
Abstract
Recent studies have suggested that prostaglandin-endoperoxide synthase-2 (PTGS2), also known as cyclo-oxygenase 2, plays an etiological role in the development of type 2 diabetes mellitus (T2DM). PTGS2 generates prostaglandins, which negatively modulate glucose-stimulated insulin secretion, and functions as a mediator of the inflammatory response, which is associated with decreased insulin sensitivity. Moreover, the gene encoding this enzyme, PTGS2, is located on 1q25.2, a region that has been linked with early onset T2DM in Pima Indians. To determine the possible role played by PTGS2 in modulating susceptibility to T2DM, we screened approximately 7.0 kb of the gene, corresponding to the promoter, coding sequence, and flanking exon-intron boundaries, and identified five variants, including three single nucleotide polymorphisms (SNPs) in the promoter, one intronic SNP, and one in the 3' untranslated region. With the exception of one rare promoter SNP (minor allele frequency <0.03), all SNPs were typed in ~1000 Pima Indians. The range of frequencies for the more common alleles was 0.65–0.88, and we found substantial linkage disequilibrium between all PTGS2 SNP pairs (D′≥0.95). Variant alleles at two markers, rs20417 and rs2066826, which are located in the promoter and intron 6, respectively, were in strong linkage disequilibrium with each other (D′=0.97) and were associated with a higher prevalence of T2DM. For marker rs20417, individuals with the variant CC genotype had a 30% higher T2DM prevalence compared with subjects with the GG genotype (odds ratio=1.6 per copy of C allele; P=0.01). The variant C allele of rs20417 has been associated with decreased PTGS2 promoter activity, thereby suggesting a possible biological consequence attributable to this polymorphism. These findings indicate that genetic variants in PTGS2 may play a role in mediating susceptibility to T2DM in Pima Indians and are consistent with the hypothesis that chronic inflammation may contribute to the development of T2DM in some individuals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent studies have suggested that chronic inflammation plays an etiologic role in the development of insulin resistance (Vozarova et al. 2002) and type 2 diabetes mellitus (T2DM; Festa et al. 2000; Schmidt et al. 1999). Cross-sectional studies have identified an association between elevated serum levels of inflammatory markers and insulin resistance and/or T2DM (Pickup and Crook 1998; Bastard et al. 2000; Festa et al. 2000; Vozarova et al. 2002). Similarly, results from prospective studies have suggested that elevated baseline levels of inflammatory markers are predictive of T2DM (Pradhan et al. 2001). Chronic activation of the immune system has been recently suggested to cause a decline in insulin sensitivity, which may influence T2DM pathogenesis (Vozarova et al. 2002).
Inflammation induces the expression of a variety of proteins, including prostaglandin-endoperoxide synthase-2 (PTGS2), also known as cyclooxygenase 2 (COX2), the key enzyme in eicosanoid metabolism. There are two PTGS isozymes that catalyze the conversion of arachidonic acid to signaling prostaglandins in response to cytokine stimulation: a constitutive PTGS1 and an inducible PTGS2, which are encoded by different genes (Hla and Neilson 1992). Both isozymes are found in inflammatory cells but differ in the regulation of their expression, tissue distribution, and promoter characteristics (Smith and Dewitt 1996). The expression of PTGS2 has been shown to increase under certain pathophysiological conditions, such as chronic pancreatitis (Schlosser et al. 2002). The products of PTGS2, such as PGE2, have been shown to inhibit glucose-induced insulin secretion (Robertson et al. 1974; Robertson 1998). Furthermore, the expression of PTGS2 in pancreatic β-cells is tightly linked with that of insulin, and PTGS2 inhibition enhances insulin secretion in a dose-dependent manner (Robertson 1998; Luo et al. 2002). The PTGS2-dependent production of PGE2 has been postulated to regulate insulin production and secretion in β-cells (Robertson and Chen 1977; Robertson 1998).
The gene encoding human PTGS2, PTGS2, is located on chromosome 1q25.2-q25.3 (Tazawa et al. 1994), where we have previously reported linkage to early onset T2DM in Pima Indians (Hanson et al. 1998). Based on the evidence supporting an etiological role for PTGS2 in the development of T2DM through its involvement in the inflammatory response and generation of prostaglandins, its concomitant expression with insulin in pancreatic β-cells, and its location within a region linked to T2DM, we hypothesized that sequence variation within the PTGS2 gene may increase T2DM susceptibility. The goal of this study has been to identify single nucleotide polymorphisms (SNPs) in the PTGS2 gene and to assess the contribution of these genetic variants to T2DM risk in Pima Indians, who have the highest reported prevalence of this disease worldwide (Knowler et al. 1978).
Subjects, materials, and methods
Subjects
Subjects selected for genomic screening, comprising approximately 1000 individuals from 332 nuclear families, are participants in ongoing longitudinal studies of T2DM conducted among members of the Gila River Indian Community since 1965 (Knowler et al. 1978). Diabetes was diagnosed using World Health Organization criteria (Alberti and Zimmet 1998). The study was approved by the Institutional Review Board of the National Institute of Diabetes and Digestive and Kidney Diseases and the Tribal Council of the Gila River Indian Community. All subjects provided written informed consent prior to participation in the study.
SNP detection
The PTGS2 genomic sequence was obtained from bacterial artificial chromosome 973M2 (National Center for Biotechnology Information: Accession AL033533) and exon-intron boundaries were mapped by alignment with the PTGS2 mRNA sequence (NM_000963). All exons, exon-intron boundaries, and 1200 bp of 5' flanking sequence were screened by direct sequencing of genomic DNA obtained from 10 Pima Indians with early onset (<25 years) T2DM. DNA was amplified in a final reaction volume of 25 μl by using 100 ng genomic DNA,10× standard PCR buffer containing 2.75 mM MgCl2 (Roche Applied Science, Mannheim, Germany), 200 μM dNTPs, 0.24 μM oligonucleotide primers, 1.25 U DNA polymerase mix (Taq/Tgo polymerases; Roche Applied Science), and 1.25 U TaqStart Antibody (BD Biosciences Clontech, Palo Alto, Calif.). Polymerase chain reaction (PCR) cycling conditions consisted of an initial denaturation at 96°C for 1 min, followed by 35 cycles of 96°C for 20 s, 57°C for 30 s, and 68°C for 45 s, ending with a final elongation step at 68°C for 5 min. PCR product concentration was estimated by gel electrophoresis, and depending on yield, 2.5–5.0 μl PCR product was treated at 37°C for 15 min/80°C for 15 min with 1–2 μl ExoSAP-IT (USB, Cleveland, Ohio) to remove unconsumed dNTPs and oligonucleotide primers. Amplicons were bidirectionally sequenced with the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, Calif.) combined in a 1:1 ratio with SeqSaver Sequencing Premix Dilution Buffer (Sigma-Aldrich, St. Louis, Mo.) as recommended by the manufacturer. Sequences were resolved on the AB3700 sequence analysis system (Applied Biosystems). Information on all sequencing primers is available upon request.
SNP genotyping
All SNPs were genotyped by using the TaqMan assay in conjunction with the ABI 7700 sequence detector and the allelic discrimination software Sequence Detector v1.7 according to the manufacturer's recommended protocol (Applied Biosystems). Primers and probes for all SNPs were obtained by means of the Assays-by-Design SNP genotyping service of Applied Biosystems, with the exception of rs20417, where the Primer Express 1.5 software (Applied Biosystems) was used to generate primer and probe sequences. Sequence information for all primers and probes used for PTGS2 SNP genotyping is shown in Table 1.
Statistical analysis
Statistical analyses were performed with the software of the SAS Institute (Cary, N.C.). Association between PTGS2 SNP alleles and T2DM prevalence was calculated by logistic regression by using generalized estimating equations to adjust for age, sex, birth year, and ethnicity, while accounting for the effect of sibship (Zeger and Liang 1986). To maximize power, associations were calculated under three different models: assuming a dominant, recessive, or additive relationship between the number of common alleles and the prevalence of T2DM. The odds ratio (OR) at a 95% confidence interval (CI) was calculated as a measure of the strength of association between allele frequencies with prevalence of T2DM.
The observed genotype frequency for each SNP was assessed for deviation from that expected under Hardy-Weinberg equilibrium by Chi-square analysis. Control measures (i.e., encrypted samples and determination of Mendelian incompatibility) were employed to assess data quality.
The degree of pairwise linkage disequilibrium (LD) between alleles was calculated by using the 2ld program: (http://www.iop.kcl.ac.uk/IoP/Departments/PsychMed/GEpiBSt/software.shtml), expressed as D′, which represents the proportion of the maximum possible allelic association given the allele frequencies and the direction of the association.
Results and discussion
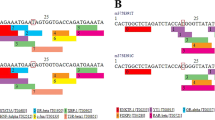
We screened approximately 7 kb of the PTGS2 gene corresponding to all 10 exons, exon-intron boundaries, and 1.2 kb of promoter sequence. In total, we identified five PTGS2 SNPs: three located in the PTGS2 promoter (rs689466, rs20417, −199); one in the extensive 3' untranslated region (rs5275), and one in intron 6 (rs2866826). Four of the markers were among the previously deposited SNPs found in the public dbSNP database (http://www.ncbi.nlm.nih.gov/SNP). A novel promoter A/G variant was detected 199 bp from the transcriptional start site, but because the variant allele was present at a frequency of less than 0.03 in 96 individuals, it was not further evaluated for association with T2DM.
All SNPs, with the exception of the rare −199 SNP, were genotyped in approximately 1000 Pima Indians, who are participants in ongoing longitudinal studies of T2DM and obesity among members of the Gila River Indian Community (Knowler et al. 1978). Sequence information for all primers and probes used for PTGS2 SNP genotyping is shown in Table 1. We first estimated the degree of pairwise LD between SNPs as quantified by the disequilibrium coefficient D′, which represents the proportion of the maximum possible disequilibrium given observed allele frequencies. As shown in Table 2, all PTGS2 SNP pairs showed substantial LD, although the frequency of the more common allele varied among the different markers. Based on the allele frequency and D′ estimate, rs20417 and rs2066826 were almost in complete genotypic concordance and were therefore expected to yield similar results in the analysis of association.
We next examined the relationship between PTGS2 SNPs and T2DM. Associations between alleles at each marker and T2DM prevalence was evaluated by using logistic regression, controlling for the effects of age, sex, birth year, and ethnicity, and accounting for familial relationships (i.e., sibship). To maximize the power of detecting an association, all analyses were conducted under three different models: one assuming a recessive common allele, one assuming a dominant common allele, and one assuming an additive relationship between the number of common alleles and prevalence of T2DM. The observed distribution of genotypes for each of the SNPs did not deviate significantly from that expected under Hardy-Weinberg equilibrium as assessed by Chi-square analysis (data not shown). As shown in Table 3, the C allele of SNP rs20417 and the T allele of rs2066826 were associated with an increased prevalence of T2DM. For marker rs20417, individuals with the variant CC genotype had 30% higher T2DM prevalence than individuals with the wild-type genotype under the additive model (OR=1.6 per copy of C allele, P=0.01). Similar results were found under the recessive model, but none of the SNPs showed strong evidence for association under the dominant model. No evidence for association between body mass index and PTGS2 genotype was found (data not shown).
PTGS2 is a key enzyme in prostaglandin biosynthesis and is constitutively expressed in pancreatic β-cells ( Robertson 1998; Luo et al. 2002). There is evidence that PTGS2 may play a role in the pathogenesis of T2DM, primarily through the generation of prostaglandins. One such PTGS2 product, PGE2, inhibits glucose-stimulated insulin release and may lead to glucose intolerance (Robertson et al. 1974; Robertson and Chen 1977). Furthermore, drugs that inhibit prostaglandin biosynthesis improve glucose disposal (Robertson 1998). Based on known biological data, it would follow that increased PTGS2 expression most likely increases susceptibility to T2DM.
Our findings do not provide a sufficient basis from which to speculate on the functional significance of the PTGS2 SNPs. However, the rs20417 polymorphism has been previously characterized and may have relevant biological consequences (Papafili et al. 2002). This polymorphism is located 765 bp upstream of the PTGS2 transcriptional start site and is contained within a putative Sp1-binding site (Papafili et al. 2002). Sp1 is a ubiquitously expressed transcription factor that is known to upregulate PTGS2 expression in conjunction with the transcription factor NF-ΚB (Xu et al. 2000). In vitro expression studies have shown that the variant C allele of rs20417 results in significantly reduced PTGS2 expression compared with the wild type G allele, and this effect has been postulated to be mediated by the loss of Sp1 transcription factor binding to its cognate element (Papafili et al. 2002). Functional studies are warranted to further delineate the role of the variant C allele of rs20417, and the other PTGS2 SNPs, in the regulation of PTGS2 mRNA expression.
PTGS2 is a key participant in the prostanoid synthesis pathway, which is initiated by the action of phospholipase A2 and results in the release of arachidonic acid from the plasma membrane. A member of the phospholipase A2 family, PLA2G4A, is also located on 1q25, approximately 148 kb telomeric to PTGS2, and expression of these genes may be coordinately regulated. We have previously identified a phenylalanine to leucine substitution at position 479 of PLA2G4A, which was associated with decreased glucose turnover and oxidation rates in Pima Indians (Wolford et al. 2003). Furthermore, the variant leucine allele was more common in subjects with diabetes compared with healthy controls. A physical distance of 274 kb separates this polymorphism and rs20417, and not surprisingly, the SNPs were not in strong LD (D′=−0.69, P=0.02).
In summary, we have found two polymorphisms that lie in the PTGS2 gene and that are associated with T2DM susceptibility in Pima Indians. One of these variants has a potential biological effect, which may help elucidate the mechanisms by which the activation of the prostaglandin biosynthetic pathway results in worsened glucose homeostasis. These findings are consistent with a role for genetic determinants of inflammation in the development of T2DM in Native Americans.
References
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 15:539–553
Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, Vidal H, Hainque B (2000) Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab 85:3338–3342
Festa A, D'Agostino R Jr, Howard G, Mykkanen L, Tracy RP, Haffner SM (2000) Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation 102:42–47
Hanson RL, Ehm MG, Pettitt DJ, Prochazka M, Thompson DB, Timberlake D, Foroud T, Kobes S, Baier L, Burns DK, Almasy L, Blangero J, Garvey WT, Bennett PH, Knowler WC (1998) An autosomal genomic scan for loci linked to type II diabetes mellitus and body-mass index in Pima Indians. Am J Hum Genet 63:1130–1138
Hla T, Neilson K (1992) Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci USA 89:7384–7388
Knowler WC, Bennett PH, Hamman RF, Miller M (1978) Diabetes incidence and prevalence in Pima Indians: a 19-fold greater incidence than in Rochester, Minnesota. Am J Epidemiol 108:497–505
Luo C, Kallajoki M, Gross R, Mulari M, Teros T, Ylinen L, Makinen M, Laine J, Simell O (2002) Cellular distribution and contribution of cyclooxygenase (COX)-2 to diabetogenesis in NOD mouse. Cell Tissue Res 310:169–175
Papafili A, Hill MR, Brull DJ, McAnulty RJ, Marshall RP, Humphries SE, Laurent GJ (2002) Common promoter variant in cyclooxygenase-2 represses gene expression: evidence of role in acute-phase inflammatory response. Arterioscler Thromb Vasc Biol 22:1631–1636
Pickup JC, Crook MA (1998) Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 41:1241–1248
Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM (2001) C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286:327–334
Robertson RP (1998) Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Diabetes 47:1379–1383
Robertson RP, Chen M (1977) A role for prostaglandin E in defective insulin secretion and carbohydrate intolerance in diabetes mellitus. J Clin Invest 60:747–753
Robertson RP, Gavareski DJ, Porte D Jr, Bierman EL (1974) Inhibition of in vivo insulin secretion by prostaglandin E1. J Clin Invest 54:310–315
Schlosser W, Schlosser S, Ramadani M, Gansauge F, Gansauge S, Beger HG (2002) Cyclooxygenase-2 is overexpressed in chronic pancreatitis. Pancreas 25:26–30
Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, Azambuja MI, Tracy RP, Heiss G (1999) Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet 353:1649–1652
Smith WL, Dewitt DL (1996) Prostaglandin endoperoxide H synthases-1 and -2. Adv Immunol 62:167–215
Tazawa R, Xu XM, Wu KK, Wang LH (1994) Characterization of the genomic structure, chromosomal location and promoter of human prostaglandin H synthase-2 gene. Biochem Biophys Res Commun 203:190–199
Vozarova B, Weyer C, Lindsay RS, Pratley RE, Bogardus C, Tataranni PA (2002) High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 51:455–461
Wolford JK, Konheim YL, Colligan PB, Bogardus C (2003) Association of a F479L variant in the cytosolic phospholipase A2 gene (PLA2G4A) with decreased glucose turnover and oxidation rate in Pima Indians. Mol Genet Metabol 79:61–66
Xu Q, Ji YS, Schmedtje JF Jr (2000) Sp1 increases expression of cyclooxygenase-2 in hypoxic vascular endothelium. Implications for the mechanisms of aortic aneurysm and heart failure. J Biol Chem 275:24583–24589
Zeger SL, Liang KY (1986) Longitudinal data analysis for discrete and continuous outcomes. Biometrics 42:121–130
Acknowledgements
We express our appreciation to the members of the Gila River Indian Community for their ongoing participation. We gratefully acknowledge the expert analytical assistance provided by Sayuko Kobes and the genotyping contribution from Peter B. Colligan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Konheim, Y.L., Wolford, J.K. Association of a promoter variant in the inducible cyclooxygenase-2 gene (PTGS2) with type 2 diabetes mellitus in Pima Indians. Hum Genet 113, 377–381 (2003). https://doi.org/10.1007/s00439-003-1000-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-003-1000-y