
Abstract
Perennial ryegrass (Lolium perenne), one of the most widely used forage and cool-season turfgrass worldwide, has a breeding history of more than 100 years. However, the current draft genome annotation and transcriptome characterization are incomplete mainly because of the enormous difficulty in obtaining full-length transcripts. To explore the complete structure of the mRNA and improve the current draft genome, we performed PacBio single-molecule long-read sequencing for full-length transcriptome sequencing in perennial ryegrass. We generated 29,175 high-confidence non-redundant transcripts from 15,893 genetic loci, among which more than 66.88% of transcripts and 24.99% of genetic loci were not previously annotated in the current reference genome. The re-annotated 18,327 transcripts enriched the reference transcriptome. Particularly, 6709 alternative splicing events and 23,789 alternative polyadenylation sites were detected, providing a comprehensive landscape of the post-transcriptional regulation network. Furthermore, we identified 218 long non-coding RNAs and 478 fusion genes. Finally, the transcriptional regulation mechanism of perennial ryegrass in response to drought stress based on the newly updated reference transcriptome sequences was explored, providing new information on the underlying transcriptional regulation network. Taken together, we analyzed the full-length transcriptome of perennial ryegrass by PacBio single-molecule long-read sequencing. These results improve our understanding of the perennial ryegrass transcriptomes and refined the annotation of the reference genome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Perennial ryegrass (Lolium perenne, 2n = 14), one of the most used cool-season turfgrass and forage plants worldwide, has a breeding history of more than 100 years (Byrne et al. 2016; Wang et al. 2017a). Because of its rapid establishment, long growing season, high yield, and high palatability, perennial ryegrass are important not only in hay production, but also in ecosystem services (Huff 1997; Wang et al. 2017a). Molecular breeding and traditional hybrid breeding approaches increasingly require a high-quality reference genome to advance the breeding process. The draft genome sequence of perennial ryegrass was first reported in 2015, covering 1128 Mb of the genome, 28,455 gene models, and 11,311 annotated genes (Byrne et al. 2016). Although this sequence is a valuable genetic resource for genetic breeding, the genome coverage is incomplete and the current reference genome is not satisfactory, preventing progress in breeding efforts (Shinozuka et al. 2017). Additionally, most existing gene models were derived from in silico prediction and lack reliable annotation on alternative isoforms and untranslated regions. Thus, studies are needed to improve genome annotation using reliable genome-wide full-length transcripts.
The Pacific Biosciences (PacBio; Menlo Park, CA, USA) single-molecule long-read sequencing technology (PacBio sequencing) can obtain full-length splice isoforms directly, without assembly, thus providing the opportunity to investigate genome-wide full-length cDNA molecules. To date, PacBio sequencing has been successfully utilized in various plant species, such as common wheat (Triticum aestivum) (Dong et al. 2015), sorghum (Sorghum bicolor) (Abdelghany et al. 2016), Arabidopsis (Arabidopsis thaliana) (Zhu et al. 2017), red clover (Trifolium pretense) (Chao et al. 2018), and alfalfa (Medicago sativa) (Chao et al. 2019). In species for which a reference genome is available, such as red clover and alfalfa, the PacBio sequencing has provided important information for improving the draft genome annotation and insight into the transcriptome. In 2018, PacBio sequencing was adopted to analyze the full-length transcriptome of red clover, identifying 2194 novel isoforms from novel genes and 29,370 novel isoforms from known genes. A total of 5492 alternative splicing events (AS) were predicted, and 8719 genes possessed poly(A) site (APA). The full-length transcriptome of alfalfa was first analyzed by PacBio sequencing in 2018, resulting in the identification of 113,321 transcripts, 1670 transcription factors, 17,740 lncRNAs, and 7568 AS events. Moreover, most AS events in alfalfa were intron retention.
For species without an available reference genome, PacBio sequencing provides a cost-effective choice for studies of gene structure and specific gene functions. PacBio sequencing was carried out to generate the full-length transcriptome dataset for bermudagrass (Cynodon dactylon), a warm-season turfgrass specie, without an available reference genome (Zhang et al. 2018a). In total, the full-length sequences comprised 78,192 unigenes, 66,409 of which were functionally annotated. Additionally, 27,946 unigenes were found to have at least 2 isoforms. These results facilitated future comparative transcriptome and gene functional studies in bermudagrass. Using a transcriptome generated by PacBio sequencing as a reference sequence, Chen et al. (2018) performed a transcriptome-referenced association study and identified 22 candidate transcripts responsible for the shape of garlic (Allium sativum) clove. Thus, this tool is efficient for association studies independent of a reference genome. Using next-generation (NGS) sequencing in error correction, PacBio sequencing may reveal full-length splicing isoforms with complete 3′- and 5′-ends more accurately, better identify differential AS events, and produce more accurate profiles of global APA compared to other methods (Wang et al. 2017b).
Drought stress is a major factor limiting the growth and persistence of plants worldwide (Shinozaki and Yamaguchi-Shinozaki 2006). Using the suppression subtractive hybridization (SSH) and NGS sequencing techniques, the expression profiles of perennial ryegrass and annual ryegrass (Lolium multiflorum) under drought stress have been reported, improving the understanding of the transcriptional regulation network in response to drought stress in ryegrass (Liu and Jiang 2010; Pan et al. 2016). However, the transcriptome remains incomplete because of the limitations of RNA sequencing and low throughput of SSH. Thus, studies are needed to investigate the underlying transcriptional network by PacBio sequencing.
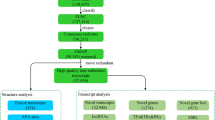
To improve the transcriptional information and refine the current draft genome annotation of perennial ryegrass, we generated a full-length transcriptome by PacBio sequencing. AS and APA events were predicted to investigate the structural characteristics of the transcripts. In addition, long non-coding RNAs and fusion transcripts were investigated. The underlying transcriptional regulation network was explored using the re-constructed reference transcriptome by PacBio sequencing. The information provides insight into the transcriptional events and refines the genome annotation of perennial ryegrass.
Materials and methods
Plant material and growth conditions
Perennial ryegrass seeds of cultivar ‘Accent’ were purchased from Jacklin Seed company (Jacklin, WA, USA). The seeds were sowed in 12 cm diameter, 15 cm deep plastic pots filled with 500 g calcined clay (Profile Products, Chicago, IL, USA) at a seeding rate of 30 g m−2. Plants were cultivated in a growth chamber (RXZ-380D-LED, Ningbo Jiang Nan Instrument Factory, Ningbo, China) at 20/16 °C (day/night), 65% relative humidity, 14-h photoperiod, and average photosynthetic active radiation (PAR) of 600 μ mol m−2 s−1 for 3 weeks. Plants were trimmed to 10 cm weekly and fertilized with half-strength Hoagland’s solution (Hoagland and Arnon 1950). For drought treatment, nine independent plots (three biological repetitions for each of the three independent treatments) were subject to water withholding for 0, 3, and 8 days. The whole plants including the leaves, sheaths, stems, roots, and tillers were collected for RNA extraction on each sampling day.
Physiological measurements
We utilized a Theta Probe soil moisture sensor (ML2; Delta-T Devices, Cambridge, UK) to monitor the soil moisture content (SWC) on each sampling day. The leaf relative water content (RWC) was calculated by examining the fresh weight, dry weight, and turgid weight. The malondialdehyde (MDA) content was determined using the trichloroacetic acid method as described in our previous report (Puyang et al. 2015). Proline content was examined using the 5-sulfosalicylic acid method (Teng et al. 2018). The total soluble sugar content was measured using the anthrone colorimetry method (Zhang et al. 2018b).
Library preparation and PacBio sequencing
Total RNA was extracted using the Plant RNA Kit (No. R6827-01, OMEGA Bio-tek, Norcross, GA, USA,). Equal amounts of RNA collected from ‘Accent’ plants on each sampling day were pooled. The quantity and integrity of RNA samples were assessed with the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Qualified RNA samples were then used to construct cDNA libraries. The SMARTer PCR cDNA Synthesis Kit (TaKaRa, Dalian, China) was utilized to synthesize full-length cDNA, and cDNA fraction and length selection (1–2 kb, 2–3 kb, and > 3 kb) was performed using the BluePippinTm Size Selection System (Sage Science, Beverly, MA, USA). These three PacBio libraries were generated using the Pacific Biosciences DNA Template Prep Kit 2.0. (Pacific Biosciences, California, USA) according to the standard protocol. PacBio sequencing was then performed on the Pacific Bioscience RSII platform at Biomarker Technology Co. (Beijing, China) according to the manufacturer’s protocol.
Illumina cDNA library construction and second-generation sequencing analysis
Total RNA was extracted from the collected samples (whole plants), with three independent biological replicates obtained for each sample. Next, nine prepared RNA samples (three each from the control, early drought treatment, and late drought treatment groups) were evaluated as described in the previous section. Strand-specific cDNA libraries were constructed using the NEBNext ® UltraTM RNA Library Prep Kit (NEB, Ipswich, MA, USA) following the manufacturer’s protocol. Qualified libraries were sent to Biomarker Technology Co. for second-generation sequencing on an Illumina HiSeq 4000 platform (San Diego, CA, USA).
The Illumina clean reads were mapped to the newly constructed reference transcriptome using the Bowite2 program (Langmead and Salzberg 2012). The expression abundance, presented as fragments per kilobase of transcript per million mapped reads (FPKM) was calculated using RSEM (Li and Dewey 2011). After evaluating the correlation of biological replicates based on the Pearson correlation coefficient, differential expression analysis was carried out using the DESeq2 program (Anders and Huber 2010). The parameters adopted in this study were fold change ≥ 2 and FDR < 0.01. Then, the functional enrichment analysis including GO enrichment analysis and KEGG pathway enrichment analysis were performed using GOseq R package and KOBAS software, respectively.
Quality filtering, error correction, and elimination of redundancy among PacBio long reads
Reads of inserts (ROIs) were generated from PacBio subreads using standard protocols in the PacBio analysis software suit (http://www.pacificbiosciences.com). Full-length non-chimeric reads (FLNC) and non-full-length (NFL) reads were classified using RS-IsoSeq v2.3 by identifying poly(A) signals and 5′ and 3′ adaptors. To identify all possible reads, a relaxed standard with a minimum full pass of 0 and accuracy of 75% was adopted in the filtering panel. The FLNC reads generated from the same isoform were clustered into one consensus isoform using the Iterative isoform-clustering program (ICE). NFL reads were clustered by the Quiver program. Subsequently, the consensus isoforms were polished, resulting in high-quality isoform (HQ) (accuracy > 99%) and low-quality isoform (LQ). The raw Illumina reads were filtered to remove adaptor sequences, ambiguous reads with ‘N’ bases, and low-quality reads. The filtered Illumina data were used to polish the LQ reads using proovread 213.841 software (Hackl et al. 2014). The polished consensus isoforms were mapped to the perennial ryegrass genomic sequence (http://185.45.23.197:5080/ryegrassgenome) using the Genomic Mapping and Alignment Program (GMAP) (Wu and Watanabe 2005) and then the redundant isoforms (identity > 0.9, coverage < 0.85) were eliminated using the ToFU package without considering the 5′ difference. Finally, a high-quality transcript dataset without redundant isoforms of perennial ryegrass was constructed.
Functional annotation of transcripts and prediction of open reading frames
Functional annotations were conducted using BLAST 2.2.26 against different protein and nucleotide databases of Nr (NCBI non-redundant proteins), Swissprot (a manually annotated, non-redundant protein database), COG (Clusters of Orthologous Groups), KOG (euKaryotic Orthologous Groups), Pfam (a database of conserved Protein families or domains), GO (Gene Ontology), and KEGG (Kyoto Encyclopedia of Genes and Genomes). For each transcript in each database search, functional information for the best matched sequence was assigned to the query transcript. TransDecoder 2.0.1 (https://transdecoder.github.io/) was utilized to define the putative coding sequences (CDS) to predict the open reading frames (ORFs). The predicted CDS were then annotated and confirmed by BLAST (E-value ≤ 1e−5). Transcripts containing complete ORFs as well as 5′- and 3′-untranslated regions (UTRs) were considered as full-length transcripts.
Analysis of alternative splicing events
Transcripts were validated against known reference transcript annotation with the python library MatchAnnot. AS events including intron retention (IR), exon skipping (ES), alternative 3′ splice site (Alt.3′), alternative 5′ splice site (Alt.5′) and mutually exclusive exon (MEE) were identified and classified using MISA (Beier et al. 2017).
Alternative polyadenylation events analysis
The TAPIS pipeline was used to identify poly(A) tails of the full-length reads. If there were at least eight adenine (A) bases and at most two non-A bases in 3′-region of the full-length reads, the first base was determined to be a poly(A) position of the full-length reads. The poly(A) positions on the reference genome were then extracted from the alignments of trimmed poly(A)-containing full-length reads.
Identification of fusion transcripts
Non-redundant HQ transcript isoforms were utilized to identify the fusion transcripts. A single transcript was classified as a fusion transcript when it simultaneously satisfied the following requirements: (1) mapped to 2 or more loci, (2) minimum coverage for each loci of 5% and minimum coverage in bp of ≥ 1 bp, (3) total coverage of ≥ 95%, and (4) distance between the loci of at least 10 kb.
Classification of long non-coding RNAs and their target genes
Four computational approaches including the Coding Potential Calculator (CPC), Coding–Non-Coding Index (CNCI), Coding Potential Assessment Tool (CPAT) and Pfam database were combined to sort non-protein-coding RNA candidates from putative protein-coding RNAs in the transcripts. Putative protein-coding RNAs were filtered out using minimum length and exon number thresholds. Transcripts longer than 200 bp with more than two exons were selected as lncRNAs candidates and further screened using CPC/CNCI/CPAT/Pfam, as these tools can distinguish protein-coding from the non-protein-coding genes. Only the transcripts identified in the four databases were regarded as lncRNAs. To investigate the target genes of the lncRNAs, the LncTar tool (Jianwei et al. 2015) was utilized following two strategies: the first was based on the localization of lncRNA and mRNA, whereas the second was based on the interactivity results of lncRNA and mRNA caused by base pairing.
Results
General properties of PacBio sequencing of perennial ryegrass
To provide a collection of transcripts, we combined the total RNA extracted from perennial ryegrass grown under three different conditions in equal amounts to obtain a full-length reference transcriptome by PacBio sequencing. Three cDNA libraries of different sizes (1–2, 2–3, and 3–6 kb) were constructed and then sequenced using the PacBio RSII sequencing platform, generating a total of 751,460 raw polymerase reads. These reads resulted in 4,554,953 post-filter subreads (length > 50 bp and accuracy > 0.75) with a mean length of 2103 bp and N50 of 2587 bp (Table 1).
Five single molecular real-time cells generated 107,187, 124,918 and 60,413 ROIs from each of the three libraries, respectively (Fig. 1a–c). The mean length of the ROIs was 2434 bp and the N50 was 3080 bp. The mean numbers of passes in the three cDNA libraries were 18, 9, and 7, respectively. The FLNC read-length distribution of each size bin agreed with the size of its cDNA library (Fig. 1d–f). In all, 292,518 ROIs were generated, more than 55.9% (163,660) of which were FLNC reads (mean length 2057 bp and N50 2409 bp) comprising the entire transcript region from the 5′ to the 3′ end, based on the inclusion of barcoded primers and 3′-poly(A) tails (Fig. 1g). Short reads with a length of < 300 bp (6.2%) and chimeric reads (0.2%) were discarded from subsequent analysis.

Summary of PacBio sequencing. Number and length distribution of 292,518 ROI sequences from 1 to 2 K a, 2–3 K b and 3–6 K c. Number and length distribution of 163,660 FLNC sequences from 1 to 2 K d, 2–3 K e and 3–6 K f. g Proportion of different types of PacBio reads in perennial ryegrass. Number and length distribution of 78,417 consensus isoforms in 0–2 K h, 2–3 K i, 3–6 K j and > 6 K k
The 78,417 consensus FLNC reads were first clustered using the ICE program and then polished using the Quiver program and NFL reads (Fig. 1h–k, Table 1). To correct the relative high error rates of single-molecule long reads compared with the Illumina platform, 215,952,015 paired-end reads (PE) were utilized to further polish the low-quality isoforms. We finally obtained 64,743 high-quality corrected isoforms from 78,417 consensus isoforms with a mean length of 2192 bp and N50 of 2550 bp.
Comparing perennial ryegrass genome and correcting previous mis-annotated gene models
The 163,660 FLNC isoforms were compared against the draft genome sequence of perennial ryegrass using GMAP. In general, 134,922 reads (82.44%) were mapped to the reference genome (Fig. 2a). Further classification of the mapped reads revealed that 4269 reads were multiple mapped, 67,362 were mapped to the positive strand (reads map to +) of the genome and 63,291 were mapped to the opposite strand (reads map to +) of the genome. Next, the corrected transcripts were mapped against the reference genome. The results showed that 64,743 HQ reads including 35,363 reads that were the same in the reference genome, 20,185 novel isoforms from known genes and 9195 novel isoforms from novel genes were successfully mapped to the genome (Fig. 2b).

GMAP analysis of PacBio sequences. a GMAP analysis of FLNC reads to reference genome. b Classification of corrected isoforms mapped to reference genome
After eliminating of redundancy, the de novo constructed transcriptome consisting of 29,175 transcripts (N50 2.53 kb) from 15,893 genic loci were finally obtained. Moreover, 3971 loci of the perennial ryegrass genome have not been annotated compared with the reference annotation. In addition, there were 9664 transcripts with identical intronic coordinates as reference annotations, and 2686 transcripts were translated from a single exon. Without assembly, these PacBio full-length transcripts are ideal resources for optimizing gene models.
Functional annotation of transcripts
To further improve the function annotation of the reference transcriptome, the updated transcripts were annotated using the Nr, Swissprot, GO, COG, KOG, Pfam and KEGG databases. Overall, 18,327 annotated transcripts were generated (Fig. 3a, Table 2). Homologous species were analyzed by comparing the transcript sequences to the Nr database; the largest number of transcripts was found in Brachypodium distachyon (31.57%) (Fig. 3b). GO classification showed that ‘Cell’, ‘catalytic activity’ and ‘metabolic process’ were ranked as the most enriched items in the ‘cellular components’, ‘molecular functions’, and ‘biological process’ categories, respectively (Fig. 3c). NOG analysis showed that 18,135 transcripts were assigned to 25 functional clusters, and the ‘function unknown’, ‘general function prediction only’, and ‘signal transduction mechanisms’ ranked as the top three largest categories (Fig. 3d).

Function annotation of corrected isoforms. a Function annotation of transcripts in all databases. b Nr homologous species distribution of transcripts. c Distribution of GO terms for all the annotated transcripts. D. NOG enriched of transcripts. Length distribution of the identified coding sequences e, 5′-UTRs f and 3′-UTRs g. h Prediction of transcription factors
TransDecoder program was used to predict ORFs and UTRs. These unique full-length transcripts contained 18,651 CDSs, including 11,738 transcripts with complete ORFs. Transcripts consist of 200–300 amino acids were most abundant and corresponded to 20.75% of the identified CDSs (Fig. 3e). Furthermore, the number and length distribution of the 5′-UTRs and 3′-UTRs were investigated (Fig. 3f–g). Transcription factors (TFs) were predicted using PacBio data finally obtained in this study. A total of 1439 putative TFs from 63 families were identified and the MYB-related, C3H, and AP2/ERF TF families were the most abundant TF families in perennial ryegrass (Fig. 3h). Next, the TFs identified by PacBio were compared to known TFs from the reference genome, which showed that 464 TFs were considered as known and 975 of the TFs were recognized as novel.
Analysis of alternative splicing events and splice isoforms
One of the most important advantages of PacBio sequencing is its ability to identify AS events by directly comparing different isoforms of the same gene. Here, we systematically analyzed AS in perennial ryegrass based on high-quality full-length isoforms. This revealed 6709 AS events among the transcripts with two or more alternative isoforms (Table S1). Specifically, five major AS events including Alt.3′, Alt.5′, ES, IR and MEE were classified, with retained intron (RI) as the most abundant type showing an occurrence rate of 58.16% (Fig. 4a). In the reference genome, 4684 AS events were identified which is much lower than the number identified by PacBio sequencing (Fig. 4b). Furthermore, five genes were randomly selected to validate the accuracy of AS events by reverse transcription polymerase chain reaction (RT-PCR). Isoforms of each gene were aligned to design primers that could amplify all predicted transcripts at the same time (Table S6). The results demonstrated that the size of each amplified fragments was consistent with that of the predicted fragment (Fig. 4c). These amplified fragments were then cloned for Sanger sequencing. The results showed sequence consistency between the cloned fragments and predicted sequences based on the PacBio sequencing data.

Transcripts and intron–exon structure in perennial ryegrass. a Classification of the AS events identified by PacBio sequencing. b The total number of AS events in detected genes by PacBio compared with the annotated gene models in reference genome. c RT-PCR verification of representative AS events. d Distribution of genes that have one or more transcripts from PacBio sequences. e Distribution of genes that have one or more introns from PacBio sequences. The nucleotide distributions flanking the donor f or acceptor sites (g)
Among the genes identified by PacBio, 9605 possessed only 1 transcript (Fig. 4d). The other 6363 genes were found to have 2 or more transcripts, producing 18,544 transcripts in total. Sixty-eight genes had more than 10 transcripts, and the victorin-binding protein gene (PB.9371) showed the largest number of spliced isoforms. Additionally, 177,489 introns were identified using the PacBio sequences, and 26,489 sequences were predicted to have introns. A total of 2686 genes showed no intron, whereas 797 genes had more than 20 introns (Fig. 4e). PB.10454.1 was found to have 41 introns, which was the largest number among all genes. In addition, the consensus donor and acceptor sites of perennial ryegrass were analyzed (Fig. 4f–g). The median number of introns in the perennial ryegrass genes containing introns was 4, whereas the numbers in alfalfa, bamboo (Phyllostachys edul) and Arabidopsis are 6, 7 and 4, respectively.
Alternative polyadenylation events analysis
PacBio sequencing enables investigation of the APA sites in perennial ryegrass; 23,789 poly(A) sites were identified among the 9546 genes in the perennial ryegrass reference genome, and 3717 genes showed 1 poly(A) sites, whereas 666 genes contained at least 6 poly(A) site (Fig. 5a, Table S2). The average number of poly(A) sites was 2.49, and average sequencing depth was 16.16. Specifically, the thiamine biosynthesis gene (ThiC, maker-scaffold_4297|ref0021721-exonerate_est2genome-gene-0.0) was found have 25 poly(A) sites, which was the largest number in this study.

APA analysis. a Distribution of the number of poly(A) sites per gene. b Nucleotide composition around poly(A) cleavage sites. The relative frequency of a nucleotide is shown as a function of genomic position across all poly(A) cleavage sites detected in our data. c MEME analysis of an over-represented motif at 50-nts upstream of the poly(A) site in perennial ryegrass transcripts
Analysis of the upstream and downstream regions of all poly(A) sites revealed that nucleotide bias around the poly(A) sites in perennial ryegrass with enrichment of U upstream and A downstream of the cleavage site in the 3′-UTRs (Fig. 5b). Using 50 nucleotides upstream of the predominant poly(A) site in all transcripts, MEME analysis was performed to identify potential cis-elements necessary for polyadenylation. The results showed that there were three conserved motifs (GGCUGG, GUGGCC, and GUGGUG) upstream of the poly(A) cleavage sites (Fig. 5c).
Identification of lncRNAs and fusion genes
Based on the prediction of CPC, CNCI, Pfam, and CPAT, 4660 transcripts were considered as putative non-coding RNAs. Finally, 218 transcripts (with length > 200 bp and > 2 exons), which could be found in all 4 prediction results, were considered as lncRNAs (Fig. 6a; Table S3). Additionally, the identified lncRNAs were further classified into four types, including 166 lncRNAs, 4 antisense lncRNAs, 8 intronic lncRNAs and 36 sense lncRNAs (Fig. 6b). The N50 value of these identified lncRNAs was 2.52 kb. Length distribution analysis of the lncRNAs revealed that their lengths ranged from 0.34 kb (PB.7219.1) to 4.49 kb (PB.15570.2). Compared to the mRNAs, the lncRNAs were predicted to have fewer exons and shorter average transcript lengths (Fig. 6c, d). Moreover, 158 target genes of the lncRNAs were predicted (Table S4).

Identification of lncRNAs. a Venn diagram of lncRNAs predicted by CNCI, CPC, CPAT and Pfam methods. b Classification of the types of the lncRNAs. c Comparison of exon number of lncRNAs and mRNAs in perennial ryegrass. d Density and length distribution of lncRNAs and mRNAs identified in this study
Gene fusion is a common feature in humans; however, few studies have been reported in plants. Here, we identified 478 fusion genes using genic alignments, which were found by merging 2 transcripts with different functions (Table S5). The major type of fusion transcript was exon fusion, and the events were more likely to occur inter-chromosomally than intra-chromosomally. The N50 value of these fusion genes was 2.58 kb, with the range of 900–1200 bp containing the most abundant transcripts. The average length of the fusion genes was 2.18 kb, and the longest transcript was 5.58 kb (PBfusion.140), whereas the shortest was 385 bp (PBfusion.312). To further investigate the fusion transcripts, the transcripts were annotated with the Nr, Swissprot, GO, COG, KOG, Pfam, and KEGG databases. Nr annotation results showed that 95.19% (455/478) of the translated transcripts were annotated. The search results of amino sequence homology demonstrated that the largest number (146) of fusion genes was distributed in Brachypodium distachyon. KEGG enrichment revealed that the fusion genes were most abundantly involved in the ‘metabolic’ and ‘biosynthesis of secondary metabolites’ pathways.
Drought treatment caused significant physiological changes in perennial ryegrass
In this study, water withholding lead to a gradual decrease in the soil water content (Fig.S1). On day 3, the SWC was decreased to 14.15%, and decreased to 2.9% on day 8. Consistent with the SWC, phenotype analysis showed that drought stress had different influences on the plants. To further investigate the influence of a dramatically decrease in SWC on the plant, we measured the changes in the leaf RWC, proline, MDA and total sugar contents. The results revealed that withholding for 3 days (early drought) and 8 days (late drought) both decreased the RWC. In detail, early and late drought treatment decreased the RWC to 86.52% and 67.03% (Fig.S2A), respectively. In contrast, the MDA, proline and total sugar contents after late drought treatment were increased significantly compared to in the well-watered groups (control) (Fig.S2B-D). The MDA and total sugar contents of the early drought group were increased compared to in the control, with the proline content showing no obvious changes. Analysis of physiological indicators supported that early drought and late drought altered physiological changes in perennial ryegrass, indicating that this period is a suitable sampling time for RNA-sequencing analysis.
Global expression analysis based on the re-constructed reference transcriptome revealed the post-transcriptional responses to drought stress of perennial ryegrass
1926, 3142, and 231 DEGs were, respectively, identified in the early drought stage compared to in the control (Drought_0-3d, T01_T02_T03_vs_T04_T05_T06), late drought stage compared to in the control (Drought_0-8d, T01_T02_T03_vs_T07_T08_T09), and late drought stage compared to in the early drought stage (Drought_3-8d, T04_T05_T06_vs_T07_T08_T09) (Fig. 7, Table S7). In terms of DEG numbers, group Drought_0-8d with 1225 up-regulated and 1917 down-regulated DEGs ranked as the most abundant group, whereas Drought_3-8d with 104 up-regulated and 127 down-regulated ranked as the least abundant group.

Global expression analysis based on the re-constructed reference transcriptome. a–c Volcano plot and KEGG enrichment of the DEGs identified in Drought_0-3d, Drought_0-8d and Drought_3-8d, respectively
GO annotation was performed to identify the putative biological processes of DEGs in response to drought stress. The top ten most significantly enriched GO terms for each group were selected among the enriched GO terms (Fig.S3). Although 80% of the GO categories were identified in all three groups, the enrichment p value showed group-specific characteristics. The specific biological processes responsible for the responses to different drought treatments were also identified. KEGG analysis revealed that ‘galactose metabolism’, ‘phenylalanine metabolism’ and ‘nitrogen metabolism’ were the top three most enriched pathways in Drought_0-3d (Fig. 7). ‘Nitrogen metabolism’, ‘phenylalanine metabolism’, and ‘phenylpropanoid biosynthesis’ ranked as the top three most enriched pathways in Drought_0-8d (Fig. 7). ‘Galactose metabolism’, ‘starch and sucrose metabolism’ and ‘glutathione metabolism’ ranked as the top three most enriched pathways in Drought_3-8d (Fig. 7).
Discussion
The current knowledge of the perennial ryegrass transcriptome is mainly based on gene expression data obtained by NGS sequencing (Studer et al. 2012; Shinozuka et al. 2017; Wang et al. 2017a). The perennial ryegrass transcriptome has not been fully explored because of a lack of full-length transcripts. Although the genome was reported in 2015, full-length mRNA, AS events, APA events, and fusion transcripts have not been well characterized. In this study, we performed PacBio sequencing to generate full-length transcripts and refine the genome annotation of perennial ryegrass. PacBio generated 163,660 FLNC sequences with a mean length of 2057 bp and N50 of 2409 bp, which are much longer than the transcripts identified in the reference genome (mean length 1.66 and N50 2.20 kb, respectively). When the red clover and alfalfa transcriptomes were characterized by PacBio sequencing in our previous studies, the mean transcript length was 405-bp longer and 1511-bp longer than those in the reference genome (Chao et al. 2018, 2019). These results demonstrate the ability of PacBio sequencing to generate longer read lengths compared to NGS sequencing. This inherent advantage makes it possible to refine the reference genome. Based on the PacBio full-length sequences, we found that 3,971 loci had not been annotated using the current process to identify genes and coding regions, and that 19,511 transcripts, including 975 novel TFs, had not been previously identified. The new loci were shown to be useful for exploring perennial ryegrass genetic resources. The mis-annotated transcripts may have resulted from the assembly of short reads from NGS technologies used in previous genome sequencing studies. In addition, prior studies focused on leaf, pollen, and stigma tissues to annotate the perennial ryegrass reference genome, which may also explain why these novel transcripts were not previously identified. The transcriptome of perennial ryegrass was significantly refined based on FLNC reads in this study, highlighting the potential of PacBio sequencing for genome annotation.
Another advantage of PacBio sequencing is its ability to detect AS events (Wang et al. 2018; Zhu et al. 2018). We found 4684 AS events using the current reference genome; however, we found 6709 AS events using PacBio sequences. Further classification of the AS types revealed that intron retention comprised most of these events. This is consistent with the results obtained using the reference genome, indicating that intron retention is important for post-transcriptional regulation in perennial ryegrass. For diverse biological responses, AS is an effective mechanism for increasing the complexity and flexibility of the transcriptome and proteome (Li et al. 2017). Recent reports suggested that 13–18% of intron-containing genes in Arabidopsis are regulated by AS and non-sense-mediated decay (Li et al. 2013). Because of its longer read capacity, PacBio sequencing facilitates accurate characterization of complex AS at the genome-wide level (Robert et al. 2015). We found that 35.10% (10,239/29,175) of transcripts were alternatively spliced, suggesting that AS is very common in perennial ryegrass; however, the frequency is still lower than AS events in strawberry (Li et al. 2017), bamboo (Wang et al. 2017b), and Arabidopsis transcripts (Zhu et al. 2017). This lower frequency may be partly related to limited reference genome assembly, and depth of PacBio sequencing, as well as the specific growth conditions used in this study. This suggests that other isoforms are expressed under various environmental stress conditions.
PacBio sequencing also outperforms NGS for profiling poly(A) sites (Ugrappa et al. 2008; Abdelghany et al. 2016; Wang et al. 2017b). Our study provides a comprehensive genome-wide APA map draft consisting of 23,789 poly(A) sites from 9546 genes, and even these results may underestimate the true number of APA genes because of the low expression of proximal poly(A) sites. We evaluated nucleotide bias and found clear enrichment of uracil (U) and adenine (A) upstream and downstream of the 3’-UTR cleavage sites, respectively. These findings are consistent with those of previous studies of sorghum (Abdelghany et al. 2016), moso bamboo (Wang et al. 2017b) and red clover (Chao et al. 2018). Unlike the maize and red clover transcriptomes (Wang et al. 2016; Chao et al. 2018), we identified three conserved polyadenylation motifs in perennial ryegrass, indicating a species-specific character of polyadenylation motifs (Li and Du 2014). Previous studies demonstrated that alternative polyadenylation of RNA is critical for gene function by increasing transcriptome complexity and adjusting gene expression (Wu et al. 2011; Li and Du 2014; Shen et al. 2014). For example, thiamine is a vitamin required for plant growth, which can be synthesized via the ThiC-mediated thiamine biosynthesis pathway (Nagae et al. 2016). The ThiC APA sites identified in this study may contribute to the regulation of perennial ryegrass development. Moreover, the map of global polyadenylation across the perennial ryegrass genome will enhance the precision of gene annotation.
LncRNAs, a recently identified class of non-coding RNAs, are essential regulators of a wide range of biological processes (Heo et al. 2013; Di et al. 2014). Most attempts to identify lncRNAs using NGS inevitably generate transcripts that lack of poly(A) tails, thereby compromising data accuracy (Yang et al. 2011). In our study, 218 lncRNAs (mean length 2.17 kb and N50 2.52 kb) and 158 corresponding target genes were predicted based on PacBio sequencing data, providing additional valid candidates for future functional characterization. Identification of fusion transcripts were reported to supplement genetic annotation (Wang et al. 2017b). In addition, we identified 478 fusion genes, most of which were involved in the ‘metabolic’ and ‘biosynthesis of secondary metabolites’ pathways. These fusion genes will help refine genetic alignment-based annotation of the perennial ryegrass transcriptome, and contribute to studies of gene models.
Consistent with the physiological changes among the three groups during different drought stages, RNA-sequencing revealed novel transcriptional regulatory mechanisms responsible for drought stress. Frequently, but not always, the number of DEGs appeared to reflect increased transcriptional activity which correlated with stress levels, and was more prevalent in the late stages of drought. GO enrichment analysis revealed that most of the top enriched biological processes consistently participated in drought stress, indicating the importance of these biological processes. Additionally, ‘starch metabolic process’ and ‘pentose-phosphate shunt’ biological processes may be crucial in severe drought stress of perennial ryegrass. Previous studies reported that galactose is involved in the tolerance to drought, high salinity and cold temperatures (Taji et al. 2002), and phenylalanine metabolism and the phenylpropanoid biosynthesis pathways modulated salt stress adaption in transgenic Arabidopsis (Teng et al. 2018). Transgenic studies demonstrated that nitrogen metabolism enhanced drought tolerance in rice (Reguera et al. 2013) and that glutathione helped Tortula ruralis adapt to rehydration following rapid desiccation (Dhindsa 1991). KEGG enrichment analysis suggested that different signaling pathways serve different roles in perennial ryegrass at different drought stages. This is the first study to reveal the transcriptional dynamics of early and late responses to drought stress in perennial ryegrass based on the updated reference transcriptome constructed by PacBio sequencing.
Taken together, our study employed PacBio single-molecule long-read sequencing to better characterize full-length transcripts of perennial ryegrass. These findings enhance the knowledge of perennial ryegrass transcriptomes and refined the annotation of the reference genome. These data will facilitate functional genomic studies and provide a foundation for further genetically engineered breeding of perennial ryegrass.
Data availability
The PacBio sequencing reads (accession number PRJNA549115) and the Illumina SGS reads (accession number PRJNA566226) generated in this study have been submitted to the BioProject database of National Center for Biotechnology Information.
Abbreviations
- APA:
-
Polyadenylation sites
- AS:
-
Alternative splicing events
- CDS:
-
Coding sequences
- CPAT:
-
Coding potential assessment tool
- CPC:
-
Coding potential calculator
- CNCI:
-
Coding–non-coding index
- FLNC:
-
Full-length non-chimeric reads
- GO:
-
Gene ontology
- HQ:
-
High-quality isoforms
- ICE:
-
Iterative isoform-clustering program
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KOG:
-
EuKaryotic orthologous groups
- lncRNA:
-
Long non-coding RNA
- LQ:
-
Low-quality isoforms
- NFL:
-
Non-full-length
- NGS:
-
Next-generation sequencing
- Nr:
-
NCBI non-redundant proteins
- ROI:
-
Reads of insert
- ORF:
-
Open reading frames
- PacBio sequencing:
-
The PacBio single-molecule long-read sequencing technology
- Pfam:
-
A database of conserved Protein families or domains
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- Swissprot:
-
A manually annotated, non-redundant protein database
References
Abdelghany SE, Hamilton M, Jacobi JL, Ngam P, Devitt N, Schilkey F, Benhur A, Reddy ASN (2016) A survey of the sorghum transcriptome using single-molecule long reads. Nat Commun 7:11706
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106
Beier S, Thiel T, Münch T, Scholz U, Mascher M (2017) MISA-web: a web server for microsatellite prediction. Bioinformatics 33:2583–2585
Byrne SL, Nagy I, Pfeifer M, Armstead I, Swain S, Studer B, Mayer K, Campbell JD, Czaban A, Hentrup S (2016) A synteny-based draft genome sequence of the forage grass Lolium perenne: for cell and molecular biology. Plant J 84:816–826
Chao Y, Yuan J, Li S, Jia S, Han L, Xu L (2018) Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol 18:300
Chao Y, Yuan J, Guo T, Xu L, Mu Z, Han L (2019) Analysis of transcripts and splice isoforms in Medicago sativa L. by single-molecule long-read sequencing. Plant Mol Biol 99:219–235
Chen X, Liu X, Zhu S, Tang S, Mei S, Chen J, Li S, Liu M, Gu Y, Dai Q, Liu T (2018) Transcriptome-referenced association study of clove shape traits in garlic. DNA Res 25:587–596
Dhindsa RS (1991) Drought stress, enzymes of glutathione metabolism, oxidation injury, and protein synthesis in Tortula ruralis. Plant Physiol 95:648–651
Di C, Yuan J, Wu Y, Li J, Lin H, Hu L, Zhang T, Qi Y, Gerstein MB, Guo Y, Lu ZJ (2014) Characterization of stress-responsive lncRNAs in Arabidopsis thaliana by integrating expression, epigenetic and structural features. Plant J 80:848–861
Dong L, Liu H, Zhang J, Yang S, Kong G, Chu JSC, Chen N, Wang D (2015) Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genom 16:1039
Hackl T, Hedrich R, Schultz J, Förster F (2014) proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30:3004–3011
Heo JB, Lee Y-S, Sung S (2013) Epigenetic regulation by long noncoding RNAs in plants. Chromosome Res 21:685–693
Hoagland DR, Arnon DI (1950) The water-culture method for growing plants without soil. Calif Agric Exp Statn 347:357–359
Huff DR (1997) RAPD characterization of heterogenous perennial ryegrass cultivars. Crop Sci 37:557–564
Jianwei L, Wei M, Pan Z, Junyi W, Bin G, Jichun Y, Qinghua C (2015) LncTar: a tool for predicting the RNA targets of long noncoding RNAs. Brief Bioinf 16:806
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf 12:323
Li X-Q, Du D (2014) Motif types, motif locations and base composition patterns around the RNA polyadenylation site in microorganisms, plants and animals. BMC Evol Biol 14:162
Li W, Lin W-D, Ray P, Lan P, Schmidt W (2013) Genome-wide detection of condition-sensitive alternative splicing in arabidopsis roots. Plant Physiol 162:1750–1763
Li Y, Dai C, Hu C, Liu Z, Kang C (2017) Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant J 90:164–176
Liu S, Jiang Y (2010) Identification of differentially expressed genes under drought stress in perennial ryegrass. Physiol Plant 139:375–387
Nagae M, Parniske M, Kawaguchi M, Takeda N (2016) The relationship between thiamine and two symbioses: root nodule symbiosis and arbuscular mycorrhiza. Plant Signal Behav 11:e1265723
Pan L, Zhang X, Wang J, Ma X, Zhou M, Huang LK, Nie G, Wang P, Yang Z, Li J (2016) Transcriptional profiles of drought-related genes in modulating metabolic processes and antioxidant defenses in Lolium multiflorum. Fron Plant Sci 7:519
Puyang X, An M, Xu L, Han L, Zhang X (2015) Antioxidant responses to waterlogging stress and subsequent recovery in two Kentucky bluegrass (Poa pratensis L.) cultivars. Acta Physiol Plant 37:197
Reguera M, Peleg Z, Abdel-Tawab YM, Tumimbang EB, Delatorre CA, Blumwald E (2013) Stress-induced cytokinin synthesis increases drought tolerance through the coordinated regulation of carbon and nitrogen assimilation in rice. Plant Physiol 163:1609–1622
Robert VB, Doug B, Edger PP, Haibao T, Diane B, Dinakar C, Kristi S, Richard H, Jenny G, Eric L (2015) Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 527:508
Shen Y, Zhou Z, Wang Z, Li W, Fang C, Wu M, Ma Y, Liu T, Kong L-A, Peng D-L, Tian Z (2014) Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 26:996–1008
Shinozaki K, Yamaguchi-Shinozaki K (2006) Gene networks involved in drought stress response and tolerance. J Exp Bot 58:221–227
Shinozuka H, Noi C, Spangenberg GC, Forster JW (2017) Reference transcriptome assembly and annotation for perennial ryegrass. Genome 60:1086
Studer B, Byrne S, Nielsen RO, Panitz F, Bendixen C, Islam MS, Pfeifer M, Lübberstedt T, Asp T (2012) A transcriptome map of perennial ryegrass (Lolium perenne L.). BMC Genom 13:140
Taji T, Ohsumi C, Iuchi S, Seki M, Kasuga M, Kobayashi M, Yamaguchi-Shinozaki K, Shinozaki K (2002) Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J 29:417–426
Teng K, Tan P, Guo W, Yue Y, Fan X, Wu J (2018) Heterologous Expression of a novel Zoysia japonica C2H2 zinc finger gene, ZjZFN1, improved salt tolerance in Arabidopsis. Front Plant Sci 9:1159
Ugrappa N, Zhong W, Karl W, Chong S, Debasish R, Mark G, Michael S (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320:1344–1349
Wang B, Tseng E, Regulski M, Clark TA, Hon T, Jiao Y, Lu Z, Olson A, Stein JC, Ware D (2016) Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat Commun 7:11708
Wang K, Liu Y, Tian J, Huang K, Shi T, Dai X, Zhang W (2017a) Transcriptional profiling and identification of heat-responsive genes in perennial ryegrass by RNA-sequencing. Front Plant Sci 8:1032
Wang T, Wang H, Cai D, Gao Y, Zhang H, Wang Y, Lin C, Ma L, Gu L (2017b) Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J 91:684–699
Wang M, Wang P, Liang F, Ye Z, Li J, Shen C, Pei L, Wang F, Hu J, Tu L, Lindsey K, He D, Zhang X (2018) A global survey of alternative splicing in allopolyploid cotton: landscape, complexity and regulation. New Phytol 217:163–178
Wu T, Watanabe C (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21:1859
Wu X, Liu M, Downie B, Liang C, Ji G, Li QQ, Hunt AG (2011) Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc Natl Acad Sci 108:12533–12538
Yang L, Duff MO, Graveley BR, Carmichael GG, Chen L-L (2011) Genomewide characterization of non-polyadenylated RNAs. Genome Biol 12:R16
Zhang B, Liu J, Wang X, Wei Z (2018a) Full-length RNA sequencing reveals unique transcriptome composition in bermudagrass. Plant Physiol Biochem 132:95–103
Zhang N, Han L, Xu LX, Zhang XZ (2018b) Ethephon seed treatment impacts on drought tolerance of kentucky bluegrass seedlings. HortTechnology 28:319–326
Zhu F-Y, Chen M-X, Ye N-H, Shi L, Ma K-L, Yang J-F, Cao Y-Y, Zhang Y, Yoshida T, Fernie AR, Fan G-Y, Wen B, Zhou R, Liu T-Y, Fan T, Gao B, Zhang D, Hao G-F, Xiao S, Liu Y-G, Zhang J (2017) Proteogenomic analysis reveals alternative splicing and translation as part of the abscisic acid response in Arabidopsis seedlings. Plant J 91:518–533
Zhu C, Li X, Zheng J (2018) Transcriptome profiling using Illumina- and SMRT-based RNA-seq of hot pepper for in-depth understanding of genes involved in CMV infection. Gene 666:123
Acknowledgements
We are very grateful to Prof. Luis A. J. Mur from Institute of Biological, Environmental and Rural Sciences, Aberystwyth University for critically discussion with the manuscript. We also thank Biomarker Technology Corporation (Beijing, China) for the facilities and expertise of PacBio platform for libraries construction and sequencing and the Editage Company (https://www.editage.com) for language editing.
Funding
This research was supported by the Scientific Technology Plan Program of Shenzhen (No. JCYJ20160331151245672)‚ the National Natural Science Foundation of China (No. 31971770 and No. 31901397) and Beijing Natural Science Foundation (No.6204039).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: LH and YC. Performed the experiments: LX, KT and PT. Data analysis and draft of the manuscript were performed by KT, YL and WG. All authors approved the final version of the manuscript for submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Research involving human participants and/or animals
This study does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2019_1635_MOESM2_ESM.jpg
Fig.S2 Physiological measurement of perennial ryegrass seedlings under different drought stresses conditions. (A) Leaf relative water content. (B) MDA content. (C) Proline content. (D) Total sugar content. Means ± SDs (n = 4). Different letters indicate significant differences at 5% level of probability. (JPG 469 kb)
438_2019_1635_MOESM3_ESM.tif
Fig.S3 GO annotation of the biological processes identified in Drought_0-3d (A), Drought_0-8d (B) and Drought_3-8d (C). (TIFF 334 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Xie, L., Teng, K., Tan, P. et al. PacBio single-molecule long-read sequencing shed new light on the transcripts and splice isoforms of the perennial ryegrass. Mol Genet Genomics 295, 475–489 (2020). https://doi.org/10.1007/s00438-019-01635-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-019-01635-y