
Abstract
The origin of supernumerary (B) chromosomes is clearly conditioned by their ancestry from the standard (A) chromosomes. Sequence similarity between A and B chromosomes is thus crucial to determine B chromosome origin. For this purpose, we compare here the DNA sequences from A and B chromosomes in the characid fish Characidium gomesi using two main approaches. First, we found 59 satellite DNA (satDNA) families constituting the satellitome of this species and performed FISH analysis for 18 of them. This showed the presence of six satDNAs on the B chromosome: one shared with sex chromosomes and autosomes, two shared with sex chromosomes, one shared with autosomes and two being B-specific. This indicated that B chromosomes most likely arose from the sex chromosomes. Our second approach consisted of the analysis of five repetitive DNA families: 18S and 5S ribosomal DNA (rDNA), the H3 histone gene, U2 snDNA and the most abundant satDNA (CgoSat01-184) on DNA obtained from microdissected B chromosomes and from B-lacking genomes. PCR and sequence analysis of these repetitive sequences was successful for three of them (5S rDNA, H3 histone gene and CgoSat01-184), and sequence comparison revealed that DNA sequences obtained from the B chromosomes displayed higher identity with C. gomesi genomic DNA than with those obtained from other Characidium species. Taken together, our results support the intraspecific origin of B chromosomes in C. gomesi and point to sex chromosomes as B chromosome ancestors, which raises interesting prospects for future joint research on the genetic content of sex and B chromosomes in this species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Supernumerary (B) chromosomes are dispensable genomic elements found in many plants, animals and fungi (Jones and Rees 1982; Camacho et al. 2000; Burt and Trivers 2006; Ahmad and Martins 2019). These elements do not follow the Mendelian law of segregation resulting in mechanisms of accumulation, although, the molecular basis of these mechanisms still not well known (Dhar et al. 2017; Houben 2017; Jones 2018; Dalla Benetta et al. 2019; Wu et al. 2019). B chromosomes are mostly composed of repetitive DNA (Ziegler et al. 2003; Carmello et al. 2017; Li et al. 2017; Ramos et al. 2017; Coan and Martins 2018; Hanlon et al. 2018; Milani et al. 2018), e.g., 95% of their DNA is repetitive in Locusta migratoria (Ruiz-Ruano et al. 2018), but they also contain protein-coding genes (Martis et al. 2012; Makunin et al. 2014, 2018; Valente et al. 2014; Huang et al. 2016; Utsunomia et al. 2016; Navarro-Domínguez et al. 2017a; Clark et al. 2018; Jehangir et al. 2019).
Interestingly, some of these B chromosome genes are actively transcribed (Banaei-Moghaddam et al. 2013; Miao et al. 1991; Ruiz-Estévez et al. 2013, 2014; Trifonov et al. 2013; Valente et al. 2014; Navarro-Domínguez et al. 2017a, b; Dalla Benetta et al. 2019) and the B chromosome of rye encodes a functional Argonaute-like protein (Ma et al. 2017), thus dismissing the idea that B chromosomes are genetically inert. This probably explains previously described effects of B chromosomes on sex determination (Yoshida et al. 2011) or antibiotic resistance (Miao et al. 1991). The additional finding of changes in the expression level of host genes, which are consistent with B chromosome effects, suggests a true transcriptomic arms race between A and B chromosomes (Navarro-Domínguez et al. 2019). Although these are interesting prospects for B chromosome research in the coming years, B chromosome origin is one of the least-known aspects of these genomic elements.
It is widely accepted that they most likely arose from standard (A) chromosomes, either intra- or interspecifically, but the A chromosome from which the B chromosome arose has rarely been identified (Camacho 2005). For instance, based on DNA sequence similarity in A and B chromosomes, it was inferred that B chromosomes derived intraspecifically from the W sex chromosome in the frog Leiopelma hochstetteri (Sharbel et al. 1998) or interspecifically in the wasp Nasonia vitripennis (McAllister and Werren 1997; Perfectti and Werren 2001) and in the bees of Partamona genus (Tosta et al. 2014). As they are dispensable, B chromosome sequences can evolve quickly (Banaei-Moghaddam et al. 2013; Clark et al. 2016; Jehangir et al. 2019), and this usually hampers the identification of the specific A-chromosome ancestor. Aimed at filling this gap, we analyze here the origin of B chromosomes in the characid fish Characidium gomesi by using next-generation sequencing (NGS) in B-carrying and B-lacking genomes and subsequent analysis of the satellitome (i.e., the full catalogue of satellite DNA) to identify those satellite DNA (satDNA) families showing overabundance in B-carrying genomes, thus indicating their presence in the B chromosomes.
SatDNA is a sort of repetitive DNA consisting of a unit of a given sequence and length (from a few to 100 bp) that is repeated in tandem arrays. SatDNA is typically located in the heterochromatic pericentromeric regions, but it can also be found in euchromatic regions (Ruiz-Ruano et al. 2016; Rodrigues et al. 2019). The analysis of satDNA distribution on A and B chromosomes using fluorescence in situ hybridization (FISH) has been shown to be highly informative about the origin of B chromosomes in grasshoppers (Ruiz-Ruano et al. 2017, 2018) and fish (Silva et al. 2017) and sex chromosomes in fish (Utsunomia et al. 2019).
Here, we extend this kind of analysis to C. gomesi (Crenuchidae, Characidiinae), a species where previous analysis using chromosome painting suggested that B chromosomes in this species arose from sex chromosomes (Pansonato-Alves et al. 2014). For this purpose, we obtained Illumina libraries from B-carrying and B-lacking genomes, and this led to the identification of 59 satDNA families using the satMiner toolkit (Ruiz-Ruano et al. 2016). We then performed FISH analysis of 18 of these families, and a thorough comparison between satDNA location on A (distinguishing between sex chromosomes and autosomes) and B chromosomes clearly showed higher similarity between B and sex chromosomes, in clear support of the hypothesis of Pansonato-Alves et al. (2014). We also analyzed the presence of five other repetitive DNA families in A and B chromosomes using chromosome microdissection, PCR amplification, cloning, Sanger sequencing and FISH, which provided additional support for the intraspecific origin hypothesis.
Materials and methods
Origin of samples, karyotypic analyses and DNA extraction
We analyzed 16 females and 8 males of C. gomesi captured at Véu da Noiva waterfall stream, Paranapanema River Basin, Botucatu city, São Paulo state (S22°59′25″ W48°25′40″). The specimens were identified and deposited at the fish collection of the Laboratório de Biologia e Genética de Peixes, Botucatu, São Paulo, Brazil, under voucher LBP16910.
For cytogenetic analyses, the animals were anesthetized, dissected, and mitotic chromosome preparations were obtained following the protocol proposed by Foresti et al. (1981). C-banding was performed according to the protocol described by Sumner (1972), and chromosome morphology was determined according to the arms ratio (Levan et al. 1964).
Genomic DNA (gDNA) was obtained from samples deposited in our DNA collection for C. gomesi, C. zebra, C. pterostictum, C. oiticicai, and C. lauroi using the Wizard Genomic DNA Purification Kit (Promega), following the manufacturer’s instructions.
Chromosome microdissection
Chromosome microdissection was performed on an Eppendorf TransferMan NK2 micromanipulator coupled to a Zeiss Axiovert 100 microscope. Ten acrocentric B chromosomes were microdissected and placed in a tube containing 9 μL of ultra-pure DNase-free water; subsequently, they were amplified using the GenomePlex Single Cell Whole Genome Amplification Kit (wga4-Sigma) (Gribble et al. 2004).
Total genome sequencing and identification of satellite DNA
To search for repetitive DNA sequences located on the B chromosomes of C. gomesi, the genomic DNA of two female individuals, one without B chromosomes (0B) and another with 0–4 mitotic unstable B chromosomes (+ B), was sequenced on the Illumina HiSeq 2500 platform, with 2 × 101 nt of paired-end reads, resulting in approximately 0.8 Gb and 0.7 Gb for the 0B sample and + B sample, respectively, representing 0.7 × and 0.6 × of genome coverage for the 0B and + B samples, respectively. Given that females carry the Z and W chromosomes, DNA of all chromosomes of this species was included in the libraries.
To characterize repetitive elements, a custom database of repetitive sequences was built using the program RepeatModeler (Smit and Hubley 2015) from data in the full genome sequencing of Astyanax mexicanus (GenBank accession number APWO00000000.1) as a complement to annotate the repetitive DNA sequences in conjunction with the RepBase database (Bao et al. 2015). It consisted of 1.243 DNA sequences comprising 589.736 bp with N50 = 613 nt.
After quality filtering of the Illumina reads with the Trimmomatic software (Bolger et al. 2014), in which reads showing 90% or less of bases with quality lower to Q20 were removed, we applied the satMiner protocol for characterization of satDNA sequences (Ruiz-Ruano et al. 2016). Then, we selected 400,000 pairs of reads (200,000 pairs of reads from each individual) for the clustering step with the RepeatExplorer software with default options (Novák et al. 2013).
After the clustering stage, we searched for clusters of satDNA families. Subsequently, the assembled contigs were extracted and processed manually using the Geneious Pro program v8.04 (http://www.geneious.com/) to visualize the dotplot graphs to detect tandem repetitions. The monomers present in each cluster were then extracted and aligned to obtain the consensus sequence of each satDNA. From these sequences, we designed primers using the Geneious Pro v8.04 program for PCR amplification of satDNA sequences with primers anchored in opposite directions (Table S1).
The monomers found were classified into variant, family or superfamily categories when sequence identity was higher than 95%, 80% or 50%, respectively. The abundance and divergence for each variant of satDNA, for the 5S rDNA and histone H3 genes, and for the 5S NTS spacer were determined by RepeatMasker (Smit et al. 2010) with the Cross_match search engine using 5.8 million reads for each genome (0B and + B). Sequences were deposited in GenBank under accession numbers MG764442–MG764499. We analyzed the satDNAs possibly present on the B chromosomes through the ratio between the abundance of satDNA in the + B and 0B genomes, as it represents an abundance fold change (FC) due to the presence of B chromosomes. In case of a satDNA being putatively B-specific (e.g., CgoSat59-103), we carried out further analysis to ascertain whether it is actually absent from the B-lacking genome. For this purpose, we searched for read pairs in each library showing homology with the satDNA using BLAT (Kent 2002) through the protocol implemented in a custom script (https://github.com/fjruizruano/ngs-protocols/blob/master/mapping_blat_gs.py).
DNA amplification, cloning and sequencing
We designed primers for PCR amplification of 48 satDNA families on gDNA of C. gomesi (Tables 1 and S1) and tried to PCR-amplify several repetitive DNA families (18S and 5S rDNA, U2 snDNA, H3 histone genes and the CgoSat01-184) using primers described here and previously (Pendas et al. 1995; Colgan et al. 1998; Utsunomia et al. 2016) on the DNA obtained from microdissected B chromosomes of C. gomesi as a template. We then used genomic DNA from 0B individuals of C. gomesi, C. zebra, C. pterostictum, C. oiticicai, and C. lauroi as templates to obtain DNA sequences for 5S rDNA, H3 histone genes and CgoSat01-184 families from the A chromosomes. The reactions were carried out with 1X PCR buffer, 1.5 mM MgCl2, 200 μM of each dNTP (dATP, dCTP, dGTP, dTTP), 0.5 U of Taq polymerase (Invitrogen), 0.1 μM of each primer and 50 ng of gDNA. The basic cycle to amplify these regions consisted of DNA denaturation at 94 °C for 5 min, followed by 30 cycles at 94 °C for 30 s, 52 °C for 30 s, 72 °C for 30 s and a final extension of 10 min at 72 °C. PCR products were visualized on a 1% agarose gel. The fragments obtained from each sample were extracted from the gel and cloned into the pGEM®-T Easy Vector System (Promega). DNA sequencing was performed using the Big Dye™ Terminator v3.1 Cycle Sequencing Ready Reaction Kit (Applied Biosystems), following the manufacturer’s instructions.
Fluorescence in situ hybridization (FISH)
Probes for 18S and 5S rDNA, U2 snDNA, H3 histone gene and 18 satDNAs were obtained by PCR from the genome of C. gomesi with the same reaction parameters described above. The probes were labeled with digoxigenin-11-dUTP or biotin-16-dUTP (Roche Applied Science) during PCR.
For chromosome painting, a probe labeled with digoxigenin-11-dUTP was generated from the microdissected DNA of the B chromosome of C. gomesi (CgB) using the GenomePlex Whole Genome Amplification Reamplification Kit (wga3-Sigma), following the manufacturer’s protocol.
For FISH experiments, the chromosomes were treated according to procedures described by Pinkel et al. (1986) under conditions of high stringency. Probe detection was performed with avidin–FITC (Sigma) or anti-digoxigenin–rhodamine (Roche), and chromosomes were counterstained with DAPI (4′,6-diamidino-2-phenylindole, Vector Laboratories). The images were digitally captured using Image Pro plus 6.0 (Media Cybernetics) software using suitable filters from the epifluorescence microscope (Olympus BX61) equipped with an Olympus DP70 camera. The final composition of the images was performed using the Adobe Photoshop CS6 image editor software.
Extraction of satDNA Cgom01-184 from Illumina reads
To obtain a more reliable and detailed score on the haplotypic abundance of CgomSat01-184 DNA in the genomic libraries of C. gomesi, several monomers were extracted directly from the Illumina reads. Since the size of reads is smaller than that of the monomers, the paired-end reads were joined using the fastq-join program (https://code.google.com/p/ea-utils/wiki/FastqJoin) with a minimum of 6-nt sequence overlap. Then, the satDNA reads were aligned against their respective dimer with the RepeatMasker software (Smit et al. 2010), and a Python script was used (https://github.com/fjruizruano/ngs-protocols/blob/master/rm_getseq.py) to extract only the aligned region. Subsequently, these sequences were mapped using the Geneious Pro v8.04 program against the satDNA dimer, and the central region of the alignment was extracted. However, in cases where it was not possible to extract at least 100 complete monomer sequences in the central region of the alignment, the sequences were edited to obtain more complete monomers in this region using a Python script (https://github.com/fjruizruano/ngs-protocols/blob/master/sat_cutter.py).
Nucleotide sequences analysis
All DNA sequences were initially analyzed with the Geneious Pro 4.8.5 software, and an alignment was performed using the Muscle algorithm (Edgar 2004) under predefined parameters. Analysis of nucleotide diversity and haplotypes was performed with DnaSP software (Librado and Rozas 2009). Minimum-spanning trees (MST) were built with Arlequin v3.5 software (Excoffier and Lischer 2010) considering only those haplotypes including more than one sequence. The sequences were used as queries for BLAST search (Altschul et al. 1990) against the NCBI nucleotide collection (http://www.ncbi.nlm.nih.gov/blast) to confirm their identity. Comparisons of synonymous substitutions per synonymous site (dS) and nonsynonymous substitutions per nonsynonymous site (dN) for the H3 histone gene of each Illumina library of C. gomesi (+ B and 0B) were performed with the DnaSP software (Librado and Rozas 2009). Statistical analyses were performed by means of Student’s t test.
Results
Basic chromosome analysis
The individuals of Characidium gomesi analyzed here showed a standard (A) chromosome set consisting of 2n = 50 chromosomes (32 metacentric and 18 submetacentric) and a heteromorphic ZW sex chromosome system (Fig. 1). In addition, 21 out of the 24 individuals analyzed showed 1–5 mitotically unstable acrocentric B chromosomes (Ba). One of these individuals carried, in addition to 0–4 Ba chromosomes, 0–2 submetacentric Bs (Bsm), which were also mitotically unstable (Fig. 1e, f). The C-banding technique showed the presence of faint C-bands on pericentromeric regions of all autosomes and a large pericentromeric C-band on the Z chromosome, whereas the W and B chromosomes were darkly C-banded across their whole length (Fig. 1b, d, f).

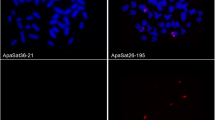
Metaphases of C. gomesi after FISH with satDNAs (a, c, e) and C-banding (b, d, f). Bar = 10 µm
SatDNA content points to B chromosome origin from the W sex chromosome
As a first step to uncover the B chromosome content of C. gomesi, we performed a high-throughput search for satellite DNA sequences on the 0B and + B Illumina libraries using RepeatExplorer (RE) (Novák et al. 2013) and satMiner (Ruiz-Ruano et al. 2016) by performing four runs of RE + filtering on 0B and + B reads. In each filtering step, previously found satDNAs were filtered out, thus increasing the likelihood of finding out other satDNAs showing lower abundance in the genomes analyzed (Ruiz-Ruano et al. 2016). We finally obtained 72 satDNA sequences that were then classified into 58 satDNA families based on their degree of identity (> 95% same variant, 80–95% same family). None of the clusters yielded by RE included the telomeric repeat, for which reason we searched for the highly conserved telomeric sequence in vertebrates (TTAGGG)n and included it for abundance and divergence comparison. The 59 families were then numbered in order of decreasing abundance, following Ruiz-Ruano et al. (2016), from CgoSat01-184 to CgoSat59-103, with the last number indicating repeat unit length (RUL) (Table 1). The telomeric repeat was the 12th family in abundance (CgoSat12-6-tel). We designed primers for PCR amplification of 48 satDNA families (Tables 1 and S1), all of which were successfully amplified, and 18 of them showed a ladder pattern in the agarose gel typical of satellite DNA (Fig. S1), for which reason they were chosen for FISH analysis. The lack of a ladder pattern for the 30 remaining satDNAs is probably a consequence of PCR itself because they all should be arranged in tandem, as indicated by the bioinformatic analysis. Nevertheless, the 18 satDNAs showing PCR ladders are representative of the whole satDNA abundance range (see Table 1).
To search for satDNA families being abundant in the B chromosomes, for each satDNA we calculated the genomic fold change (gFC) due to B chromosome presence as log2 of the quotient between + B and 0B abundances, with gFC > 1 values indicating twofold or more higher satDNA amount in the B-carrying genome (Table 1). To test whether the satDNAs showing gFC > 1 form arrays large enough to be visualized on B chromosomes, we performed FISH analysis of 18 satDNA families representing the whole abundance range and found that six of them showed conspicuous FISH signals on B chromosomes (Figs. 1 and S2), whereas the remaining 12 did not (see examples in Fig. S3). Table 1 shows that the two satDNAs with the highest gFC values, CgoSat52-143 and CgoSat59-103 (6.3 and 109, respectively), revealed conspicuous FISH signals on the B chromosome (Fig. 1c, e). The remaining 16 satDNAs, however, showed gFC values from 0.7 to 1.7, but only four of them (CgoSat01-184, CgoSat07-67, CgoSat25-281 and CgoSat44-177) showed FISH signals on the B chromosome (Table 1 and Fig. S2). This indicates that only extremely high gFC values are indicative of satDNAs showing conspicuous signals by FISH.
Remarkably, the two satDNAs showing extreme gFC values (CgoSat52-143 and CgoSat59-103) were the only satDNAs apparently absent from the A genome, as indicated by the absence of FISH signals on the Z and W sex chromosomes and the autosomes (Fig. 1c, e). We thoroughly tested this possibility for CgoSat59-103 by searching for read pairs showing sequence homology with it in the B-carrying and B-lacking libraries and found 4100 read pairs in the former but only nine in the latter, thus suggesting the extreme scarcity (or absence) of this satDNA in the 0B individuals.
The scarce amount of cytogenetic material obtained from the individual harboring the Bsm chromosome only allowed FISH for CgoSat59-103 (Fig. 1e) and CgoSat07-67 (Fig. S2a), and it showed that these two satDNAs are also present on this B chromosome, suggesting that both B variants share common descent.
Additional evidence for the intraspecific origin of B chromosomes
We performed a second approach to investigate B chromosome origin in this species. For this purpose, we performed PCR on five repetitive DNA families on the DNA obtained from B chromosomes by microdissection (μB-DNA): 18S and 5S rDNA, U2 snDNA, the H3 histone gene and CgoSat01-184. FISH analysis showed that only the two latter DNA probes yielded signals on the B chromosome (Fig. S4). PCR amplification on μB-DNA was successful for these two DNAs and, unexpectedly, for 5S rDNA despite not having been visualized on the B chromosome by FISH. It is thus conceivable that the B chromosome actually contains some 5S rDNA copies in arrays shorter than the minimum detected by FISH, i.e., approximately 1 kb (Schwarzacher and Heslop-Harrison 2000). We also amplified these three repetitive DNAs on genomic DNA from B-lacking C. gomesi individuals and from several other Characidium species. All PCR-amplified DNA fragments were cloned and sequenced by the Sanger method. In the case of CgoSat01-184, we also extracted 57 repetitive units of 176–194 nt from the Illumina read pairs. The DNA sequences were aligned (370 nt for H3, 97–99 nt for 5S rDNA, and 176–194 nt for CgoSat01-184), primer sequences were cut from the alignment, and singletons were not considered for further analyses.
Among the 20 clones sequenced for the H3 histone gene, those obtained from the μB-DNA showed about similar nucleotide diversity than those obtained from the 0B genome (Table 2), and the rate of synonymous substitution (dS) was higher than the nonsynonymous (dN) rate in both types of chromosomes, but this result was based on very low number of sequences and Student’s t tests did not find significant these differences (Table 3). In fact, the predicted amino acid sequence was identical for the gene copies from the A and B chromosomes, which would be consistent with recent B chromosome origin. An MST built with H3 sequences showed that those coming from the B chromosome showed high similarity with those coming from the A chromosomes in the same species (C. gomesi), but they showed lower similarity with those from three other Characidium species (Fig. 2). This is also consistent with the intraspecific origin of these B chromosomes. Estimates of abundance and sequence divergence for reads showing homology with the H3 gene in the Illumina libraries showed that the B-carrying genome harbored higher abundance than the B-lacking genome (0.00265% and 0.00205%, respectively) but lower divergence (2.69 and 3.07, respectively) (Table 4).

Minimum-spanning tree showing the relationships between the different haplotypes of H3 histone sequences from the gDNA of Characidium species and from the DNA of a microdissected B chromosome of C. gomesi (µB-DNA). The diameter of the circles is logarithmically proportional to their abundance and the numbers represent the number of mutational steps
A total of 38 clones were sequenced for the 5S rDNA coding region, 14 from B-carrying and B-lacking genomes of C. gomesi and 24 from B-lacking genomes of four other Characidium species. An MST showed that the C. gomesi sequences obtained from the 0B genome were placed in two different branches separated by many mutational steps, suggesting the possible existence of pseudogenes in the 0B genome (Fig. 3). In another way, all haplotypes coming from B chromosome microdissection were placed in the same branch but showed 1–9 nucleotide differences between them, consistent with the higher nucleotide diversity of B-derived sequences (see Table 2). However, the abundance and divergence of 5S rDNA sequences in the Illumina reads were similar in the 0B and + B genomes (Table 4). The most remarkable finding was that all B chromosome haplotypes shared a common branch with two relatively abundant haplotypes in 0B C. gomesi, as well as with one C. zebra haplotype showing residual abundance in this species (Fig. 3). This also supports the intraspecific origin of B chromosomes in C. gomesi.

Minimum-spanning showing the relationships between the different haplotypes of 5S rDNA sequences from the gDNA of Characidium species and from the DNA of a microdissected B chromosome of C. gomesi (µB-DNA). The diameter of the circles is logarithmically proportional to their abundance and the numbers represent the number of mutational steps
Finally, we sequenced 54 clones for CgoSat01-184 and extracted 57 monomer sequences from the Illumina paired-reads obtained from 0B and + B genomes (Table 2). The nucleotide diversity observed in the DNA sequences obtained by B chromosome microdissection (thus coming from the B chromosomes) was only 44% of that observed in the 0B genome (thus coming from the A chromosomes) (Table 2). This might be due to amplification of this satellite in the B chromosome yielding many repeat units with identical sequences. In contrast, + B Illumina reads only showed a slight decrease in nucleotide diversity (8%) compared with 0B reads (Table 2), which is logical since this library contains sequences from both A and B chromosomes. An MST built with the haplotypes found in all former sequences in C. gomesi and those obtained by PCR amplification in C. zebra (Fig. 4) showed a clear separation of the sequences coming from both species (excepting a single shared haplotype showing low abundance in both species). Again, this result points to the intraspecific origin of B chromosomes in C. gomesi.

Minimum-spanning tree showing the relationships between the different haplotypes of CgoSat01-184 sequences from the gDNA of Characidium species, DNA of a microdissected B chromosome of C. gomesi (µB-DNA) and sequences extracted from Illumina reads of gDNA with (+ B) and without (0B) B chromosome. The diameter of the circles is logarithmically proportional to their abundance and the numbers represent the number of mutational steps
Discussion
The analysis of the molecular content of B chromosomes has been suggested to be a very informative tool to investigate their origin in several kinds of organisms (Teruel et al. 2010; Martis et al. 2012; Valente et al. 2014; Silva et al. 2014, 2017; Utsunomia et al. 2016; Ruiz-Ruano et al. 2017, 2018). In C. gomesi, chromosome painting suggested that B chromosomes are most likely derived from sex chromosomes (Pansonato-Alves et al. 2014). However, the anonymity of the repetitive DNA sequences presumably included in the painting probes weakened this conclusion. To shed new light on this subject, we followed two different strategies to investigate B chromosome origin in C. gomesi. The PCR amplification of repetitive DNA sequences on DNA obtained by microdissection of B chromosomes (following Teruel et al. 2010) was attempted for five different families of repetitive sequences but was successful only for three of them: the H3 histone genes, 5S rDNA and CgoSat01-184 satDNA. MSTs showed, in all three cases, remarkable haplotype sharing between the sequences located on chromosomes A and B in C. gomesi and lower similarity of B-sequences with those in other Characidium species. This gives strong support to the intraspecific origin of B chromosomes in this species. Our second strategy consisted of the thorough analysis of the satellitome in C. gomesi (following Ruiz-Ruano et al. 2016, 2017) and the FISH analysis of the 18 satDNA families, which yielded a ladder pattern in gel electrophoresis of the PCR products. We thus found six satDNA families showing conspicuous FISH signals on the B chromosome, two of which failed to show signals on A chromosomes and thus were B-specific (CgoSat52-143 and CgoSat59-103). One of the four remaining satDNAs (CgoSat07-67) was also found on Z and W chromosomes and on autosomes; two other (CgoSat01-184 and CgoSat25-281) were also found on Z and W chromosomes but not on the autosomes, and the latter (CgoSat44-177) was shared with the autosomes but not with sex chromosomes. We interpret these results as evidence for B chromosome derivation from sex chromosomes in C. gomesi, as it shares more satDNAs with sex chromosomes than with any of the 24 autosome pairs. The exclusive presence of SF1 and SF2 satDNA superfamilies on autosomes, and their consequent absence on Z, W and B chromosomes (see Table 1), reinforces this conclusion.
Satellitome analysis has thus proven to be a useful tool for unveiling B chromosome origin in C. gomesi, as previously shown in other species. For instance, in the characid fish genus Astyanax, 12 out of 18 satDNA families analyzed in three species showed FISH signals on the B chromosomes of at least two species, thus suggesting the common origin of these B chromosomes (Silva et al. 2017). Likewise, satellitome analysis in the grasshopper Eumigus monticola pointed out the origin of the B chromosome from the S8 autosome, as it is the only A chromosome bearing all six satDNA families found on the B chromosome (Ruiz-Ruano et al. 2017). Finally, in the migratory locust, the S9 autosome is the only A chromosome carrying all five satDNA families found on the B chromosome, presumably because this autosome was involved in B origin (Ruiz-Ruano et al. 2018).
At first glance, the presence of H3 histone genes (visualized on the B chromosome by FISH) and 5S rDNA (not visualized by FISH but inferred from PCR amplification of microdissected Bs) in the B chromosome might cast some doubt on B derivation from sex chromosomes, as both DNA sequences were not observed on the sex chromosomes by FISH. However, it is conceivable that one of the sex chromosomes might harbor these sequences in arrays below the FISH detection level. Alternatively, it is also conceivable that the B chromosome might have received a few copies of these sequences after its origin. The presence of virtually all B-sequences in a single branch for the three markers (H3, 5S and CgoSat01-184) would support the possibility that B chromosomes started with only a few copies of these three markers, and two of them (H3 and CgoSat01-184) were subsequently amplified on the B chromosome so that they are detectable by FISH. This is also supported by the higher abundance and lower divergence for these two repetitive DNAs in the Illumina libraries obtained from the B-carrying genome compared with the sequences obtained from the B-lacking one. Finally, the high dynamism of satDNA sequences, indicated by the two B-specific families presumably arose in the B chromosome after its origin (CgoSat52-143 and CgoSat59-103), in addition to the usual enrichment of B chromosome content in repetitive DNA (see Ruiz-Ruano et al. 2018), make the amplification of several repetitive DNA families highly likely in the C. gomesi B chromosome.
Additional evidence was provided by CgoSat01-184, as its nucleotide diversity was one order of magnitude higher in the monomers extracted from the Illumina reads than in those obtained by PCR and cloning (see Table 2). This is likely due to a logical PCR bias resulting from amplification using a single primer pair, compared with Illumina sequencing where PCR is performed on sequence adaptors presumably distributed at random across the genome. This predicts that samples obtained by Illumina sequencing represent a higher range of sequence variation than those obtained by PCR and cloning, as previously noted by Utsunomia et al. (2016, 2017), although recent research has revealed that conventional Illumina sequencing is not free from PCR bias (Wei et al. 2018). Our additional observation that nucleotide diversity in the DNA sequences obtained by PCR on DNA obtained from microdissected B chromosomes was only 44% that observed in the sequences obtained from Illumina sequencing of a 0B genome (thus coming from the A chromosomes) is also consistent with the amplification of this satDNA in the B chromosome, although the PCR bias mentioned above cannot be ruled out. The nucleotide diversity observed in + B and 0B Illumina reads also support the amplification of this satDNA in the B chromosome, as it is 8% lower in the former (see Table 2). We thus conclude that Illumina reads provide more reliable information on intragenomic sequence diversity for satDNA than PCR and cloning.
In summary, our present research has provided strong support for the hypothesis of B chromosome origin from the sex chromosomes in C. gomesi, and the large collection of satDNA families, reported here, provides new markers that may further future research in the field of B and sex chromosome evolution. For instance, B-specific satDNAs will make it easy to identify B chromosomes in any kind of cell, albeit in interphase, mitosis or meiosis, which may be useful in future analyses of B chromosome transmission or any other research pursuing sex chromosome monitoring during development. Our present results can also serve as a foundation for future analysis of genic content on these B chromosomes, as recent research has revealed the presence of protein-coding genes on B chromosomes from rye (Martis et al. 2012), mammals (Makunin et al. 2014), fish (Valente et al. 2014; Utsunomia et al. 2016) and grasshoppers (Navarro-Domínguez et al. 2017a). Derivation from sex chromosomes predicts high similarity in gene content between B and sex chromosomes, which would make a joint analysis of gene content in both kinds of chromosomes highly logical. Finally, the possible activity of some of these genes might be crucial to determine the biological role of B chromosomes, and this is one of the main prospects remaining for future research in C. gomesi B chromosomes. In addition, the presence of H3 histone genes suggests the possibility that these genes might be active in the B chromosomes, an interesting possibility demanding future genomic and transcriptomic studies.
Data availability
We deposited the 0B and + B genomic libraries in the Sequence Read Archive (SRA) database with accession numbers SRR9613702 and SRR9613703, respectively.
References
Ahmad SF, Martins C (2019) The modern view of B chromosomes under the impact of high scale omics analyses. Cells 8(2):156
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Banaei-Moghaddam AM, Meier K, Karimi-Ashtiyani R, Houben A (2013) Formation and expression of pseudogenes on the B chromosome of rye. Plant Cell 25:2536–2544
Bao W, Kojima KK, Kohany O (2015) Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6:11
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Burt A, Trivers R (2006) Genes in conflict: the biology of selfish genetic elements. Harvard University Press, Cambridge
Camacho JPM (2005) B chromosomes. The evolution of the genome. Elsevier, Amsterdam, pp 223–286
Camacho JPM, Sharbel TF, Beukeboom LW (2000) B-chromosome evolution. Philos Trans R Soc Lond B 355:163–178
Carmello BO, Coan RLB, Cardoso AL et al (2017) The hnRNP Q-like gene is retroinserted into the B chromosomes of the cichlid fish Astatotilapia latifasciata. Chromosome Res 25:277–290
Clark FE, Conte MA, Ferreira-Bravo IA et al (2016) Dynamic sequence evolution of a sex-associated B chromosome in Lake Malawi cichlid fish. J Hered 108:53–62
Clark FE, Conte MA, Kocher TD (2018) Genomic characterization of a B chromosome in Lake Malawi cichlid fishes. Genes 9:610
Coan RLB, Martins C (2018) Landscape of transposable elements focusing on the B chromosome of the cichlid fish Astatotilapia latifasciata. Genes 9:269
Colgan DJ, McLauchlan A, Wilson GDF et al (1998) Histone H3 and U2 snRNA DNA sequences and arthropod molecular evolution. Aust J Zool 46:419–437
Dalla Benetta E, Akbari OS, Ferree PM (2019) Sequence expression of supernumerary B chromosomes: function or fluff? Genes 10:123
Dhar MK, Gurmeet Kour G, Kaul S (2017) B chromosome in Plantago lagopus Linnaeus, 1753 shows preferential transmission and accumulation through unusual processes. Comp Cytogenet 11:375–392
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Foresti F, Almeida-Toledo LF, Toledo-Filho SA (1981) Polymorphic nature of nucleolus organizer regions in fishes. Cytogenet Genome Res 31:137–144
Gribble S, Ng BL, Prigmore E et al (2004) Chromosome paints from single copies of chromosomes. Chromosome Res 12:143–151
Hanlon SL, Miller DE, Eche S et al (2018) Origin, composition, and structure of the supernumerary B chromosome of Drosophila melanogaster. Genetics 210(4):1197–1212
Houben A (2017) B chromosomes—a matter of chromosome drive. Front Plant Sci 8:210
Huang W, Du Y, Zhao X et al (2016) B chromosome contains active genes and impacts the transcription of A chromosomes in maize (Zea mays L.). BMC Plant Biol 16:88
Jehangir M, Ahmad SF, Cardoso AL et al (2019) De novo genome assembly of the cichlid fish Astatotilapia latifasciata reveals a higher level of genomic polymorphism and genes related to B chromosomes. Chromosoma 128(2):81–96
Jones RN (2018) Transmission and drive involving parasitic B chromosomes. Genes 9:388
Jones RN, Rees H (1982) B chromosomes. Academy Press, London
Kent WJ (2002) BLAT—the BLAST-like alignment tool. Genome Res 12:656–664
Levan A, Fredga K, Sandberg AA (1964) Nomenclature for centromeric position on chromosomes. Hereditas 52:201–220
Li Y, Jing XA, Aldrich JC et al (2017) Unique sequence organization and small RNA expression of a “selfish” B chromosome. Chromosoma 126:753–768
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Ma W, Gabriel TS, Martis MM et al (2017) Rye B chromosomes encode a functional Argonaute-like protein with in vitro slicer activities similar to its A chromosome paralog. New Phytol 213:916–928
Makunin AI, Dementyeva PV, Graphodatsky AS et al (2014) Genes on B chromosomes of vertebrates. Mol Cytogenet 7:99
Makunin AI, Romanenko SA, Beklemisheva VR et al (2018) Sequencing of supernumerary chromosomes of red fox and raccoon dog confirms a non-random gene acquisition by B chromosomes. Genes 9:405
Martis MM, Klemme S, Banaei-Moghaddam AM et al (2012) Selfish supernumerary chromosome reveals its origin as a mosaic of host genome and organellar sequences. Proc Natl Acad Sci 109:13343–13346
McAllister BF, Werren JH (1997) Hybrid origin of a B chromosome (PSR) in the parasitic wasp Nasonia vitripennis. Chromosoma 106:243–253
Miao VP, Covert SF, VanEtten HD (1991) A fungal gene for antibiotic resistance on a dispensable (“B”) chromosome. Science 254:1773–1776
Milani D, Bardella VB, Ferretti ABSM et al (2018) Satellite DNAs unveil clues about the ancestry and composition of B chromosomes in three grasshopper species. Genes 9:523
Navarro-Domínguez B, Ruiz-Ruano FJ, Cabrero J et al (2017a) Protein-coding genes in B chromosomes of the grasshopper Eyprepocnemis plorans. Sci Rep 7:45200
Navarro-Domínguez B, Ruiz-Ruano FJ, Camacho JPM et al (2017b) Transcription of a B chromosome CAP-G pseudogene does not influence normal Condensin Complex genes in a grasshopper. Sci Rep 7:17650
Navarro-Domínguez B, Martín-Peciña M, Ruiz-Ruano FJ et al (2019) Gene expression changes elicited by a parasitic B chromosome in the grasshopper Eyprepocnemis plorans are consistent with its phenotypic effects. Chromosoma 128:53–67
Novák P, Neumann P, Pech J et al (2013) RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29:792–793
Pansonato-Alves JC, Serrano EA, Utsunomia R et al (2014) Single origin of sex chromosomes and multiple origins of B chromosomes in fish genus Characidium. PLoS One 9:e107169
Pendas AM, Moran P, Martinez JL, Garcia-Vazquez E (1995) Applications of 5S rDNA in Atlantic salmon, brown trout, and in Atlantic salmon brown trout hybrid identification. Mol Ecol 4:275–276
Perfectti F, Werren JH (2001) The interspecific origin of B chromosomes: experimental evidence. Evolution 55:1069–1073
Pinkel D, Straume T, Gray JW (1986) Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci 83:2934–2938
Ramos É, Cardoso AL, Brown J et al (2017) The repetitive DNA element BncDNA, enriched in the B chromosome of the cichlid fish Astatotilapia latifasciata, transcribes a potentially noncoding RNA. Chromosoma 126:313–323
Rodrigues PHM, Santos RZ, Silva DMZA et al (2019) Chromosomal and genomic dynamics of satellite DNAs in Characidae (Characiformes, Teleostei) Species. Zebrafish 16:408–414
Ruiz-Estévez M, López-León M, Cabrero J et al (2013) Ribosomal DNA is active in different B chromosome variants of the grasshopper Eyprepocnemis plorans. Genetica 141:337–345
Ruiz-Estévez M, Badisco L, Broeck JV et al (2014) B chromosomes showing active ribosomal RNA genes contribute insignificant amounts of rRNA in the grasshopper Eyprepocnemis plorans. Mol Genet Genom 289:1209–1216
Ruiz-Ruano FJ, López-León MD, Cabrero J, Camacho JPM (2016) High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci Rep 6:28333
Ruiz-Ruano FJ, Cabrero J, López-León MD, Camacho JPM (2017) Satellite DNA content illuminates the ancestry of a supernumerary (B) chromosome. Chromosoma 126:487–500
Ruiz-Ruano FJ, Cabrero J, López-León MD et al (2018) Quantitative sequence characterization for repetitive DNA content in the supernumerary chromosome of the migratory locust. Chromosoma 127:45–57
Schwarzacher T, Heslop-Harrison P (2000) Practical in situ hybridization. BIOS Scientific Publishers, Milton Park
Sharbel TF, Green DM, Houben A (1998) B-chromosome origin in the endemic New Zealand frog Leiopelma hochstetteri through sex chromosome devolution. Genome 41:14–22
Silva DMZA, Pansonato-Alves JC, Utsunomia R et al (2014) Delimiting the origin of a B chromosome by FISH mapping, chromosome painting and DNA sequence analysis in Astyanax paranae (Teleostei, Characiformes). PLoS One 9:e94896
Silva DMZA, Utsunomia R, Ruiz-Ruano FJ et al (2017) High-throughput analysis unveils a highly shared satellite DNA library among three species of fish genus Astyanax. Sci Rep 7:12726
Smit AFA, Hubley R (2008–2015) RepeatModeler Open-1.0. http://www.repeatmasker.org
Smit AFA, Hubley R, Green P (1996–2010) RepeatMasker Open-3.0. http://www.repeatmasker.org
Sumner AT (1972) A simple technique for demonstrating centromeric heterochromatin. Exp Cell Res 75:304–306
Teruel M, Cabrero J, Perfectti F, Camacho JPM (2010) B chromosome ancestry revealed by histone genes in the migratory locust. Chromosoma 119:217–225
Tosta VC, Marthe JB, Tavares MG et al (2014) Possible introgression of B chromosomes between Bee species (Genus Partamona). Cytogenet Genome Res 144:220–226
Trifonov VA, Dementyeva PV, Larkin DM et al (2013) Transcription of a protein-coding gene on B chromosomes of the Siberian roe deer (Capreolus pygargus). BMC Biol 11:90
Utsunomia R, Silva DMZA, Ruiz-Ruano FJ et al (2016) Uncovering the ancestry of B chromosomes in Moenkhausia sanctaefilomenae (Teleostei, Characidae). PLoS One 11:e0150573
Utsunomia R, Ruiz-Ruano FJ, Silva DMZA et al (2017) A glimpse into the satellite DNA library in characidae fish (Teleostei, Characiformes). Front Genet 8:103
Utsunomia R, Silva DMZA, Ruiz-Ruano FJ et al (2019) Satellitome landscape analysis of Megaleporinus macrocephalus (Teleostei, Anostomidae) reveals intense accumulation of satellite sequences on the heteromorphic sex chromosome. Sci Rep 9:5856
Valente GT, Conte MA, Fantinatti BEA et al (2014) Origin and evolution of B chromosomes in the cichlid fish Astatotilapia latifasciata based on integrated genomic analyses. Mol Biol Evol 31:2061–2072
Wei KHC, Lower SE, Caldas IV et al (2018) Variable rates of simple satellite gains across the Drosophila phylogeny. Mol Biol Evol 35:925–941
Wu D, Ruban A, Fuchs J et al (2019) Nondisjunction and unequal spindle organization accompany the drive of Aegilops speltoides B chromosomes. New Phytol 223:1340–1352
Yoshida K, Terai Y, Mizoiri S et al (2011) B chromosomes have a functional effect on female sex determination in lake victoria cichlid fishes. PLoS Genet 7(8):e1002203
Ziegler CG, Lamatsch DK, Steinlein C et al (2003) The giant B chromosome of the cyprinid fish Alburnus alburnus harbours a retrotransposon-derived repetitive DNA sequence. Chromosome Res 11:23
Acknowledgements
We are thankful Renato Devidé for his technical assistance and reviewers for the very helpful suggestions and corrections for improving the paper.
Funding
This study was funded by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (Grant Number 2013/02143-3) and by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no conflict of interest.
Ethical approval
The animals were collected in accordance with Brazilian environmental protection legislation (Collection Permission MMA/IBAMA/SISBIO—number 3245), and the procedures for fish sampling, maintenance and analysis were performed in compliance with the Brazilian College of Animal Experimentation (COBEA) and approved (protocol 504) by the Bioscience Institute/Unesp Ethics Committee on the Use of Animals (CEUA).
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2019_1615_MOESM2_ESM.tif
Supplementary material 2 Karyotypes of C. gomesi after FISH with 18S (red) and 5S rDNA (green) (a), U2 snDNA (b) and H3 histone (c) probes and metaphases of C. gomesi after chromosome painting with the probe from the microdissected B chromosome of the same species (d). Bar = 10 µm (TIFF 43 kb)
438_2019_1615_MOESM3_ESM.jpg
Supplementary material 3 Metaphases of C. gomesi after FISH with satDNAs (a, c, e) and C-banding (b, d, f). Bar = 10 µm (JPEG 461 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Serrano-Freitas, É.A., Silva, D.M.Z.A., Ruiz-Ruano, F.J. et al. Satellite DNA content of B chromosomes in the characid fish Characidium gomesi supports their origin from sex chromosomes. Mol Genet Genomics 295, 195–207 (2020). https://doi.org/10.1007/s00438-019-01615-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-019-01615-2