow D (2001) Polymorphism of the human Muc genes. Front Biosci 6: D1207–D1215
Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, Pemberton L, Lalani EN, Wilson D (1990) Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem 265:15286–15293
Hanisch FG, Reis CA, Clausen H, Paulsen H (2001) Evidence for glycosylation-dependent activities of polypeptide N-acetylgalactosaminyltransferases rGalNAc-T2 and -T4 on mucin glycopeptides. Glycobiology 11:731–740
Irimura T, Denda K, Iida S, Takeuchi H, Kato K (1999) Diverse glycosylation of MUC1 and MUC2: potential significance in tumor immunity. J Biochem (Tokyo) 126:975–985
Jeffreys AJ, Neumann R, Wilson V (1990) Repeat unit sequence variation in minisatellites: a novel source of DNA polymorphism for studying variation and mutation by single molecule analysis. Cell 60:473–485
Jeffreys AJ, MacLeod A, Tamaki K, Neil DL, Monckton DG (1991) Minisatellite repeat coding as a digital approach to DNA typing. Nature 354:204–209
Kardon R, Price RE, Julian J, Lagow E, Tseng SC, Gendler SJ, Carson DD (1999) Bacterial conjunctivitis in Muc1 null mice. Invest Ophthalmol Vis Sci 40:1328–1335
Kirkbride HJ, Bolscher JG, Nazmi K, Vinall LE, Nash MW, Moss FM, Mitchell DM, Swallow DM (2001) Genetic polymorphism of MUC7: allele frequencies and association with asthma. Eur J Hum Genet 9:347–354
Kyo K, Parkes M, Takei Y, Nishimori H, Vyas P, Satsangi J, Simmons J, Nagawa H, Baba S, Jewell D, Muto T, Lathrop GM, Nakamura Y (1999) Association of ulcerative colitis with rare VNTR alleles of the human intestinal mucin gene, MUC3. Hum Mol Genet 8:307–311
May CA, Jeffreys AJ, Armour JA (1996) Mutation rate heterogeneity and the generation of allele diversity at the human minisatellite MS205 (D16S309). Hum Mol Genet 5:1823–1833
Müller S, Alving K, Peter-Katalinic J, Zachara N, Gooley AA, Hanisch FG (1999) High density O-glycosylation on tandem repeat peptide from secretory MUC1 of T47D breast cancer cells. J Biol Chem 274:18165–18172
Pratt WS, Islam I, Swallow DM (1996) Two additional polymorphisms within the hypervariable MUC1 gene: association of alleles either side of the VNTR region. Ann Hum Genet 60:21–28
Price MR, Rye PD, Petrakou E, Murray A, Brady K, Imai S, Haga S, Kiyozuka Y, Schol D, Meulenbroek MF, Snijdewint FG, Mensdorff-Pouilly S von, Verstraeten RA, Kenemans P, Blockzjil A, Nilsson K, Nilsson O, Reddish M, Suresh MR, Koganty RR, Fortier S, Baronic L, Berg A, Longenecker MB, Hilgers J, et al (1998) Summary report on the ISOBM TD-4 Workshop: analysis of 56 monoclonal antibodies against the MUC1 mucin. Tumour Biol 19:1-20
Siddiqui J, Abe M, Hayes D, Shani E, Yunis E, Kufe D (1988) Isolation and sequencing of a cDNA coding for the human DF3 breast carcinoma-associated antigen. Proc Natl Acad Sci USA 85:2320–2323
Silva F, Carvalho F, Peixoto A, Seixas M, Almeida R, Carneiro F, Mesquita P, Figueiredo C, Nogueira C, Swallow DM, Amorim A, David L (2001) MUC1 gene polymorphism in the gastric carcinogenesis pathway. Eur J Hum Genet 9:548–552
Taylor-Papadimitriou J, Burchell J, Miles DW, Dalziel M (1999) MUC1 and cancer. Biochim Biophys Acta 1455:301–313
Toribara NW, Gum JR Jr, Culhane PJ, Lagace RE, Hicks JW, Petersen GM, Kim YS (1991) MUC-2 human small intestinal mucin gene structure. Repeated arrays and polymorphism. J Clin Invest 88:1005–1013
Vinall LE, Fowler JC, Jones AL, Kirkbride HJ, Bolos C de, Laine A, Porchet N, Gum JR, Kim YS, Moss FM, Mitchell DM, Swallow DM (2000a) Polymorphism of human mucin genes in chest disease: possible significance of MUC2. Am J Respir Cell Mol Biol 23:678–686
Vinall LE, Pratt WS, Swallow DM (2000b) Detection of mucin gene polymorphism. Methods Mol Biol 125:337–350
Vinall LE, King M, Novelli M, Green CA, Daniels G, Hilkens J, Sarner M, Swallow DM (2002) Altered expression and allelic association of the hypervariable membrane mucin MUC1 in Helicobacter pylori gastritis. Gastroenterology 123:41–49
Yonezawa S, Byrd JC, Dahiya R, Ho JJ, Gum JR, Griffiths B, Swallow DM, Kim YS (1991) Differential mucin gene expression in human pancreatic and colon cancer cells. Biochem J 276:599–605
Zrihan-Licht S, Baruch A, Elroy-Stein O, Keydar I, Wreschner DH (1994) Tyrosine phosphorylation of the MUC1 breast cancer membrane proteins. Cytokine receptor-like molecules. FEBS Lett 356:130–136
Acknowledgements
We thank Dr. John Armour and Dr. John Stead for helpful advice.
Author information
Authors and Affiliations
Corresponding author
Additional information
J.F. was supported by an MRC studentship, and A.T. by the Portuguese Foundation for Science and Technology (reference no. SFRH\BD\2743\2000). This work was partly supported by the MRC as part of the programme of the former MRC Human Biochemical Genetics Unit.
Rights and permissions
About this article
Cite this article
Fowler, J.C., Teixeira, A.S., Vinall, L.E. et al. Hypervariability of the membrane-associated mucin and cancer marker MUC1. Hum Genet 113, 473–479 (2003). https://doi.org/10.1007/s00439-003-1011-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-003-1011-8