
Abstract
Regulatory factors function by modulating a variety of cascade mechanisms in cells. RBM4 is a multifunctional RNA-binding protein in post-transcriptional gene regulation. Cytoplasmic RBM4 interacts with Ago2 to regulate inflammatory responses by affecting mRNA decay and cap-dependent translation. However, it is unclear whether RBM4 functions in inflammation regulation by its splicing factor role. Here, the cell biology, gene expression profile and alternative splicing pattern of HeLa cells with RBM4 overexpression (RBM-OE) were compared with the control. The results showed that RBM4-OE inhibited proliferation. RBM4-OE extensively affects the transcriptional level of genes involved in cell surface receptor signalling pathway, inflammatory responses and the response to lipopolysaccharide. RBM4 broadly regulated the alternative splicing of hundreds of genes with functions of protein binding, helicase activity, DNA binding and transcription co-activator. RBM4-regulated splicing of these genes plays an important role in apoptotic process and gene transcription regulation. As an example, exon inclusion of TNIP1 mediated by RBM4 affects the expression of its targets in inflammatory pathways. These results indicated that RBM4 can mediate the inflammatory response via splicing regulation, which adds to the understanding of the critical role of RBM4 in cancer complicated by inflammation. In conclusion, this study indicated a mechanism in which the dysregulation of alternative splicing can influence cellular biology and lead to various immune-related diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
How gene expression is regulated is one of the most important problems in the field of biology research. Cellular gene expression is generally regulated by post-transcriptional mechanisms. RNA-binding proteins (RBPs) play key roles in post-transcriptional gene regulation by interacting with RNAs. At present, more than 1000 RBPs have been identified based on high-throughput omics technologies (Hentze et al. 2018). These RBPs affect all RNA metabolic processes, including splicing, transport, translation and decay. As a result, RBPs regulate the mRNA or protein level expression of numerous protein-coding genes by binding with pre-mRNA, mRNA and noncoding RNA in various cell types. Moreover, RBPs have been reported to play important regulator roles in different biological processes, including germline, embryo development and diverse immune-related diseases. In particular, RBPs exert their biological regulatory function in the immune responses and inflammatory reactions in cancer and other diseases (Tiedje et al. 2016; Fu and Blackshear 2017; Turner and Díaz-Muñoz 2018). Therefore, it is still meaningful to explore the post-transcriptional regulatory role of RBPs in cell lines or tissue samples, which will provide a basis to monitor and treat diseases involving inflammatory responses.
Alternative splicing of precursor mRNA is often regulated by serine/arginine-rich proteins (SR protein). RNA-binding motif protein 4 (RBM4) was first identified as a non-SR protein that modulates alternative pre-mRNA splicing but acts opposite of some SR proteins in splice sites and during exon selection (Lai et al. 2003). RBM4 is a multifunctional RNA-binding protein involved in various post-transcriptional regulation processes, including alternative splicing and translation control (Lin and Tarn 2005, 2009; Kar et al. 2006; Markus et al. 2006; Hock et al. 2007; Lin et al. 2007; Brooks et al. 2009; Markus and Morris 2009). RBM4 modulates the differentiation of various cell types by regulating alternative splicing (Brooks et al. 2009; Lin and Tarn 2009; Lin et al. 2013, 2016b; Lin 2015; Tarn et al. 2016; Su et al. 2017). For example, RBM4 functions by different splicing cascades to regulate adipogenesis (RBM4a-SRSF3-MAP4K4), differentiation (RBM4-MEF2C) and development (RBM4-Nova1-SRSF6) in brown adipocytes (Lin 2015; Lin et al. 2016a, b; Chi and Lin 2018; Peng et al. 2018). Additionally, RBM4 suppresses the proliferation and migration of various cancer cells by specifically controlling cancer-related splicing (Lin et al. 2014, 2017, 2018; Wang et al. 2014; Liang et al. 2015; Markus et al. 2016; Qi et al. 2016; Yong et al. 2016; Chen et al. 2017; Huang et al. 2017). It is notable that RBM4 is also cytoplasmic and can shuttle rapidly between the cytoplasm and the nucleus (Lai et al. 2003). Under stress conditions, RBM4 participates in the formation of a translation initiation complex to promote the expression of stress-response genes and evade stress-induced repression of protein translation initiation (Lin et al. 2007; Uniacke et al. 2012), which indicates an important role in translation control. However, RBM4 was initially reported as a tumour suppressor with a decreased expression levels in breast and pancreatic carcinomas (Wang et al. 2014), but was also reported to promote cancer progression with increased overexpression in cervical, breast, lung, colon, ovarian and rectal cancers (Lin et al. 2014; Markus et al. 2016). Therefore, RBM4 functions as a tumour suppressor or promoter depending on variable expression in different types of cancer.
In fact, there was an association between cancer and the inflammatory response (Karin et al. 2006; Mantovani et al. 2008; Grivennikov et al. 2010; Jones and Jenkins 2018). As previously reported, cancer chemotherapy will result in the simultaneous onset of an acute inflammatory response and sepsis-like symptoms (Tsuji et al. 2003). Infection is an important complication in cancer patients, leading to acute organ dysfunction (severe sepsis) and eventually death (Williams et al. 2004; Fox et al. 2010; Mokart et al. 2014; El Haddad et al. 2018; Liu et al. 2018b). Alteration of immune surveillance is a common link between cancer and sepsis (Nana-Sinkam and Crouser 2010). Therefore, it is important to explore the mechanisms mediating the immune response in cancers with sepsis. RBP will affect the expression of innate immune response genes by regulating splicing and translation, which alert the immune system to danger (Fu and Ares 2014; Gowen et al. 2015; Fu and Blackshear 2017; Pereira et al. 2017; Turner and Díaz-Muñoz 2018). As reported, RBM4 plays a key role by interacting with Ago2 in the regulation of both mRNA decay and the translation disruption of TNF-α during TLR4-induced gene reprogramming of acute systemic inflammatory responses in a THP-1 sepsis cell model (El Gazzar et al. 2011; Brudecki et al. 2013a, b; McClure et al. 2015). Therefore, we speculate that RBM4 also regulates inflammatory responses in cancer patients with infection complications, which resulted in inconsistent RBM4 expression detection in previous studies (Lin et al. 2014; Wang et al. 2014; Markus et al. 2016). However, it is still unclear how RBM4 modulates the expression of immune response genes via the regulation of alternative splicing.
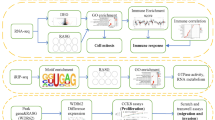
Here, to further explore the regulatory roles of RBM4, this gene was overexpressed in HeLa cells, which are primarily derived from cervical cancer tissue. Then, high-throughput RNA sequencing (RNA-Seq) was performed for overexpressed cells and controls to define comprehensive gene expression profiles and to identify genome-wide alternative splicing events regulated by RBM4. The results showed clear changes in transcript profiles and splicing patterns of specific subsets of genes after RBM4 overexpression in HeLa cells. Changes in alternative splicing of transcription factors and transcription co-activators mediate the differential expression of inflammatory factors. Our study provides an important basis and data platform to clarify the role of RBM4 in mediating the mechanisms for immune responses linking cancer with inflammation.
Results
RBM4 overexpression inhibits proliferation in HeLa cells
To explore the roles of RBM4 in cancer, RBM4 was overexpressed in the HeLa cell line. HeLa cells were transfected by vector expressing the RBM4 gene (RBM4-OE) or empty vector (not expressing the RBM4 gene) (control). After transfecting the HeLa cells with the RBM4 expression vector, both the qPCR and Western blot results showed that RBM4 was significantly overexpressed in HeLa cells (Fig. 1a, b). We found that RBM4 overexpression significantly inhibited the proliferation of HeLa cells (Fig. 1c). Then, we detected cell apoptosis using Annexin V in RBM4-OE and control HeLa cells. Although there was no significant difference between RBM4-OE and the control, the apoptosis level increased after RBM4 overexpression in HeLa cells (Fig. 1d). These results indicated that RBM4 played an important role in the proliferation but not apoptosis regulation in HeLa cells.

Effect of RBM4 overexpression on proliferation and apoptosis in HeLa cells. a RBM4 was overexpressed and verified by qPCR. b RBM4 was overexpressed and verified by Western blotting. c RBM4 overexpression inhibits the proliferation of HeLa cells. d RBM4 overexpression did not significantly affect the apoptosis level of HeLa cells; data are represented as the mean ± SD. Student’s t test was performed to compare control and RBM4-OE HeLa cells with significance set at a P value of less than 0.05. *P < 0.05, ***P < 0.001
RBM4 overexpression changes the gene expression profiles of HeLa cells
To explore the molecular mechanisms by which RBM4 overexpression functions in HeLa cells, the gene expression profiles of RBM4-OE cells (transfected with vector expressing the RBM4 gene) and control cells (transfected with empty vector not expressing the RBM4 gene) were detected by RNA-seq. A total of four RNA-seq libraries were constructed and sequenced for RBM4-OE and control HeLa cells, with two biological replicates for each group (RBM4_1st, RBM4_2nd, Ctrl_1st, Ctrl_2nd). After removing sequence adaptors and low-quality reads, an average of 87.5 million clean pair-end reads per sample was obtained. These reads were mapped to the human genome, which resulted in an average of 65.8 million uniquely mapped read pairs per sample (Table S1).
Then, these uniquely mapped reads were used to calculate the gene expression. Fragments per kilobase of transcript per million fragments mapped (FPKM) were calculated by an in-house pipeline and used to represent the levels of gene expression. The results showed 25, 167 genes expressed (FPKM > 0) and 12,529 genes expressed at an expression level of FPKM > 1 in at least one sample (Table S2 and Table S3). FPKM values for RBM4 further supported that this gene was effectively overexpressed in HeLa cells (Fig. 2a). A correlation matrix was calculated based on the FPKM values of expressed genes in all four samples. There was a high Pearson’s correlation value between RBM4-OE and the control (more than 0.98), which indicated the similar expression of most genes. However, unsupervised hierarchical clustering of the correlation matrix showed a clear separation of RBM4-OE and control samples, with two biological replicates in a cluster (Fig. 2b). This result demonstrated that RBM4 overexpression had an effect on the gene expression profile in HeLa cells.

Analysis of the change in gene expression in response to RBM4 overexpression. a RBM4 expression was quantified by RNA sequencing data. b Heat map shows the hierarchically clustered Pearson’s correlation matrix resulting from comparing the transcript expression values for control and RBM4 overexpression samples. c Identification of RBM4-regulated genes. Upregulated genes are labelled in red, whereas downregulated genes are labelled in blue in the volcano plot. d Hierarchical clustering of DEGs in control and RBM4 overexpression samples. FPKM values are log2-transformed and then median-centred by each gene (color figure online)
To further compare the gene expression profile, edgeR was performed to identify the differentially expressed genes (DEGs) between the RBM-OE and control cells (Robinson et al. 2010), with a cut-off as fold change ≥ 2 or ≤ 0.5 and a 5% false discovery rate (FDR). There were 1027 upregulated and 561 downregulated genes between RBM4-OE and control cells, indicating that RBM4 overexpression extensively regulates gene expression in HeLa cells (Fig. 2c). Detailed information for these DEGs, including FPKMs and fold changes, is presented in Table S4. Moreover, hierarchical clustering of normalized FPKM values of DEGs showed a clear separation of the RBM4-OE and control samples and a high consistency for the two replicate data sets (Fig. 2d). These results indicated that RBM4 overexpression significantly changed the transcript expression level of a set of genes.
RBM4 overexpression affects the expression of inflammatory factors in HeLa cells
To reveal the potential roles of DEGs, GO function analysis was performed to annotate all 1588 DEGs. The results revealed 301 upregulated and 202 downregulated genes annotated with GO categories biological process terms, respectively. The upregulated DEGs were enriched in 51 GO terms and the downregulated DEGs in 39 GO terms (Table S5). Both the upregulated and downregulated genes were enriched in terms related to proliferation and apoptosis. These results supported RBM4 overexpression affecting the proliferation of HeLa cells.
It is notable that both the upregulated and down-regulated genes in RBM4-OE cells were significantly enriched in the inflammatory response, which belongs to the top 10 terms (Fig. 3a, b). To further reveal the functional roles of these DEGs, KEGG pathway analysis was performed for the 1588 DEGs. The results showed that DEGs were enriched in ECM-receptor interaction, peroxisome, legionellosis, rheumatoid arthritis and cytokine–cytokine receptor interaction (Fig. 3c, d). These results indicated that RBM4 also plays an important role in many immune- and inflammatory-associated diseases by affecting the expression of inflammatory factors.

Functional analysis of DEGs after RBM4 overexpression in HeLa cells. The top 10 GO biological processes of RBM4-upregulated (a) and downregulated genes (b). The top 10 KEGG pathways of RBM4-upregulated (c) and downregulated genes (d). e Relative expression levels of selected DEGs measured by RNA-seq (FPKM) (left) and qPCR (right). For qPCR, GAPDH was used as the reference gene. Student’s t test was performed to compare RBM-OE and control cells with significance set at a P value of less than 0.05. *P < 0.05, **P < 0.01
To verify the effect of RBM4-OE on the expression of these DEGs, qPCR was conducted to quantify the changes in mRNA levels of these genes. Sixteen upregulated DEGs were randomly selected for qPCR analysis, including JMJD6, RRP8, HSPA1B, HSPA6, ITGA6, ITGA2, HSPA1L, CSF2, HMGA2, AOC2, F2RL1, GAST, CD101, MATN3, APLN and AOC3. All selected DEGs were annotated in the GO or KEGG analysis, and the FPKM of these genes was higher than one in at least one sample. The results showed that 15 of the 16 selected DEGs showed a significant increase after RBM4 overexpression in HeLa cells, which was in agreement with the RNA-seq analysis (Fig. 3e; Fig S1).
RBM4 overexpression regulated the alternative splicing of transcription factor in HeLa cells
As reported, RBM4 plays an important role in regulating alternative splicing (AS). Therefore, we explored whether RBM4 affects the expression of DEGs at the mRNA level via RBM4-dependent AS regulation in HeLa cells. The splice reads from RBM4-OE and control HeLa cells (Table S1) were mapped to the reference genome, and 233,418 annotated exons (63.5% of total annotated ones) were detected. Then, a total of 159,184 known and 179,084 novel splice junctions were detected using TopHat2. AS events were identified from the splice junctions by the ABLAS pipeline (Xia et al. 2017), which resulted in 19,223 known alternative splicing events (ASEs) and 59,855 novel ASEs (Table S6).
To identify the high-confidence RBM4-regulated alternative splicing events (RASEs), a custom pipeline was used to compare the changes the AS ratio between RBM4-OE and control cells at a cut-off of P value ≤ 0.05 and changes in the AS ratio ≥ 0.15. Under this condition, a total of 606 RASEs were identified (Table S7), including 122 intron-retention (IR) and 484 non-IR (NIR) RASEs. Among these RASEs, the number of alternative 3′ splice sites (A3SS), alternative 5′ splice sites (A5SS), exon skipping (ES) and cassette exons was relatively high (Fig. 4a). After mapping and counting, a total of 531 genes contributed to these RASEs were defined as RBM4-regulated alternative splicing genes (RASGs). However, there were nine genes overlapping between DEGs and RASGs (Fig. 4b). In particular, the alternative splicing of DEGs enriched in the inflammatory response was not affected by RBM4 overexpression. These results indicated that RBM4-OE did not simultaneously affect the expression at the transcript level or alternative splicing of most DEGs or RASGs in HeLa cells.

Identification and functional analysis of RBM4-regulated splicing events. a Classification of different AS types regulated by RBM4 protein. b The top 10 GO molecular function analyses c. The top 10 GO biological process analyses. d The overlap analysis between RBM4-regulated differentially expressed genes (DEG) and RBM4-regulated alternative splicing genes (RASGs)
To explore the potential function of RASGs, GO terms analysis was conducted. The results showed that RASGs were highly enriched in GO molecular functional terms, including protein binding, helicase activity, RNA-binding, nucleotide binding, DNA binding and transcription coactivator activity (Fig. 4b; Table S8). There were 207 RASEs annotated with protein binding and 53 RASEs with DNA binding. Moreover, GO biological process annotation showed that RASGs were enriched in chromatin modification, mitotic cell cycle, apoptotic process, translational initiation, regulation of translational initiation and positive regulation of apoptotic process (Fig. 4c, Table S8). These results indicated that RBM4 overexpression affected the expression of DEGs enriched in pathways, including the inflammatory response, by regulating the alternative splicing of transcription factors and co-activators.
To validate the effects of RBM4 overexpression on alternative splicing, qPCR was conducted to quantify the ratios of 14 RASEs in RBM4-OE and control cells. The results showed that changes in the ratio of 13 detected RASEs in qPCR were in agreement with the results of the transcriptome analysis (Fig. 5; Fig. S2). These RASEs occurred in MORF4L2, EIF4A2, SAP130, MAFG, UBE2I, RBM4, PLEC, TNIP1, NSL1, FBXW5, SRA1 and ZNF213. These genes were annotated with GO molecular functional terms, including DNA binding and transcription coactivator activity and GO biological process terms, including apoptotic process and negative regulation of transcription (Table S8). In particular, RBM4 was spliced at an alternative 3′ splice site after overexpression, which would affect its RNA-binding activity (Fig. 5a; Table S8). Exon exclusion of TNIP1 will affect the inflammatory response by changes in protein binding activity and ubiquitin-specific protease activity (Fig. 5b; Table S8). The alternative 5′ splice site of MAFG will change DNA binding and protein heterodimerization activity, which affect transcription and cell proliferation (Fig. 5c; Table S8).

Validation of RBM4-regulated alternative splicing events. a Alternative 3′ splice site in RBM4. b Exon skipping of TNIP1. c Alternative 5′ splice site of MAFG. AIGV-sashimi plots show AS changes in RBM4 overexpression cells and control cells (left panel), and the transcripts for the gene are shown below. The schematic diagrams depict the structures of ASEs, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by boxes and intron sequences by the horizontal line (right panel, top). RNA-seq quantification and qPCR validation of ASEs are shown in the bottom right panel. The altered ratio of AS events in RNA-seq was calculated using the formula: AS1 junction reads/(AS1 junction reads + AS2 junction reads), while the altered ratio of AS events in qPCR was calculated using the formula: AS1 transcript level/AS2 transcript level. Student’s t test was performed to compare RBM-OE and control cells with significance set at a P value of less than 0.05. *P < 0.05, **P < 0.01 (color figure online)
Discussion
RBM4 plays important roles in post-transcriptional gene regulation by regulating alternative splicing and translation, which affect the differentiation of adipocytes, the proliferation and apoptosis of cancer cells and the inflammatory response in immune cells (Lin and Tarn 2012; Brudecki et al. 2013a; Lin et al. 2014, 2017; Wang et al. 2014; Markus et al. 2016). In this study, we showed that RBM4 overexpression inhibits cell proliferation in HeLa cells. RBM4-OE affects the expression of inflammatory response genes at the transcript level, and the alternative splicing patterns of genes that have RNA-binding, nucleotide binding, DNA binding and transcription coactivator activities. Our results suggested that RBM4 affects the proliferation and expression of inflammatory factors by regulating the alternative splicing of regulatory factors in HeLa cells.
In many previous studies, RBM4 has been reported to affect the proliferation and apoptosis of various cancer cells by specifically controlling cancer-related splicing (Wang et al. 2014; Yong et al. 2016). Notably, there are inconsistent results regarding the expression levels of RBM4 in cancer cells and tissues (Lin et al. 2014; Wang et al. 2014; Markus et al. 2016). Lin et al. and Markus et al. showed elevated levels of RBM4 in breast cancer cells and tissue (Lin et al. 2014; Markus et al. 2016). However, Wang et al. reported that RBM4 is a tumour suppressor and that its overexpression inhibits cancer cell growth (Wang et al. 2014). In our study, we showed that RBM4 overexpression inhibits the proliferation of HeLa cells, which is consistent with previous study (Wang et al. 2014). Moreover, our transcriptome analysis showed that RBM4 affects the alternative splicing of genes from pathways including the mitotic cell cycle. These results suggest that RBM4 affects the proliferation in HeLa cells by regulating splicing. In particular, we experimentally validated that RBM4 regulates the alternative splicing of SRA1. In fact, alternative splicing of the first intron of SRA1 participates in the generation of coding and noncoding RNA isoforms in cancer cells (Hube et al. 2006). Decreased expression of SRA1 has clinical significance in hepatocellular carcinoma (Luo et al. 2017). Thus, further work could be conducted to explore how RBM4 regulates the alternative splicing of SRA1.
RBM4 promotes the expression of stress-response genes by forming a translation initiation complex with other proteins (Lin et al. 2007; Uniacke et al. 2012). In fact, RBM4 regulates the acute systemic inflammatory response in THP-1 sepsis cells by interacting with Ago2 to regulate translation (Brudecki et al. 2013a). In this study, we explored whether RBM4 affects the expression and alternative splicing of immune response genes in cancer cell lines. In fact, infection is an important complication in cancer patients, leading to sepsis (Nana-Sinkam and Crouser 2010; Liu et al. 2018b). Therefore, it is meaningful to explore the mechanisms underlying the alteration of immune surveillance, which is a common link between cancer and sepsis. Our results showed that RBM4-OE upregulated or downregulated expression of many genes enriched in the inflammatory response. Interestingly, we found that RBM4 upregulated the expression of amine oxidase, copper containing 3 (AOC3) and coagulation factor II (thrombin) receptor-like 1 (F2RL1). F2RL1 and AOC3 have been reported to regulate the inflammatory response in cancer and infection (Bettin et al. 2016; Ward et al. 2016). Therefore, it is interesting to explore whether RBM4 regulates the inflammatory response by alternative splicing.
RBM4 is a multifunctional RNA-binding protein that regulates both alternative splicing and translation (Lin and Tarn 2009; Markus and Morris 2009). In our study, transcriptome analysis showed that AS events occurred in genes involved in the inflammatory response. We have observed RBM4-dependent alternative splicing of transcription factors and splice factors. For example, TNIP1 has protein binding activity and is increasingly being recognized as a key repressor of inflammatory signalling and a potential factor in multiple autoimmune diseases (Shamilov and Aneskievich 2018). In fact, toll-like receptor signalling was considered an important target of TNIP1 (Shamilov and Aneskievich 2018). These results suggest that RBM4 regulates the inflammatory response via toll-like receptor signalling with alternative splicing as well as translation control. In addition, RBM4 controls the alternative splicing of MAFG, which regulates liver cancer (Liu et al. 2018a). Antisense RNA of MAFG facilitates the migration and invasion of non-small-cell carcinoma cells via sponging miR-339-5p from MMP15 (Cui et al. 2018). Notably, our results also showed that RBM4 controls the expression of itself by alternative splicing after overexpression in HeLa cells. In fact, a previous study reported that hnRNPDL regulates the alternative splicing of cassette exons at their 3′ UTRs, which mediates the expression of this gene itself (Kemmerer et al. 2018). Thus, RBM4 could broadly regulate the alternative splicing of transcription factors and splicing factors, which further affects the expression of their targets.
In summary, the RNA-binding properties of RBM4 suggest that this protein could have multifunctional roles in RNA processing and translation. In this study, we conducted RNA-seq technology to study the role of RBM4 in HeLa cells. We concluded that RBM4 regulates the expression of genes involved in the inflammatory response by regulating alternative splicing patterns of regulator factors, including transcription factors and co-activators. The dysregulated alternative splicing of transcription factors and co-activators will result in functional loss or gain, which mediates the expression of downstream genes (Li et al. 2017). These results indicated that RBM4 regulates proliferation and immune response via alternative splicing in HeLa cells, which provides a basis for exploring the role of RBM4 in cancer complicated by inflammation.
Materials and methods
Cloning and plasmid construction
Primer pairs used for hot fusion were designed with CE Design V1.04 (Vazyme). Each of the primers comprises a fragment of RBM4-specific sequences and a 17–30 bp sequence of the pIRES-hrGFP-1a vector (Stratagene).
The pIRES-hrGFP-1a vector was digested by EcoRI and XhoI (NEB) at 37 °C for 2–3 h. Then, the enzyme-digested vector was run on a 1.0% agarose gel and purified by a Qiagen column kit. Total RNA was isolated from HeLa cells with Trizol. Purified RNA was transcribed for cDNA by oligo dT primer. Then, the insert fragment of RBM4 was synthesized by PCR amplification. A linearized vector digested by EcoRI and XhoI (NEB) and PCR insert fragment of RBM4 were added to a PCR microtube for ligation with the ClonExpress® II One Step Cloning Kit (Vazyme). Plasmids were introduced into Escherichia coli by chemical transformation. The cells were plated onto LB agar plates containing 1 µl/ml ampicillin and incubated overnight at 37 °C. Colonies were screened by colony PCR (28 cycles) with universal primers (located on the backbone vector). The insert sequence of RBM4 was verified by Sanger sequencing.
Cell culture and transfection
HeLa cells were cultured under standard conditions with Dulbecco’s modified Eagle’s medium (DMEM) with supplemented 10% foetal bovine serum (FBS), 100 µg/ml streptomycin and 100 U/ml penicillin. The transfection of the pIRES-hrGFP-1a vector containing the RBM4 gene (RBM4-OE) or not containing the RBM4 gene (control) into HeLa cells was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The transfected cells were harvested after 48 h for qPCR analysis.
Assessment of gene overexpression
GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a control gene to assess the effects of RBM4 overexpression. cDNA synthesis was performed by standard procedures, and qPCR was performed on a Bio-Rad S1000 with Bestar SYBR Green RT-PCR Master Mix (DBI Bioscience, Shanghai, China). The concentration of each transcript was then normalized to the GAPDH mRNA level using 2−ΔΔCT method (Livak and Schmittgen 2001).
Apoptosis and proliferation analysis
HeLa cells were cultured to a confluence of 70–80% after 48 h post-transfection with the pIRES-hrGFP-1a vector containing the RBM4 gene (RBM4) or not containing the RBM4 gene (control) and were collected for apoptosis and proliferation analysis.
For apoptosis analysis, the transfected HeLa cells were collected and then stained with the Annexin V-PE/7-AAD Apoptosis Detection kit (BD Pharmingen Biosciences, San Diego, CA). Apoptotic cells were detected using the Beckman MoFlo XDP, and the data were analysed with Flowjo (TreesStar) software.
A MTT assay was used to evaluate cell proliferation according to a previous study (Wang et al. 2019). Cultured cells were pretreated with 20 µl MTT solution. The treated cells were cultured under the same conditions for 30 min. Then, the medium was removed. Next, 0.15 ml dimethylsulfoxide (DMSO) was used to solubilize the resulting formazan crystal. An ELISA reader was used to detect the optical density at 490 nm.
Western blotting
Total protein lysate was extracted from RBM4 overexpression/control cells with RIPA buffer. Protein extracts (40–60 μg) were separated by SDS-PAGE and transferred to PVDF membranes (Bio-Rad, cat. 1620177, Foster, California, USA). The membranes were blocked with 5% non-fat milk, followed by overnight incubation with primary antibodies against either RBM4 (CUSABIO, Shanghai) overnight at RT. After incubation with rhodamine (TRITC), goat anti-rabbit IgG (Abclonal, cat. A5040) (1:10,000) for 1 h at room temperature, the signals were detected with Clarity Max™ Western ECL Substrate (Bio-Rad, cat. 1705062). β-actin was used as a loading control for the Western blot analysis.
RNA extraction and sequencing
The HeLa cells were ground into fine powder before RNA extraction. Total RNA was extracted by the hot phenol method. The RNA was further purified with two phenol–chloroform treatments and then treated with RQ1 DNase (Promega, Madison, WI, USA) to remove DNA. The quality and quantity of the purified RNA were determined by measuring the absorbance at 260 nm/280 nm (A260/A280) using Smartspec Plus (Bio-Rad, USA). The integrity of RNA was further verified by 1.5% agarose gel electrophoresis. For each sample, 10 μg of total RNA was used for RNA-seq library preparation. Polyadenylated mRNAs were purified and concentrated with oligo(dT)-conjugated magnetic beads (Invitrogen, Carlsbad, CA, USA) before directional RNA-seq library preparation. The purified mRNAs were then iron fragmented at 95 °C followed by end repair and 5′ adaptor ligation. Then, reverse transcription was performed with RT primers harbouring a 3′ adaptor sequence and a randomized hexamer. The cDNAs were purified, amplified, and stored at − 80 °C until sequencing. For high-throughput sequencing, the libraries were prepared following the manufacturer’s instructions.
An Illumina Nextseq 500 system was used to collect data from 151 bp pair-end sequencing (ABlife Inc., Wuhan, China).
Differentially expressed genes (DEG) analysis
FPKM (paired-end fragments per kilobase of exon per million fragments mapped) was used to evaluate the expression level of genes. To screen for the differentially expressed genes (DEGs), we applied the software edgeR, which was specifically used to analyse the differential expression of genes using raw RNA-Seq reads. To determine whether a gene was differentially expressed, we analysed the results based on the fold change (fold change ≥ 2 or ≤ 0.5) and false discovery rate (FDR < 0.05).
To predict the gene function and calculate the functional category distribution frequency, gene ontology (GO) analyses and enriched KEGG pathways were identified using KOBAS 2.0 server (Xie et al. 2011). The hypergeometric test and Benjamini–Hochberg FDR controlling procedure were used to define the enrichment of each pathway (corrected P value < 0.05).
Alternative splicing analysis
The alternative splicing events (ASEs) and regulated alternative splicing events (RASEs) between the samples were defined and quantified by using the ABLas pipeline as described previously (Xia et al. 2017). In brief, the detection of seven types of ASEs was based on splice junction reads. The eight types of ASE included cassette exon (Cassette Exon), exon skipping (ES), mutually exclusive exon skipping (MXE), A5SS, A3SS, the MXE combined with an alternative 5′ promoter (5pMXE) and an alternative polyadenylation site (3pMXE). Intron retention was calculated based on the average base in the intronic and adjacent exonic regions.
After detecting the ASEs in each RNA-seq sample, Fisher’s exact test was chosen to calculate the significant P value, with alternative reads and model reads of samples as input data. The changed ratio of alternatively spliced reads and constitutively spliced reads between compared samples was defined as the RASE ratio. The P value < 0.05 and RASE ratio > 0.2 were set as the threshold for RASE detection.
Real-time qPCR validation of DEGs and AS events
In this study, to elucidate the validity of the RNA-seq data, quantitative real-time PCR (qPCR) was performed for some selected DEGs, and normalized with the reference gene GAPDH. The same RNA samples for RNA-seq were used for qPCR. The PCR conditions consisted of denaturing at 95 °C for 10 min, 40 cycles of denaturing at 95 °C for 15 s, and annealing and extension at 60 °C for 1 min. PCR amplifications were performed in triplicate for each sample. The primers for qPCR analysis are listed in Table S9.
Statistical analysis
An unpaired two-tailed t test (between two groups) was performed for the cell biology and qPCR data. Probability (P) values < 0.05 were considered statistically significant. The data are presented as the mean ± standard deviation (SD). Each experiment was conducted in at least three biological replicates, except for the RNA-seq.
Data availability
All data generated or analysed during this study have been included in this published article and its supplementary information files. The datasets supporting the results of this article are available in the NCBI Gene Expression Omnibus and are accessible through GEO series accession number GSE134402.
References
Bettin A, Reyes I, Reyes N (2016) Gene expression profiling of prostate cancer-associated genes identifies fibromodulin as potential novel biomarker for prostate cancer. Int J Biol Markers 31:e153–e162
Brooks YS, Wang G, Yang Z, Smith KK, Bieberich E, Ko L (2009) Functional pre-mRNA trans-splicing of coactivator CoAA and corepressor RBM4 during stem/progenitor cell differentiation. J Biol Chem 284:18033–18046
Brudecki L, Ferguson DA, McCall CE, El Gazzar M (2013a) MicroRNA-146a and RBM4 form a negative feed-forward loop that disrupts cytokine mRNA translation following TLR4 responses in human THP-1 monocytes. Immunol Cell Biol 91:532–540
Brudecki L, Ferguson DA, McCall CE, El Gazzar M (2013b) Mitogen-activated protein kinase phosphatase 1 disrupts proinflammatory protein synthesis in endotoxin-adapted monocytes. Clin Vaccine Immunol 20:1396–1404
Chen JY, Liu LP, Xu JF (2017) Decrease of RBM4 indicates poor prognosis in patients with hepatocellular carcinoma after hepatectomy. Onco Targets Ther 10:339–345
Chi YL, Lin JC (2018) RBM4a modulates the impact of PRDM16 on development of brown adipocytes through an alternative splicing mechanism. Biochim Biophys Acta 1865:1515–1525
Cui S, Yang X, Zhang L, Zhao Y, Yan W (2018) LncRNA MAFG-AS1 promotes the progression of colorectal cancer by sponging miR-147b and activation of NDUFA4. Biochem Biophys Res Commun 506:251–258
El Gazzar M, Church A, Liu T, McCall CE (2011) MicroRNA-146a regulates both transcription silencing and translation disruption of TNF-alpha during TLR4-induced gene reprogramming. J Leukoc Biol 90:509–519
El Haddad H, Chaftari AM, Hachem R, Chaftari P, Raad II (2018) Biomarkers of sepsis and bloodstream infections: the role of procalcitonin and proadrenomedullin with emphasis in patients with cancer. Clin Infect Dis 67:971–977
Fox AC, Robertson CM, Belt B, Clark AT, Chang KC, Leathersich AM, Dominguez JA, Perrone EE, Dunne WM, Hotchkiss RS, Buchman TG, Linehan DC, Coopersmith CM (2010) Cancer causes increased mortality and is associated with altered apoptosis in murine sepsis. Crit Care Med 38:886–893
Fu XD, Ares M Jr (2014) Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15:689–701
Fu M, Blackshear PJ (2017) RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat Rev Immunol 17:130–143
Gowen BG, Chim B, Marceau CD, Greene TT, Burr P, Gonzalez JR, Hesser CR, Dietzen PA, Russell T, Iannello A, Coscoy L, Sentman CL, Carette JE, Muljo SA, Raulet DH (2015) A forward genetic screen reveals novel independent regulators of ULBP1, an activating ligand for natural killer cells. Elife. https://doi.org/10.7554/eLife.08474
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140:883–899
Hentze MW, Castello A, Schwarzl T, Preiss T (2018) A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol 19:327–341
Hock J, Weinmann L, Ender C, Rudel S, Kremmer E, Raabe M, Urlaub H, Meister G (2007) Proteomic and functional analysis of Argonaute-containing mRNA–protein complexes in human cells. EMBO Rep 8:1052–1060
Huang GW, Zhang YL, Liao LD, Li EM, Xu LY (2017) Natural antisense transcript TPM1-AS regulates the alternative splicing of tropomyosin I through an interaction with RNA-binding motif protein 4. Int J Biochem Cell Biol 90:59–67
Hube F, Guo J, Chooniedass-Kothari S, Cooper C, Hamedani MK, Dibrov AA, Blanchard AA, Wang X, Deng G, Myal Y, Leygue E (2006) Alternative splicing of the first intron of the steroid receptor RNA activator (SRA) participates in the generation of coding and noncoding RNA isoforms in breast cancer cell lines. DNA Cell Biol 25:418–428
Jones SA, Jenkins BJ (2018) Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol 18:773–789
Kar A, Havlioglu N, Tarn WY, Wu JY (2006) RBM4 interacts with an intronic element and stimulates tau exon 10 inclusion. J Biol Chem 281:24479–24488
Karin M, Lawrence T, Nizet V (2006) Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 124:823–835
Kemmerer K, Fischer S, Weigand JE (2018) Auto-and cross-regulation of the hnRNPs D and DL. RNA 24:324–331
Lai MC, Kuo HW, Chang WC, Tarn WY (2003) A novel splicing regulator shares a nuclear import pathway with SR proteins. EMBO J 22:1359–1369
Li J, Wang Y, Rao X, Wang Y, Feng W, Liang H, Liu Y (2017) Roles of alternative splicing in modulating transcriptional regulation. BMC Syst Biol. https://doi.org/10.1186/s12918-12017-10465-12916
Liang YC, Lin WC, Lin YJ, Lin JC (2015) The impact of RNA binding motif protein 4-regulated splicing cascade on the progression and metabolism of colorectal cancer cells. Oncotarget 6:38046–38060
Lin JC (2015) RBM4-MEF2C network constitutes a feed-forward circuit that facilitates the differentiation of brown adipocytes. RNA Biol 12:208–220
Lin JC, Tarn WY (2005) Exon selection in alpha-tropomyosin mRNA is regulated by the antagonistic action of RBM4 and PTB. Mol Cell Biol 25:10111–10121
Lin JC, Tarn WY (2009) RNA-binding motif protein 4 translocates to cytoplasmic granules and suppresses translation via argonaute2 during muscle cell differentiation. J Biol Chem 284:34658–34665
Lin JC, Tarn WY (2012) Multiple roles of RBM4 in muscle cell differentiation. Front Biosci 4:181–189
Lin JC, Hsu M, Tarn WY (2007) Cell stress modulates the function of splicing regulatory protein RBM4 in translation control. Proc Natl Acad Sci USA 104:2235–2240
Lin JC, Yan YT, Hsieh WK, Peng PJ, Su CH, Tarn WY (2013) RBM4 promotes pancreas cell differentiation and insulin expression. Mol Cell Biol 33:319–327
Lin JC, Lin CY, Tarn WY, Li FY (2014) Elevated SRPK1 lessens apoptosis in breast cancer cells through RBM4-regulated splicing events. RNA 20:1621–1631
Lin JC, Chi YL, Peng HY, Lu YH (2016a) RBM4-Nova1-SRSF6 splicing cascade modulates the development of brown adipocytes. Biochim Biophys Acta 1859:1368–1379
Lin JC, Lu YH, Liu YR, Lin YJ (2016b) RBM4a-regulated splicing cascade modulates the differentiation and metabolic activities of brown adipocytes. Sci Rep. https://doi.org/10.1038/srep20665
Lin JC, Lee YC, Liang YC, Fann YC, Johnson KR, Lin YJ (2017) The impact of the RBM4-initiated splicing cascade on modulating the carcinogenic signature of colorectal cancer cells. Sci Rep. https://doi.org/10.1038/srep44204
Lin JC, Lee YC, Tan TH, Liang YC, Chuang HC, Fann YC, Johnson KR, Lin YJ (2018) RBM4-SRSF3-MAP4K4 splicing cascade modulates the metastatic signature of colorectal cancer cell. Biochim Biophys Acta 1865:259–272
Liu T, Yang H, Fan W, Tu J, Li TWH, Wang J, Shen H, Yang J, Xiong T, Steggerda J, Liu Z, Noureddin M, Maldonado SS, Annamalai A, Seki E, Mato JM, Lu SC (2018a) Mechanisms of MAFG dysregulation in cholestatic liver injury and development of liver cancer. Gastroenterology 155:557–571.e514
Liu Z, Mahale P, Engels EA (2018b) Sepsis and risk of cancer among elderly adults in the United States. Clin Infect Dis. 5:717–724
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Luo P, Jing W, Zhu M, Li ND, Zhou H, Yu MX, Liang CZ, Tu JC (2017) Decreased expression of LncRNA SRA1 in hepatocellular carcinoma and its clinical significance. Cancer Biomark 18:285–290
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454:436–444
Markus MA, Morris BJ (2009) RBM4: a multifunctional RNA-binding protein. Int J Biochem Cell Biol 41:740–743
Markus MA, Heinrich B, Raitskin O, Adams DJ, Mangs H, Goy C, Ladomery M, Sperling R, Stamm S, Morris BJ (2006) WT1 interacts with the splicing protein RBM4 and regulates its ability to modulate alternative splicing in vivo. Exp Cell Res 312:3379–3388
Markus MA, Yang YH, Morris BJ (2016) Transcriptome-wide targets of alternative splicing by RBM4 and possible role in cancer. Genomics 107:138–144
McClure C, Brudecki L, Yao ZQ, McCall CE, El Gazzar M (2015) Processing body formation limits proinflammatory cytokine synthesis in endotoxin-tolerant monocytes and murine septic macrophages. J Innate Immun 7:572–583
Mokart D, Saillard C, Sannini A, Chow-Chine L, Brun J-P, Faucher M, Blache J-L, Blaise D, Leone M (2014) Neutropenic cancer patients with severe sepsis: need for antibiotics in the first hour. Intensive Care Med 40:1173–1174
Nana-Sinkam SP, Crouser ED (2010) Altered immune surveillance: a common link between cancer and sepsis? Crit Care Med 38:1000–1001
Peng H-Y, Liang Y-C, Tan T-H, Chuang H-C, Lin Y-J, Lin J-C (2018) RBM4a-SRSF3-MAP4K4 splicing cascade constitutes a molecular mechanism for regulating brown adipogenesis. Int J Mol Sci. https://doi.org/10.3390/ijms19092646
Pereira B, Billaud M, Almeida R (2017) RNA-Binding proteins in cancer: old players and new actors. Trends Cancer 3:506–528
Qi Y, Yu J, Han W, Fan X, Qian H, Wei H, Tsai YH, Zhao J, Zhang W, Liu Q, Meng S, Wang Y, Wang Z (2016) A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat Commun. https://doi.org/10.1038/ncomms11840
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Shamilov R, Aneskievich BJ (2018) TNIP1 in autoimmune diseases: regulation of toll-like receptor signaling. J Immunol Res. https://doi.org/10.1155/2018/3491269
Su CH, Hung KY, Hung SC, Tarn WY (2017) RBM4 regulates neuronal differentiation of mesenchymal stem cells by modulating alternative splicing of pyruvate kinase M. Mol Cell Biol. https://doi.org/10.1128/mcb.00466-00416
Tarn WY, Kuo HC, Yu HI, Liu SW, Tseng CT, Dhananjaya D, Hung KY, Tu CC, Chang SH, Huang GJ, Chiu IM (2016) RBM4 promotes neuronal differentiation and neurite outgrowth by modulating Numb isoform expression. Mol Biol Cell 27:1676–1683
Tiedje C, Diaz-Munoz MD, Trulley P, Ahlfors H, Laass K, Blackshear PJ, Turner M, Gaestel M (2016) The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res 44:7418–7440
Tsuji E, Hiki N, Nomura S, Fukushima R, Kojima J, Ogawa T, Mafune K, Mimura Y, Kaminishi M (2003) Simultaneous onset of acute inflammatory response, sepsis-like symptoms and intestinal mucosal injury after cancer chemotherapy. Int J Cancer 107:303–308
Turner M, Díaz-Muñoz MD (2018) RNA-binding proteins control gene expression and cell fate in the immune system. Nat Immunol 19:120–129
Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, Payette J, Holcik M, Pause A, Lee S (2012) An oxygen-regulated switch in the protein synthesis machinery. Nature 486:126–129
Wang Y, Chen D, Qian H, Tsai YS, Shao S, Liu Q, Dominguez D, Wang Z (2014) The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell 26:374–389
Wang C-J, Zhu C-C, Xu J, Wang M, Zhao W-Y, Liu Q, Zhao G, Zhang Z-Z (2019) The lncRNA UCA1 promotes proliferation, migration, immune escape and inhibits apoptosis in gastric cancer by sponging anti-tumor miRNAs. Mol Cancer 18:115
Ward ST, Weston CJ, Shepherd EL, Hejmadi R, Ismail T, Adams DH (2016) Evaluation of serum and tissue levels of VAP-1 in colorectal cancer. BMC Cancer. https://doi.org/10.1186/s12885-12016-12183-12887
Williams MD, Braun LA, Cooper LM, Johnston J, Weiss RV, Qualy RL, Linde-Zwirble W (2004) Hospitalized cancer patients with severe sepsis: analysis of incidence, mortality, and associated costs of care. Crit Care 8:R291–R298
Xia H, Chen D, Wu Q, Wu G, Zhou Y, Zhang Y, Zhang L (2017) CELF1 preferentially binds to exon-intron boundary and regulates alternative splicing in HeLa cells. Biochim Biophys Acta 1860:911–921
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:W316–W322
Yong H, Zhu H, Zhang S, Zhao W, Wang W, Chen C, Ding G, Zhu L, Zhu Z, Liu H, Zhang Y, Wen J, Kang X, Zhu J, Feng Z, Liu B (2016) Prognostic value of decreased expression of RBM4 in human gastric cancer. Sci Rep. https://doi.org/10.1038/srep28222
Acknowledgements
This study was partially supported by ABLife (ABL-7702098).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that he/she has no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, WY., Quan, W., Yang, F. et al. RBM4 modulates the proliferation and expression of inflammatory factors via the alternative splicing of regulatory factors in HeLa cells. Mol Genet Genomics 295, 95–106 (2020). https://doi.org/10.1007/s00438-019-01606-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-019-01606-3