
Abstract
Since the growth traits of chickens are largely related to the production of meat and eggs, it is definitely important to understand genetic basis of growth traits. Although many quantitative trait loci (QTLs) that affect growth traits have recently been reported in chickens, little is known about genetic architecture of growth traits across all growth stages. Therefore, we conducted a longitudinal QTL study of growth traits measured from 0 to 64 weeks of age using 134 microsatellite DNA markers on 26 autosomes from 406 F2 females, which resulted from an intercross of Oh-Shamo and White Leghorn chicken breeds. We found 27 and 21 independent main-effect QTLs for body weight and shank length, respectively. Moreover, 15 and 4 pairs of epistatic QTLs were found for body weight and shank length, respectively. Taken together, the present study revealed 48 QTLs for growth traits on 21 different autosomes, and these loci clearly have age-specific effects on phenotypes throughout stages that are important for meat and egg productions. Approximately 60% of Oh-Shamo-derived alleles increased the phenotypic values, corresponding to the fact that Oh-Shamo traits were higher than those of White Leghorn. On the other hand, remaining Oh-Shamo alleles decreased the phenotypic values. Our results clearly indicated that the growth traits of chickens are regulated by several main and epistatic QTLs that are widely distributed in the chicken genome, and that the QTLs have age-dependent manners of controlling the traits. This study implies importance of not only cross-sectional but also longitudinal growth data for further understanding of the complex genetic architecture in animal.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The growth traits exhibited by animals (typically quantitative traits) are largely related meat, egg, and milk production in livestock. Since growth traits will be good indicator to predict the amount of meat (Venturini et al. 2014) and timing of sexual maturity (Podisi et al. 2011) which are important traits for meat and egg production, respectively, it is definitely important to understand genetic basis of growth traits. Quantitative genetic studies that examined model organisms such as yeast, flies, and mice recently revealed that quantitative traits are controlled by several quantitative trait loci (QTLs) with small effects and epistatic interacting loci, and are influenced by environmental factors (Mackay 2014). Although QTL analyses and genome-wide association studies (GWAS) have identified many QTLs and GWAS loci in animals, the overall genetic variations associated with most traits cannot be explained by the detected loci (i.e. missing heritability). Therefore, it is necessary to address the cause of missing heritability to accumulate results of genetic mapping studies that use several animal resource types for better understanding of genetic architecture in animal complex traits.
Long-term longitudinal studies revealed some information that is critical to clinical and psychological research fields. For instance, Petersen et al. (2004) reported the long-term relationship between physical activity and obesity in humans. The study indicated that obesity may lead to physical inactivity, but the results did not support the hypothesis that long-term physical inactivity was associated with the development of obesity. Therefore, long-term periodic investigations have the potential to reveal novel insights based on comparisons with short-term investigations. Until now, most QTL analyses of livestock animals focused on trait data collected at a few time points. In the present study, we measured the growth traits of an F2 resource population at multiple time points (from 0 to 64 weeks of age) that covered entire growth period of chickens. Thus, our longitudinal QTL study could potentially identify novel data that could aid our understanding of the genetic basis of growth traits in chickens.
Several QTLs that affect the growth traits of chickens have recently been reported. According to the chicken QTLdb of the Animal QTLdb (Hu et al. 2013), over 2500 QTLs have been listed as growth trait types, and the present study focused on body weight and shank length as growth traits. Although there are approximately 90 research papers reporting QTLs for body weight and shank length (Hu et al. 2013), most studies used commercial broiler- and layer-related breeds as the parental breeds of resource populations. To understand the genetic basis of the traits in chicken populations that are widely distributed around the world, it is important to utilize several genetically divergent resources such as traditional and village chicken breeds rather than the sole use of specific breeds. Therefore, we used the Oh-Shamo breed (a Japanese indigenous breed; Tsudzuki 2003) as a parental breed to create a QTL mapping population. Since our QTL mapping studies using the Oh-Shamo breed have revealed a large number of loci for growth, egg, and meat traits (Tsudzuki et al. 2007; Goto et al. 2011, 2014a, b; Yoshida et al. 2013), the present longitudinal QTL mapping of growth traits will probably identify several loci. In the present study, we aimed to detect QTLs based on a longitudinal study of QTL mapping for growth traits (body weight and shank length) in chickens using a unique F2 resource population of Oh-Shamo and White Leghorn (a traditional layer breed).
Materials and methods
Animals
A Japanese Large Game (Oh-Shamo) male was mated to three White Leghorn (CB strain; Reynaud et al. 1987) females to obtain an F2 resource population (Goto and Tsudzuki 2017). F2 females (n = 406) were produced via the full-sib mating of four F1 males and 19 F1 females (Goto et al. 2011). All birds were maintained under previously described conditions (Tsudzuki et al. 2007). The birds were cared for according to the rules of the Standards Related to the Care and Management of Experimental Animals (Prime Ministers’ Office, Japan, 1980) and the Guide for the Use of Experimental Animals in Universities (The Ministry of Education, Science, Sports, and Culture, Japan, 1987).
Phenotypic measurements
Body weight and shank length measurements of the birds were recorded from hatching to 64 and 52 weeks of age, respectively. These traits were measured as previously described (Tsudzuki et al. 2007) every week from 0 to 16 weeks of age. Trait data were then collected once every 4 weeks from 20 to 64 weeks of age for body weight and from 20 to 52 weeks of age for shank length.
Genotyping and linkage map
As described by Goto et al. (2014a, b), 134 microsatellite DNA markers on 26 autosomes were genotyped based on genomic DNA extracted from blood samples. These markers were fully informative because of the exclusion of markers that had common alleles between the parental breeds (one Oh-Shamo male vs three White Leghorn females; Goto and Tsudzuki 2017). Map Manager QTX b20 software (Manly et al. 2001) was used to construct a marker linkage map of the F2 population using the Kosambi map function, and marker orders were determined using ArkDB of the Roslin Bioinformatics Group (http://www.thearkdb.org/). Our linkage map covered approximately 67% of the chicken genome (Goto et al. 2014a).
Statistical analysis
A one-way analysis of variance (ANOVA), followed by Tukey’s HSD test, was conducted to compare trait data among four groups, which are the Oh-Shamo, White Leghorn, F1, and F2, in each age of traits (29 stages for BW and 26 stages for SL) using JMP software version 5.0.5a (SAS Institute Inc., Cary, NC, USA). To analyze the phenotypic relationship among traits, Pearson’s correlation coefficients in the F2 were calculated using the JMP (P < 0.05).
QTL analysis
Before QTL analyses were conducted, two environmental factors (date of hatch and F1 dams) were tested using the least squares method in JMP to determine whether these factors affected phenotypic values. When these factors exhibited significant effects (P < 0.05), the affected phenotypic values were corrected. Shapiro-Wilk’s test was used to assess whether the phenotypic distribution was normal, and the Box–Cox transformation was used to approximate a normal distribution when a non-normal phenotypic distribution was detected.
For QTL analyses, residuals after correction were standardized using JMP, and growth traits were analyzed using the Bayesian model selection method (Yi et al. 2005) found in R/qtlbim software (Yandell et al. 2006). Bayesian QTL mapping was performed for each trait using previously described methods (Yoshida et al. 2013). Briefly, all chromosomes were partitioned with a 1-cM grid, and a main effect and four epistatic-interaction effects (additive by additive, additive by dominance, dominance by additive, and dominance by dominance) were modelled for each QTL. A Markov chain Monte-Carlo (MCMC) algorithm was used to test the posterior distribution on the parameters (Yi et al. 2005), and the graphical and numerical methods of R/qtlbim confirmed the convergence of the Markov chain. The Bayes factor (BF) for a locus was defined as the ratio of the posterior odds to the prior odds for inclusion against exclusion of the locus (Ankra-Badu et al. 2009). The BF threshold of 2 log (BF) was used to declare genome-wide significant QTLs, according to Kass and Raftery (1995), and the BF threshold of epistatic interactions was set at 20 (Yi et al. 2005).
Results
Phenotypic analysis
Supplementary Table 1 shows the mean values of body weight (BW) at 0–64 weeks of age (BW0–BW64) and the differences between the Oh-Shamo, White Leghorn, F1, and F2 populations. Throughout all stages (BW0–BW64), the body weights of Oh-Shamo were significantly higher than those of the others. Regarding BW3–BW11, body weights of the F1 were significantly higher than those of the White Leghorn and F2, and the phenotypic order for stages BW12–BW32 was F1 > F2 > White Leghorn. However, the order changed to F1 = F2 > White Leghorn during stages BW36–BW64, with the exception of BW56 (F1 > F2).
Supplementary Table 2 indicates the mean values of shank length (SL) at 0–52 weeks of age (SL0–SL52) and the differences between the Oh-Shamo, White Leghorn, F1, and F2 populations. Regarding shank traits, the phenotypic values of the Oh-Shamo were significantly higher than those of the others throughout all stages (SL0–SL44). The phenotypic order during stages SL2–SL5 was F1 = White Leghorn > F2. The order during stages SL6–SL13 was F1 > White Leghorn > F2, whereas the order was F1 > White Leghorn = F2 during stages SL15–SL16. However, the order changed to F1 > F2 > White Leghorn during stages SL20–SL44, with the exception of stage SL32 (F1 = F2).
Phenotypic correlations among all BW stages are shown in Supplementary Fig. 1. Correlation coefficients between adjacent stages were estimated as more than 0.79, despite a lower value (0.22) estimated between BW0 and BW1. Moreover, relatively higher estimated correlation values were observed among stages at and before 4 weeks (> 0.58), with the exception of BW0. On the other hand, estimated correlations among early stages (BW0–BW16) and late stages (BW20–BW64) were less than 0.50.
Supplementary Fig. 2 shows the phenotypic correlations for SL. Correlation coefficients between adjacent stages were estimated as more than 0.68, although there were relatively lower values [e.g. 0.49 (SL0 and SL1) and 0.36 (SL16 and SL20)]. Correlations among traits at late stages (SL20–SL52) exhibited higher values (> 0.90), because the extension of the shank bone would have plateaued. Correlations were estimated as more than 0.65 and 0.70 among early stages (SL3–SL9) and middle stages (SL11–SL16), respectively. There were no significant correlations among traits at both early and middle stages (SL0–SL13) and traits at late stages (SL20–SL52), but correlations among SL1 and traits at late stages (SL20–SL52) were significant.
Supplementary Fig. 3 indicates the phenotypic correlations between BW and SL. Correlations between body weight (BW2–BW20) and shank length (SL2–SL16) at early and middle stages were estimated from 0.24 to 0.93, whereas there were relatively high correlations ranged from 0.72 to 0.92 between body weight and shank length at the same stages during 2–16 weeks of age [e.g. 0.72 (BW16 and SL16) and 0.92 (BW7 and SL7)]. There were lower correlations ranged from − 0.04 to 0.19 between body weight at all stages (BW0–BW64) and shank length at late stages (SL20–SL52).
QTL analysis
QTLs were found based on the positions of flanking markers, so QTLs co-located at adjacent regions with shared flanking markers were treated as identical QTL.
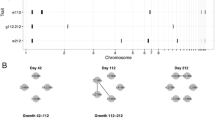
Main-effect QTLs Twenty-seven main-effect QTLs associated with body weight traits (between hatching 64 weeks of age) were identified on chromosomes 1, 2, 3, 4, 5, 6, 7, 9, 13, 18, 19, 23, 24, 26, 27, and 28 (Fig. 1 and Supplementary Tables 3 and 4). Each QTL contributed 1.4–18.2% to phenotypic variances (Supplementary Table 3). The QTLs were identified: seven on chromosome 1; three on chromosome 2; two each on chromosomes 7, 13, and 28; and one each on chromosomes 3–6, 9, 18, 19, 23, 24, 26, and 27. The Oh-Shamo alleles at most detected QTLs increased body weight phenotypes, and the White Leghorn alleles at eight QTLs (1D, 1E, 1F, 7A, 7B, 13C, 24A, and 28B) increased the phenotypic value.

Summary of age-specific main-effect QTLs associated with body weight and shank length traits. Main-effect QTLs detected at each age in hens are indicated for both body weight (BW) and shank length (SL) traits. Colored squares indicate the detection of QTLs at the specified ages. The density of squares reflects Bayes factor values. S and W denote that the Oh-Shamo and White Leghorn alleles increased the phenotypic value, respectively, based on the additive effects. Diagonal lined cells in shank length are out of analyses. See Supplementary Tables 3, 4, 5, and 6 for QTL details
Twenty-one main-effect QTLs were identified for shank length (between hatching and 52 weeks of age) on chromosomes 1, 2, 4, 5, 6, 7, 9, 11, 13, 19, 27, and 28 (Fig. 1 and Supplementary Tables 5 and 6). Each locus explained 1.6–32.8% of the phenotypic variance (Supplementary Table 5). The following QTLs were detected: four each on chromosomes 1 and 2; two each on chromosomes 5, 9, and 19; one each on chromosomes 4, 6, 7, 11, 13, 27, and 28. The Oh-Shamo alleles at 13 QTLs increased shank length, whereas the White Leghorn alleles at eight QTLs (2A, 2B, 2C, 5A, 5B, 6B, 7A, and 11A) increased the phenotypic value.
In Fig. 1, we can see main-effect QTLs detected for both traits across all time points. It enabled us to see when hotspots for both traits arose. In ten QTLs (1A, 1G, 1H, 2C, 4A, 7A, 9A, 19A, 27A, and 28A), nine QTLs were detected at the similar stages for both traits, except for QTL 7A. A hotspot for both traits seems to be around 16–28 weeks because seven QTLs were co-located around the stage.
Epistatic QTLs 15 pairs of epistatic QTLs were detected for body weight traits (Supplementary Tables 7 and 8). These pairs were categorized into three types based on the following interaction manners: (1) interaction between two main-effect QTLs; (2) interaction between a main-effect QTL and a non-main-effect QTL; (3) interaction between two non-main-effect QTLs. In Supplementary Table 8, the “QTL no.” that has a “-” between the chromosome number and a letter (e.g. 1-I, 1-J, and 3-B) indicates QTLs that have an epistatic-interaction effect only. On the other hand, QTLs having both main effect and epistatic interaction were named by chromosome number and a letter with no “-” (e.g. 1A, 1B, and 1C).
The following six pairs of epistatic QTLs were identified as type (1): 4A and 13C for BW0; 5C and 6B for BW5; 1D and 7A for BW12; 3A and 19A for BW15; 1B and 6B for BW52; and 2C and 27A for BW52. The following seven pairs of epistatic QTLs were identified as type (2): 28B and 5-D for BW8; 28A and 1-J for BW9, BW14, and BW20; 26A and 1-I for BW12; 28A and 10-A for BW13; 2E and 15-A for BW20; 13A and 3-B for BW28; and 27A and 14-A for BW52. The following two pairs of epistatic QTLs were identified as type (3): 10-A and 15-A for BW0; and 4-B and 9-C for BW1.
Regarding shank length traits, four pairs of epistatic QTLs were discovered (Supplementary Tables 9 and 10). Pair 5B and 6B was categorized as type (1), whereas pair 2B and 15-A was identified as type (2). Interestingly, main-effect QTL 28A, which was associated with body weight and shank length traits, was also identified as an epistatic QTL for shank traits in combination with 1-J. A type (3) interaction was discovered between 8-A and 14-A.
Summary of all QTLs detected Table 1 shows a summary of QTLs identified in the present study. Taken together, this study identified 48 QTLs associated with growth traits (body weight and shank length) on 21 different chromosomes.
Discussion
The present long-term longitudinal study identified 48 main and epistatic QTLs that affect growth traits in chickens. These QTLs were located on 21 different chromosomes, thus suggesting that growth traits are regulated by several QTLs that are widely distributed across genomic regions. Furthermore, these loci clearly have age-specific effects on the phenotypes in 0–64 weeks, which are associated with important stages of meat and egg production in broiler and layer industries, respectively. The study enabled us to develop a holistic idea of genetic control of chicken growth traits, which includes the location of loci, mode of inheritance, and age-specificity (Goto and Tsudzuki 2017).
The present study revealed that some QTLs were found repeatedly at several stages. For instance, four QTL regions (1B, 4A, 27A, and 28A) should be robust QTLs, because they were found for both body weight and shank length traits at 7–19 different ages. Our previous QTL study of 245 F2 birds based on Oh-Shamo and White Leghorn (population focused on growth and meat-related traits) reported QTLs that affected body weight and shank length traits at 20 weeks of age in these regions on chromosomes 1, 4, and 27 (Tsudzuki et al. 2007). Since these three QTL regions were also discovered in the present study that examined 406 F2 birds based on the Oh-Shamo and White Leghorn breeds (population focused on growth and egg-related traits; Goto et al. 2011, 2014a, b), these results should be reproducible. Furthermore, the contributions of these QTLs to phenotypic variance were relatively high (5.0–21.2%, 2.2–32.8%, and 2.1–15.3% for 1C, 4A, and 27A, respectively in Supplementary Tables 3 and 5). In addition, the QTLs on chromosomes 1, 4, 27, and 28 were supported by several previous studies that used crosses of White Leghorn × Red Jungle Fowl, White Leghorn × New Hampshire, White Leghorn × Rhode Island Red, White Leghorn × Fayoumi, White Leghorn × broiler, layer × broiler, broiler × broiler, White Plymouth Rock × White Plymouth Rock, and White Plymouth Rock × Silky (e.g. Nassar et al. 2015; Podisi et al. 2013; Demeure et al. 2013; Gu et al. 2011) on the Animal QTLdb (Hu et al. 2013). Since these QTLs were found in a wide variety of breeds, they should be important common loci that regulate the growth of chickens. Therefore, it would be useful to identify causal genes underlying growth traits at these QTL regions in chickens using high-resolution mapping with many individuals in populations that have undergone many recombination events and/or using whole genome sequencing technology with a wide variety of breeds.
Our study revealed 13 QTLs (1A, 1G, 1H, 1-J, 2C, 4A, 6B, 7A, 9A, 15-A, 19A, 27A, and 28A) that affected both body weight and shank length traits, but the remaining 36 QTLs affected either body weight or shank length only. The former case implies that there may be closely linked multiple genes and/or pleiotropic genes affecting the growth of muscle and bone. On the other hand, the latter case indicates that there may be different genes that affect the growth of either muscle or bone. It is well known that there is a robust genetic correlation between body weight and shank length (Demeure et al. 2013), suggesting that body weight increases along with increased shank length. The present study also indicated relatively high phenotypic correlations between body weight and shank length at early and middle stages. To develop chickens with high body weight and low shank length, the conventional statistical breeding system will require much more time. The information about the 36 QTLs for either trait will be useful to achieve these improvements in a more direct fashion using genomic information and within a shorter time period. Moreover, we detected numerous age-specific QTLs for growth in chickens. Therefore, the spatio-temporal features of genetic control mechanisms found in the present study should be adopted by the poultry industry and basic science fields.
Under the condition of relatively high phenotypic correlations between body weight and shank length, we have a question that how independent are QTLs affecting shank length rather than body weight. Actually, since the present single-trait QTL mapping of shank length did not use body weight as a covariate, some QTLs for shank length may have no direct effect on shank length (due to direct and indirect effects on body weight). To distinguish direct and indirect effects of the QTLs, QTL-directed dependency graph (QDG) mapping is more suitable statistical method using the corrected multiple phenotypes (Chaibub Neto et al. 2008). QDG mapping can discover the causal interrelationship among correlated phenotypes and distinguish whether the QTL has direct and/or indirect effect to the trait by comparing the results between QDG mapping and single-trait QTL mapping (Yang et al. 2011). Therefore, further analysis such as QDG mapping using correlated phenotypes will be needed to know independent effect on shank length and deeper interrelationship between shank length and body weight.
Approximately 60% of the alleles derived from the Oh-Shamo breed increased the phenotypic values, corresponding to the observation that the phenotypic Oh-Shamo values were higher than those of the White Leghorn breed. However, there were QTLs with effects in the opposite direction in approximately 40% of the main-effect QTLs, even in the condition that Oh-Shamo phenotypes were significantly higher than White Leghorn phenotypes in both traits. Regardless of large phenotypic differences in parental breeds, the ratio of allelic effect increasing phenotypic value was approximately 60:40, which shows close to random (50:50). This phenomenon of quantitative traits was seen in other organisms, including plants and animals, and it will be identified as one of the causes of “transgressive segregation” (Rieseberg et al. 1999). Therefore, the phenomenon could represent a general biological principal in which some loci increase the trait value and the others decrease it, regardless of the differences between the phenotypes of parental breeds. Generally speaking, most breeds will have a large variety of alleles that increase and decrease the traits of interest. Therefore, untapped genetic resources in the world such as indigenous chicken breeds, and traditional and village chickens should be analyzed more frequently because of their great potential to have several alleles influencing trait variation (Goto and Tsudzuki 2017).
All QTLs detected in the present study exhibited age-specific effects on the growth traits of chickens. Most of the loci have a continuous effect on the direction of the phenotype, but some loci had different effects on the phenotype (e.g. 24A for body weight and 2B and 2C for shank length). For instance, at 24A QTL, the Oh-Shamo allele increased BW40 but decreased BW44, and we believe that there are two possible explanations for this phenomenon. One is that there may be two closely linked loci with effects on the phenotype in both positive and negative directions, and this was also reported in body weight QTLs in mice (Mollah and Ishikawa 2011). The other is that the locus has a pleiotropic effect in opposite directions on the trait value associated with age specificity. However, there is no suitable example of this case in the literature, to the best of our knowledge. In addition, there are some QTLs (1E and 1F for body weight and 1H and 2C for shank length) that were detected non-consecutively during developmental stages. For instance, the 2C QTL affected shank length in the early (4 weeks), middle (15 and 16 weeks), and late (44 weeks) stages, and this phenomenon could be caused by either of the two possibilities described below. One is that closely linked and age-specific QTLs may be located in the region, and the other is that a QTL may influence several growth stages. Since our results showed relatively large confidence intervals for each QTL, additional high-resolution mapping with more individuals and population genomics data from other chicken populations (Goto and Tsudzuki 2017) are required to obtain concrete results.
Fourteen pairs of epistatic QTLs were detected in the present study, and these loci were categorized into three interaction types that were examined in our previous studies of chickens and mice (Goto et al. 2011, 2014b; Yoshida et al. 2013; Ishikawa et al. 2005). The results indicated some limitations and advances of the present study, and one limitation was the lack of a sufficient number of individuals. We think that the present mapping population (406 F2 individuals) has less power to detect epistatic QTL because epistatic QTL studies of populations are at their most powerful if they use good-quality data from 500 or more F2 individuals (Carlborg and Haley 2004). Carlborg et al. (2004) described that high power to detect epistasis can only be expected in reasonably large datasets with high-quality phenotypic measurements and highly informative markers. The present study fits at least one factor because we used only highly informative markers which are completely traceable from the F2 to the parental breeds. And also, Carlborg et al. (2004) mentioned that (1) an exotic cross such as red jungle fowl × White Leghorn has some advantages than a cross of broiler × layer, (2) their study using 466 F2 individuals in the broiler × layer cross shows potential to detect epistasis in the moderately sized experiments, and (3) mapping epistatic QTLs can be valuable for experimental datasets of limited population size that are initially not designed for detection of epistasis. Therefore, we decided to conduct the present QTL mapping of main-effect QTL and subsequently epistatic QTL using the exotic type of cross (Oh-Shamo × White Leghorn) and the limited population size. In addition, recent extreme QTL (X-QTL) analyses of flies have used several thousand individuals (Shorter et al. 2015), but there are several problems associated with domestic animal research in comparison with studies of model organisms. For instance, greater rearing space, feeding costs, and time are needed in domestic animal research because of body size and reproductive cycle differences. To overcome these problems, novel experimental setups and statistical methods that target higher-order epistasis are needed to unravel the complex genetic architecture of quantitative traits.
On the other hand, an advantage of the present study is that the tip of a complex genetic interaction network linked to the growth traits of chickens was uncovered (Fig. 2). The network consisting of allelic combinations of 28 QTLs clearly indicated a complex and highly polygenic genetic architecture of growth in chickens. Therefore, it is notable that a single QTL mapping population enabled us to discover a holistic genetic network. Moreover, the present study may provide an opportunity to experimentally validate gene–gene interactions in the future. Thus, further studies of the genetic interactions underlying quantitative variation in growth will provide useful insights for advancing animal breeding methods, and it could help us understand the causes of missing heritability.

Genetic network of QTLs associated with body weight (BW) and shank length (SL) traits detected in the present study. Forty-eight QTLs were mapped on 21 different chromosomes. QTLs highlighted with circles and squares indicate main-effect QTLs and epistatic QTLs with no main effect, respectively. Light and dark gray lines indicate epistatic QTL pairs associated with body weight and shank length traits, respectively. See Table 1 and Supplementary Tables 7–10 for details of each QTL
The present longitudinal QTL study with a population resulting from Oh-Shamo and White Leghorn breeds clearly indicated that chicken growth is controlled by numerous main and epistatic QTLs with age-specific effects. This study also revealed the importance of using of indigenous genetic resources for QTL analyses in chickens. The results of this study represent importance of not only cross-sectional but also longitudinal growth data for further understanding of the complex genetic architecture in animal.
References
Ankra-Badu GA, Pomp D, Shriner D, Allison DB, Yi N (2009) Genetic influences on growth and body composition in mice: multilocus interactions. Int J Obes 33:89–95
Carlborg O, Haley CS (2004) Epistasis: too often neglected in complex trait studies? Nat Rev Genet 5:618–625
Carlborg O, Hocking PM, Burt DW, Haley CS (2004) Simultaneous mapping of epistatic QTL in chickens reveals clusters of QTL pairs with similar genetic effects on growth. Genet Res 83:197–209
Chaibub Neto E, Ferrara CT, Attie AD, Yandell BS (2008) Inferring causal phenotype networks from segregating populations. Genetics 179:1089–1100
Demeure O, Duclos MJ, Bacciu N, Le Mignon G, Filangi O, Pitel F, Boland A, Lagarrigue S, Cogburn LA, Simon J, Le Roy P, Le Bihan-Duval E (2013) Genome-wide interval mapping using SNPs identifies new QTL for growth, body composition and several physiological variables in an F2 intercross between fat and lean chicken lines. Genet Sel Evol 45:36
Goto T, Tsudzuki M (2017) Genetic mapping of quantitative trait loci for egg production and egg quality traits in chickens: a review. J Poult Sci 54:1–12
Goto T, Ishikawa A, Onitsuka S, Goto N, Fujikawa Y, Umino T, Nishibori M, Tsudzuki M (2011) Mapping quantitative trait loci for egg production traits in an F2 intercross of Oh-Shamo and White Leghorn chickens. Anim Genet 42:634–641
Goto T, Ishikawa A, Yoshida M, Goto N, Umino T, Nishibori M, Tsudzuki M (2014a) Quantitative trait loci mapping for external egg traits in F2 chickens. J Poult Sci 51:375–386
Goto T, Ishikawa A, Goto N, Nishibori M, Umino T, Tsudzuki M (2014b) Mapping of main-effect and epistatic quantitative trait loci for internal egg traits in an F2 resource population of chickens. J Poult Sci 51:118–129
Gu X, Feng C, Ma L, Song C, Wang Y, Da Y, Li H, Chen K, Ye S, Ge C, Hu X, Li N (2011) Genome-wide association study of body weight in chicken F2 resource population. PLoS One 6:e21872
Hu ZL, Park CA, Wu XL, Reecy JM (2013) Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res 41:D871–D879
Ishikawa A, Hatada S, Nagamine Y, Namikawa T (2005) Further mapping of quantitative trait loci for postnatal growth in an inter-sub-specific backcross of wild mus musculus castaneus and C57BL/6J mice. Genet Res 85:127–137
Kass RE, Raftery AE (1995) Bayes Factors. J Am Stat Assoc 90:773–795
Kenny-Hunt JP, Vaughn TT, Pletscher LS, Peripato A, Routman E, Cothran K, Durand D, Norgard E, Perel C, Cheverud JM (2006) Quantitative trait loci for body size components in mice. Mamm Genome 17:526–537
Mackay TFC (2014) Epistasis and quantitative traits: using model organisms to study gene–gene interactions. Nat Rev Genet 15:22–33
Manly KF, Cudmore RH, Meer JM (2001) Map manager QTX, cross-platform software for genetic mapping. Mamm Genome 12:930–932
Mollah MBR, Ishikawa A (2011) Intersubspecific subcongenic mouse strain analysis reveals closely linked QTLs with opposite effects on body weight. Mamm Genome 22:282–289
Nassar MK, Goraga ZS, Brockmann GA (2015) Quantitative trait loci segregating in crosses between new hampshire and white leghorn chicken lines: IV. Growth performance. Anim Genet 46:441–446
Petersen L, Schnohr P, Sørensen TIA (2004) Longitudinal study of the long-term relation between physical activity and obesity in adults. Int obes Relat Metab Disord 28:105–112
Podisi BK, Knott SA, Dunn IC, Law AS, Burt DW, Hocking PM (2011) Overlap of quantitative trait loci for early growth rate, and for body weight and age at onset of sexual maturity in chickens. Reproduction 141:381–389
Podisi BK, Knott SA, Burt DW, Hocking PM (2013) Comparative analysis of quantitative trait loci for body weight, growth rate and growth curve parameters from 3 to 72 weeks of age in female chickens of a broiler-layer cross. BMC Genet 14:22
Reynaud CA, Anquez V, Grimal H, Weill JC (1987) A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell 48:379–388
Rieseberg LH, Archer MA, Wayne RK (1999) Transgressive segregation, adaptation and speciation. Heredity 83:363–372
Shorter J, Couch C, Huang W, Carbone MA, Peiffer J, Anholt RR, Mackay TF (2015) Genetic architecture of natural variation in Drosophila melanogaster aggressive behavior. Proc Natl Acad Sci 112:E3555–E3563
Tsudzuki M (2003) Japanese native chickens, In: Chang HL, Huang YC (eds). The relationship between indigenous animals and humans in APEC region, The Chinese Society of Animal Science, Tainan, p 91–116
Tsudzuki M, Onitsuka S, Akiyama R, Iwamizu M, Goto N, Nishibori M, Takahashi H, Ishikawa A (2007) Identification of quantitative trait loci affecting shank length, body weight and carcass weight from the Japanese cockfighting chicken breed, Oh-Shamo (Japanese Large Game). Cytogenet Genome Res 117:288–295
Venturini GC, Cruz VAR, Rosa JO, Baldi F, El Faro L, Ledur MC, Peixoto JO, Munari DP (2014) Genetic and phenotypic parameters of carcass and organ traits of broiler chickens. Genet Mol Res 13:10294–10300
Yandell BS, Mehta T, Banerjee S, Shriner D, Venkataraman R, Moon JY, Neely WW, Wu H, von Smith R, Yi N (2006) R/qtlbim: QTL with bayesian interval mapping in experimental crosses. Bioinformatics 23:641–643
Yang B, Navarro N, Noguera JL, Munoz M, Guo TF, Yang KX, Ma JW, Folch JM, Huang LS, Perez-Enciso M (2011) Building phenotypic networks to improve QTL detection: a comparative analysis of fatty acid and fat traits in pigs. J Anim Breed Genet 128:329–343
Yi N, Yandell BS, Churchill GA, Allison DB, Eisen EJ, Pomp D (2005) Bayesian model selection for genome-wide epistatic quantitative trait loci analysis. Genetics 170:1333–1344
Yoshida M, Ishikawa A, Goto T, Goto N, Nishibori M, Tsudzuki M (2013) QTL Mapping for meat color traits using the F 2 intercross between the Oh-Shamo (Japanese Large Game) and White Leghorn Chickens. J Poult Sci 50:198–205
Acknowledgements
We thank all the members of Laboratory of Animal Breeding and Genetics in Hiroshima University for their support of data collection.
Funding
This work was supported in part by a grant in aid for Scientific Research (B) (#19380159) from the Japan Society for the Promotion of Science awarded to M.T. and A.I.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Tatsuhiko Goto declares that he has no conflict of interest. Akira Ishikawa declares that he has no conflict of interest. Masahide Nishibori declares that he has no conflict of interest. Masaoki Tsudzuki declares that he has no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Goto, T., Ishikawa, A., Nishibori, M. et al. A longitudinal quantitative trait locus mapping of chicken growth traits. Mol Genet Genomics 294, 243–252 (2019). https://doi.org/10.1007/s00438-018-1501-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-018-1501-y