
Abstract
Infectious diseases pose significant threats to the catfish industry. Enteric septicemia of catfish (ESC) caused by Edwardsiella ictaluri is the most devastating disease for catfish aquaculture, causing huge economic losses annually. Channel catfish and blue catfish exhibit great contrast in resistance against ESC, with channel catfish being highly susceptible and blue catfish being highly resistant. As such, the interspecific backcross progenies provide an ideal system for the identification of quantitative trait locus (QTL). We previously reported one significant QTL on linkage group (LG) 1 using the third-generation backcrosses, but the number of founders used to make the second- and third-generation backcross progenies was very small. Although the third-generation backcross progenies provided a greater power for fine mapping than the first-generation backcrosses, some major QTL for disease resistance may have been missing due to the small numbers of founders used to produce the higher generation backcrosses. In this study, we performed a genome-wide association study using first-generation backcrosses with the catfish 690 K SNP arrays to identify additional ESC disease resistance QTL, especially those at the species level. Two genomic regions on LG1 and LG23 were determined to be significantly associated with ESC resistance as revealed by a mixed linear model and family-based association test. Examination of the resistance alleles indicated their origin from blue catfish, indicating that at least two major disease resistance loci exist among blue catfish populations. Upon further validation, markers linked with major ESC disease resistance QTL should be useful for marker-assisted introgression, allowing development of highly ESC resistant breeds of catfish.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Disease resistance describes the overall capacity and efficiency of the host to cope with infections, which is composed of the innate and adaptive immunities. The innate immune system provides physical and chemical barriers, senses pathogens through various receptors to activate microbial defense and stimulate the adaptive immune response (Medzhitov 2007). The adaptive immune system comprises of T and B lymphocytes and their mediated processes. T lymphocytes, including T-helper (Th) cells (CD4+ T cells) and cytotoxic T cells (CD8+ T cells), recognize antigenic peptides bound to major histocompatibility complex (MHC) via antigen-specific extracellular receptors. Cytotoxic T cells induce apoptosis of infected cells or kill target cells (Andersen et al. 2006), while Th cells differentiate into effector cells (Th1, Th2, and Th17) upon antigen stimulation, producing distinct sets of cytokines for antigen-specific activation of innate host-defense cells such as macrophage and granulocytes as well as for the activation of B cells to generate antigen-specific antibodies (Medzhitov 2007; Annunziato et al. 2009). In addition, B cells can recognize antigens by directly binding to them. The various modules of innate immunity cooperate with cell-mediated and humoral immune responses of adaptive immunity, providing effective protection against infection for vertebrates.
Immunogenetic analysis in fish species has been rare. The practical approaches have focused on the identification of disease resistance-associated DNA markers for marker-assisted selection. The interest for the identification of genetic markers and genomic regions for resistance against diseases in aquaculture species is increasing because rapid progress can be made in this area to reduce disease problems and enhance aquaculture production without deep analysis of the immunological and molecular mechanisms. Among various approaches, quantitative trait locus (QTL) mapping and genome-wide association study (GWAS) have been the most useful approaches for disease resistance studies with aquaculture species.
Disease resistance QTL have been identified in various aquaculture species (For a recent review, see Abdelrahman et al. 2017). For example, QTL have been identified in rainbow trout (Oncorhynchus mykiss) for resistance against infectious pancreatic necrosis (Ozaki et al. 2007), infectious hamatopoietic necrosis (Campbell et al. 2014), whirling disease (Baerwald et al. 2011), and bacterial cold water disease (Liu et al. 2015; Palti et al. 2015). In Asian seabass (Lates calcarifer), QTL have been detected for viral nervous necrosis disease resistance (Liu et al. 2016a), and for iridovirus resistance (Wang et al. 2017a). For Japanese flounder (Paralichthys olivaceus), a single major genetic locus was found to explain 50% of the total variation of disease resistance to lymphocystis disease (Fuji et al. 2006), and the linked DNA marker was successfully used for marker-assisted selection (Fuji et al. 2007). In Atlantic salmon (Salmo salar), QTL associated with resistance to Salmon Rickettsial Syndrome were identified (Correa et al. 2015). Moreover, a major QTL in Atlantic salmon that explained most of the genetic variance for resistance to infectious pancreatic necrosis has been commercially utilized by breeding companies (Houston et al. 2008; Moen et al. 2009).
Catfish is the primary aquaculture species in the United States. Channel catfish (Ictalurus punctatus) used to be the major cultured species. In recent years, the interspecific hybrid [channel catfish female × blue catfish (Ictalurus furcatus) male] is increasingly used by the catfish industry because of its superiority for disease resistance, growth rate, fillet yield, and harvestability (Dunham et al. 2008; Lucas and Southgate 2012). The interspecific hybrids not only promote the aquaculture production but also serve as an excellent model for the analysis of QTL associated with traits such as disease resistance. With enteric septicemia of catfish (ESC), blue catfish is very resistant while channel catfish is highly susceptible (Wolters and Johnson 1994; Wolters et al. 1996). Therefore, the interspecific hybrid provides an ideal system for genetic analysis of ESC disease resistance.
ESC is the most prevalent and devastating disease in catfish farming (Hawke 1979; Hawke et al. 1981; Plumb and Hanson 2011), leading to economic loss of $40–60 million in the US catfish industry annually (Shoemaker et al. 2009). In addition to channel catfish, cases have been reported worldwide in white catfish (Ameiurus catus), brown bullhead catfish (Ameiurus nebulosus), yellow catfish (Pylodictis olivaris), striped catfish (Pangasianodon hypophthalmus), walking catfish (Clarias batrachus), and species of fish other than catfish (Hawke et al. 1981; Kasornchandra et al. 1987; Ye et al. 2009; Dong et al. 2015). ESC occurs most often within a specific temperature range between 20 and 28 °C (Hawke et al. 1998). This disease causes enteritis and septicemia, characterized by ulcerative skin lesions, petechial hemorrhages around the mouth and abdomen, and pimples or holes between eyes (Areechon and Plumb 1983; Hawke et al. 1998). ESC is caused by Edwardsiella ictaluri, a Gram-negative, rod-shaped bacterium of the family Enterobacteriacae. E. ictaluri can enter catfish through the intestinal tract, the nares, the gills, and the skin (Hawke et al. 1998; Menanteau-Ledouble et al. 2011). E. ictaluri is internalized in leukocytes of channel catfish, particularly in macrophages (Miyazaki and Plumb 1985; Shotts et al. 1986; Baldwin and Newton 1993). The initial detection of E. ictaluri in the internal organs can occur as early as 15 min post infection by gastric intubation (Baldwin and Newton 1993). Bacteria are released in the blood, and colonize other organs within 48 h (Menanteau-Ledouble 2009). The earliest lesions were observed microscopically at 2 days after an immersion challenge of channel catfish (Newton et al. 1989). Although the disease progression and pathology of ESC are well characterized, little is known about the molecular mechanism underlying the pathogenesis of E. ictaluri and associated disease resistance of the host.
Genomic resources of catfish have been made available to facilitate GWAS in catfish, including a large number of SNPs (Liu et al. 2011; Sun et al. 2014), the high-density SNP arrays (Liu et al. 2014; Zeng et al. 2017), and the reference genome sequence (Liu et al. 2016b). Using the 250 K SNP array, several GWAS analyses have been conducted with performance and production traits of catfish, including disease resistance against columnaris (Geng et al. 2015), hypoxia tolerance (Wang et al. 2017b; Zhong et al. 2017), heat tolerance (Jin et al. 2017), albinism (Li et al. 2017a), body weight (Li et al. 2017b), body conformation (Geng et al. 2017a) and head size (Geng et al. 2016). With ESC disease resistance, GWAS analysis was conducted using the third-generation backcrosses (Zhou et al. 2017). The use of higher generation backcrosses could have provided greater powers for the detection of QTL in a smaller genomic region because of accumulated chromosomal recombinations. However, some significant QTL may have been undetected if such QTL were not included in the few individuals of the first-generation backcrosses used as founders of the higher generation backcrosses.
The QTL by definition must be there in the first generation. However, it would go undetected if the appropriate allelic variation was missing. In this study, we utilized the first-generation backcrosses for determining the genomic regions associated with ESC disease resistance, to ensure detection of species-level QTL, especially those offered from blue catfish.
Materials and methods
Experimental fish, bacterial challenge and sample collection
The bacterial challenge experiment was performed with 1-year-old catfish (average 14.8 cm) produced by crossing male F1 hybrid catfish (channel catfish female × blue catfish male) with female channel catfish (Fig. 1). The female channel catfish were Marion strain (Dunham and Smitherman 1984), including the maternal parent of the F1 hybrid catfish. Four backcross families were used, and the grandparents among families were different. A total of 1200 experiential fish were randomly selected (300 fish per family) at the Auburn University Fish Genetics Facility and were distributed in four tanks (300 L water) by family. A control group containing 200 fish was reared in another tank. The average temperature was maintained at 28 °C, and oxygen level was controlled at optimal level by aeration.

Backcross strategy for the development of the experimental population
The infestation with E. ictaluri was carried out after acclimation. The bacteria challenge procedure was conducted as previously described (Wang et al. 2013). The bacteria were cultured from a single colony, re-isolated from a symptomatic fish, and biochemically confirmed. A single colony was cultured in brain heart infusion (BHI) medium and incubated at 28 °C overnight. The concentration of the bacteria was determined using colony forming unit (CFU) per mL by plating tenfold serial dilution onto BHI agar plates. The infestation was conducted by immersion exposure for 2 h at a final bacterial concentration of 2 × 107 CFU/mL, whereas control fish were treated with an equal volume of sterile BHI medium. Moribund catfish were continuously sampled by taking blood, and the sampling time was recorded. Fish for further genotyping were selected from the extremes of the disease resistance distribution for each family based on the selective genotyping method (Darvasi and Soller 1992).
DNA isolation and genotyping
DNA was isolated using standard protocols. Briefly, the blood samples in cell lysis solution were incubated at 55 °C overnight. Protease K and protein precipitation solution were used to remove proteins. Extracted DNA was precipitated with isopropanol and collected by brief centrifugation, washed twice with 70% ethanol, air-dried, and rehydrated in TE buffer (pH 8.0). DNA was quantified by spectrophotometry (NanoDrop Technologies, Wilmington, DE, USA) and checked by 1% agarose gel electrophoresis stained with ethidium bromide for integrity. DNA was diluted to 50 ng/µL with a final volume of 10 µL. Genotyping with the catfish 690 K SNP array was performed at GeneSeek (Lincoln, Nebraska, USA). Genotype callings were generated using the Axiom Analysis Suite software.
Statistical analysis
To identify association between SNPs and resistance to E. ictaluri, the single SNP test was performed using the SVS software package (SNP & Variation Suite, Version 8.3) and PLINK (Version 1.9) (Purcell et al. 2007). Quality control was performed for samples and markers. Samples with genotype missingness larger than 5% were removed. The markers with a minor allele frequency (MAF) lower than 0.05 or a call rate lower than 95% were excluded from the analysis. Linkage disequilibrium (LD) pruning was then conducted to achieve a set of independent SNPs with a window size of 50 SNPs, window increment of 5, and r2 threshold of 0.5. With the independent SNP markers, identity-by-state (IBS) distances were estimated to reflect relatedness between pairs of samples, and the population structure was analyzed by principal component analysis (PCA).
Efficient Mixed-Model Association eXpedited (EMMAX) analyses using all qualified SNPs were conducted with the first three principal components and body weight as covariates (Kang et al. 2010). The model is listed as follows:
where Y is the vector of phenotype; β is the coefficient vector of fixed effects including first three principal components and fish body weight; u is the vector of the random effect, Var(u) = Gσ2 g, where σ2 g is the additive genetic variance and G is the genomic kinship matrix using the IBS; e is the vector of random residuals; X is the matrix of fixed effects and Z is the matrix of random additive genetic effects.
The Manhattan plot of the − log10 (P value) was generated using the SVS software, with the genetic marker map according to channel catfish genome sequence (Version Coco1.2, Liu et al. 2016a, b). The threshold P value for genome-wide significance was calculated based on Bonferroni correction with estimated number of independent markers and LD blocks.
Family-based association test for quantitative traits (QFAM) was conducted using PLINK (Purcell et al. 2007). QFAM partitions the genotypes into between- and within- family components, and the within-family component is free of sample structure (Fulker et al. 1999; Abecasis et al. 2000). The QFAM-Within procedure in PLINK performed a simple linear regression of phenotype on the within-family component of genotype, and then used permutations to correct for family structure. The model is
where yij denotes the phenotype of individual j in family i; µ is the population mean; βb is the vector of between-family effect and βw is the vector of within-family effect; bi = (Σgij)/ni if parental genotypes are unknown (gij: genotype of the jth offspring in the ith family), and bi = (giF + giM)/2 if parental genotypes are available (giF: genotype of father, giM: genotype of mother); wij = gij − bi.
Sequence analysis
Genes within ± 1 Mb of associated regions were predicted from catfish genome sequence (Liu et al. 2016b) using FGENESH (Solovyev et al. 2006) and annotated by BLAST against the NCBI-nr database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/). Synteny analyses and catfish genome information from the NCBI were utilized to provide supporting evidence for the proper annotation.
Results
Experimental fish and sample structure
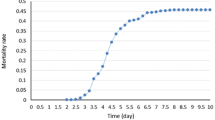
A total of 1200 catfish from four families were challenged with E. ictaluri, and 288 fish with extreme phenotype were chosen for genotyping. The mortality rate after infection is shown in Fig. 2. The information of catfish samples utilized in this study is summarized in Table 1. PCA analysis was conducted using eigenvalues as coordinates to visualize the sample structure. As shown in Fig. 3, each family was grouped into a separate cluster and the four families were distantly related.

Mortality rate of the first-generation backcrosses after Edwardsiella ictaluri infection

Sample structure identified by principal component analysis with two major principal components
QTL for ESC resistance
A total of 407,196 SNPs were kept after filtering out SNP markers with genotyping errors, MAF < 5%, or call rate < 95%. The number of independent SNPs and LD blocks was 15,061 after LD pruning, and the genome-wide threshold for statistical significance was calculated to be 3.32e−6 (0.05/15,061) with − log10 (P value) = 5.48. The threshold of − log10(P value) for suggestive association was arbitrarily set as 5 (Geng et al. 2016).
The Manhattan plots generated from EMMAX and QFAM are shown in Fig. 4a, b, respectively. In general, the association results of EMMAX and QFAM were positively correlated. EMMAX showed one genome-wide significant region on linkage group (LG) 23 and two suggestively associated regions on LG1 and LG14, while QFAM revealed two genome-wide significant regions on LG23 and LG1 as well as one suggestive region on LG1. Generally, QFAM is more robust in correcting family structure when using family-based samples than EMMAX (Geng et al. 2017b). In the following sections, we mainly describe the identified regions according to the results generated by QFAM.

Manhattan plot of genome-wide association analysis for ESC disease resistance generated from EMMAX (a) and QFAM (b). The black solid line indicates the threshold P value for genome-wide significance. The grey solid line indicates the threshold P value for significance of “suggestive association”. The red box represents the shared QTL using two methods. (Color figure online)
SNPs associated with ESC resistance are listed in Table S1. Forty-five genome-wide significant SNPs were found on LG1 from 33,233,001 to 33,712,554, spanning approximately 479.6 Kb. The most significant SNP (AX-157732970) on LG1 reached the genome-wide significance level with − log10 (P value) = 6.05. Similarly, three significantly associated SNPs were located on LG23 in genomic region from 7,793,018 to 8,136,119, spanning approximately 343.1 Kb. The − log10 (P value) of the most significant SNP (AX-157695826) was 6.25. In addition, 20 SNPs were identified within the suggestive QTL on LG1 from 5,052,936 to 9,817,492. Based on the phenotype and the SNPs placed on the catfish 690 K SNP array (Zeng et al. 2017), the resistance allele of associated SNPs (Table S1) as well as their origins could be determined. Four significant SNPs and four suggestively associated SNPs are all interspecific, which means they are SNPs from interspecific origin and not intraspecific variants. For these interspecific SNPs, all resistance alleles originated from blue catfish. EMMAX was utilized to investigate the contribution of significantly associated QTL to the phenotypic variance. The most significant SNP was used to represent the corresponding significant region (Geng et al. 2015), with AX-157732970 on LG1 and AX-157695826 on LG23 accounting for 6.8 and 10.4% of the phenotypic variance, respectively. These two loci alone would indicate a minimum heritability of 0.17 in a first-generation backcross population. According to the phenotypic variance (3827.5) calculated in SVS, fish with the “good” allele of AX-157732970 and AX-157695826 will live 16.1 and 20.0 h longer, respectively.
Genes within the associated QTL for ESC resistance
To explore the potential genes involved in ESC resistance, the ± 1 Mb regions around associated SNPs were examined. Synteny analyses were conducted to compare the extend regions around the significant SNPs. The conserved synteny was observed between catfish and zebrafish, as shown in Figs. 5 and 6. A set of 43 genes were identified within the significantly associated region on LG1 (Table S2). Of these genes, seven genes were found to have known functions in immunity (Table 2), including NCK adaptor protein 1 (nck1), angiotensin II receptor type 1 (agtr1), transient receptor potential cation channel subfamily C member 1 (trpc1), abl interactor 1 (abi1), Rap1-GTP-interacting adaptor molecule (apbb1ip), ARP3 actin-related protein 3 homolog B (actr3b), and vav guanine nucleotide exchange factor 3 (vav3). Within the significant QTL on LG23, 45 genes were detected (Table S3) and three genes had immune-related functions (Table 2), including mannose receptor C-type 1 like (mrc1l), protein kinase C theta (prkcq), and GATA binding protein 3 (gata3). Moreover, 49 genes were immune-related, of a total of 271 genes identified in the suggestive region on LG1 (Table S4).

Genes within the significant region associated with ESC resistance on LG1. a Regional Manhattan plot for the QTL on LG1. The yellow horizontal line indicates the threshold P value for genome-wide significance. The blue horizontal line indicates the threshold P value for significance of “suggestive association”. b Synteny analysis between catfish and zebrafish. Immune-related genes in channel catfish are highlighted in red. (Color figure online)

Genes within the significant region associated with ESC resistance on LG23. a Regional Manhattan plot for the QTL on LG23. The yellow horizontal line indicates the threshold P value for genome-wide significance. The blue horizontal line indicates the threshold P value for significance of “suggestive association”. b Synteny analysis between catfish and zebrafish. Immune-related genes in channel catfish are highlighted in red. (Color figure online)
Correlation of the SNPs with ESC resistance
Conditioned analyses were performed to determine whether the associations detected on a linkage group were independent (Nishimura et al. 2012). The lead SNP on each associated QTL was included as a covariate in the mix linear model. After conditioning, associations of the surrounding SNP on the same linkage group disappeared, while SNPs on other linkage groups generally remained the same, indicating there was no independently associated SNP marker within the same linkage group.
Discussion
Identification of polymorphisms affecting ESC disease resistance could facilitate effective breeding through marker-assisted selection and introgression, and thereby reduce losses for the catfish aquaculture industry. Here, GWAS was conducted using the channel catfish × blue catfish backcross progenies genotyped with the newly designed catfish 690 K SNP arrays. We successfully identified QTL associated with ESC resistance on LG1 and LG23. Channel catfish and blue catfish exhibit great contrast in the resistance to ESC, with blue catfish being highly resistant, almost never becoming infected naturally and having low mortality when challenged artificially (Bilodeau-Bourgeois et al. 2008; Hanson 2006; Wolters and Johnson 1994; Wolters et al. 1996). In the backcross progenies, both genes and the disease resistance traits are segregating, allowing mapping of disease resistance to chromosomal locations. Although the F2 intercrosses of hybrids between the two species with phenotypic divergence are appropriate populations for mapping QTL (Cnaani et al. 2003; Poompuang and Hallerman 1997), it is extremely difficult to produce the interspecific F2 hybrids due to the very low hatching rate (Dunham and Argue 2000). Therefore, the backcross progenies provide a best available alternative for the study of interspecific ESC resistance/susceptibility-associated QTL.
Two methods, EMMAX and QFAM, were utilized for family-based samples in the present study. Both are effective for correcting population stratification which can lead to biased or spurious results. The statistical approaches of EMMAX and QFAM to control the population stratification are different. EMMAX uses a pairwise relatedness matrix as random effect to correct for sample structures including hidden relatedness and population stratification (Kang et al. 2010). QFAM partitions the genotypes into between- and within- family components (Fulker et al. 1999; Abecasis et al. 2000). The within-family analysis is robust to population stratification, which assesses transmission of alleles within a family, but without making use of allelic association observed across families. When using family-based samples, QFAM was found to be more robust in correcting family structure than EMMAX (Geng et al. 2017b). Family-based samples were utilized in the present study and the suggestive QTL on LG14 found by EMMAX but not by QFAM was suggested to be false positive due to family structure. Furthermore, the suggestive region on LG1 was not found by EMMAX but identified by QFAM in this study. Geng et al. (2016) reported that QFAM possessed more power compared with EMMAX in a family-based study. Recent work has shown that inclusion of candidate markers in the relatedness matrix could lead to decreased power due to double-fitting of candidate markers in the mix linear model (Yang et al. 2014). Therefore, QFAM results were chosen for further analysis.
Examination of the associated SNPs revealed that the superior disease resistance alleles were from blue catfish. Four suggestively associated SNPs and four significant SNPs including the most significant SNP (AX-157732970) on LG1 were all interspecific. All of the resistance alleles were found to be derived from blue catfish, explaining the very strong resistance of blue catfish against the ESC disease. For LG23, all associated SNPs were channel catfish-specific, which means the single nucleotide variation on these loci occurred only in channel catfish. Further investigation showed that all the SNPs that could be mapped to blue catfish genome sequence are homologous with the two resistance alleles. The results implied that certain blue catfish alleles were associated with increased ESC resistance.
Within the two significantly associated regions on LG1 and LG23 (indicated by red boxes in Fig. 4), an examination of genes and related pathways provided insights into the molecular mechanisms underlying ESC resistance. The most studied, closest relative of catfish, zebrafish, has 696 genes involved in the immune system process in PANTHER database (http://www.pantherdb.org). The channel catfish has 27,143 genes based on NCBI annotation. The approximate proportion of immune genes in the genome is 0.026. The proportion of immune genes within our QTL region for LG1 and LG23 is 0.163 (7/43) and 0.067 (3/45), which in the case of LG1 is more than expected (Chi-square test, P < 0.001) compared to the rest of the genome. The immune-related genes observed in the significant QTL were found to be mainly involved in phagocytosis (Fig. 7) and T-cell activation (Fig. 8). Phagocytosis is a principal component of the innate immunity in which phagocytes, including macrophages, neutrophils and dendritic cells, internalize targets in an actin-dependent manner (Botelho and Grinstein 2011). E. ictaluri is internalized in catfish phagocytes after infection, especially in macrophages (Miyazaki and Plumb 1985; Shotts et al. 1986; Baldwin and Newton 1993), suggesting that phagocytic activities play crucial roles in immunity for combating ESC. T cells have a central role in adaptive immunity by regulating immune responses of various phagocytes and B cells through cytokine secretion or by directly destroying antigen-bearing cells (Medzhitov 2007). Russo et al. (2009) demonstrated that macrophages from vaccinated fish were more efficient in rapid clearance of infection upon re-exposure to virulent E. ictaluri, reflecting that memory lymphocytes were involved and lymphocytes in adaptive immunity were important in enhancing macrophage activities during immune responses.

Signal transduction pathway involved in Fcγ receptor-mediated phagocytosis. Corresponding proteins of immune-related genes identified in this study are highlighted in red. SFKs, Src family tyrosine kinase; Src, proto-oncogene tyrosine-protein kinase Src; SLP76 Src homology 2 domain-containing leukocyte protein of 76 kilodaltons, VAV, VAV guanine nucleotide exchange factor, Rac, small GTPase Rac; Cdc42, small GTPase Cdc42; NCK, NCK adaptor protein; WASP, Wiskott–Aldrich syndrome protein; WAVE, WASP family Verprolin-homologous protein; ABI1, abl interactor 1; Arp2/3, the actin nucleation complex Arp2/3; ADAP, Adhesion and degranulation-promoting adapter protein; SKAP55, Src kinase-associated phosphoprotein of 55 kDa; RIAM, Rap1-GTP-interacting adaptor molecule; Rap1, small GTPase Rap1. (Color figure online)

Signal transduction pathway leading to T-cell activation. Corresponding proteins of immune-related genes identified in this study are highlighted in red. LCK, LCK proto-oncogene; Src family tyrosine kinase, ZAP70, zeta chain of T-cell receptor associated protein kinase 70; SLP76, Src homology 2 domain-containing leukocyte protein of 76 kilodaltons; VAV, VAV guanine nucleotide exchange factor; Rac, small GTPase Rac; Cdc42, small GTPase Cdc42; NCK, NCK adaptor protein; WASP, Wiskott–Aldrich syndrome protein; WAVE, WASP family Verprolin-homologous protein; ABI1, abl interactor 1; Arp2/3, the actin nucleation complex Arp2/3; ADAP, Adhesion and degranulation-promoting adapter protein; SKAP55, Src kinase-associated phosphoprotein of 55 kDa; RIAM, Rap1-GTP-interacting adaptor molecule; Rap1, small GTPase Rap1; PLCγ1 ,phospholipase C-γ1; DAG, diacylglycerol; PKCθ, protein kinase C theta; PKD1, polycystin 1; transient receptor potential channel interacting, TRPC1, transient receptor potential cation channel subfamily C member 1; CaN, calcineurin; NF-κB, nuclear factor-kappa B; AP-1, activator protein-1; NFAT, nuclear factor of activated T cells. (Color figure online)
One of the similar process of both phagocytosis and T-cell activation is cytoskeleton rearrangement. In phagocytes, local polymerization of actin filaments supports the protrusion of pseudopodia that facilitates the engulfment of pathogens or particles. In T cells, cytoskeleton reorganization is crucial for the formation of immunological synapse, which is crucial for cell adhesion and T-cell activation. Four genes within the significantly associated region on LG1, including nck1, vav3, abi1, and actr3b, were involved in actin cytoskeleton reorganization. Nck proteins are adapter proteins which play pivotal role in the phagocytic process (Coppolino et al. 2001) and T-cell receptor (TCR) induced actin cytoskeleton reorganization during T-cell activation (Lettau et al. 2009). Upon the phosphorylation of tyrosine kinases, SH2 domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76) recruits Nck. Nck then promotes the recruitment of the multidomain adapter protein Wiskott–Aldrich syndrome protein (WASP) and WASP family Verprolin-homologous protein (WAVE), whose activations facilitate the actin-related protein 2/3 (Arp2/3) complex to drive actin filament formation. SLP-76 also recruit the Vav proteins, promoting the GTP-loading of Rho GTPases to facilitate the activation of WASP family proteins. The Vav family proteins (Vav1, Vav2, and Vav3) are guanine nucleotide exchange factors (GEFs) for Rho family GTPases (Hornstein et al. 2004; Tybulewicz 2005), playing crucial roles in the regulation of actin dynamics during phagocytosis (Patel et al. 2002; Hall et al. 2006) and T-cell activation (Villalba et al. 2000, 2002). Abl interactor 1 (Abi1) is crucial in actin cytoskeleton dynamics due to its participation in the WAVE complex, which stimulates Arp2/3 complex-dependent actin assembly (Innocenti et al. 2004; Kheir et al. 2005). Arp2/3 complex plays a major role in the regulation of the actin cytoskeleton (Machesky and Gould 1999), essential for actin polymerization in the phagocytic cup and subsequent particle engulfment in both FcγR- and CR3-mediated phagocytosis (May et al. 2000). Moreover, the Arp2/3 complex was involved in actin cytoskeleton remodeling during T-cell activation (Krause et al. 2000). Actin-Related Proteins ARP3 (actr3) is one subunit of the Arp2/3 complex. In the present study, the homology of actr3, actr3b, was in the significantly associated region on LG1 that associated with ESC disease resistance.
Another shared process during T-cell activation and phagocytosis is the integrin activation, where Rap1-GTP-interacting adaptor molecule (RIAM, apbb1ip) is implicated. Integrins are transmembrane heterodimers responsible for mediating cell/cell or cell/matrix adhesions (Hynes 2002), which are crucial for development, tissue maintenance and repair, immune response and hemostasis (Hynes 2002; Harburger and Calderwood 2009). Binding affinity and avidity of integrins for their ligands facilitate T-cell activation by providing stable contact with APCs and extracellular proteins (Burbach et al. 2007). A key integrin regulator is the small GTPase Ras-proximity-1 (Rap1), which enhances T-cell activation by mediating TCR-induced adhesion to intercellular adhesion molecule (ICAM) (Katagiri et al. 2000; Sebzda et al. 2002). In addition, FcγR-mediated responses have been shown to require the presence of β2 integrins, include adhesion to immune complexes and enhanced Fc-dependent phagocytosis (Ortiz-Stern and Rosales 2003).
The calcium ion (Ca2+) is the main common second messenger involved in signaling transduction following immunoreceptor activation. For the T-cell activation, TCR-induced increase in intracellular Ca2+ level is crucial for many cellular functions, including the translocation of NFAT (nuclear factor of activated T cells) to the nucleus through calcineurin (CaN) activation (Macian 2005). TRPC1, a membrane-spanning subunit of cation channel, was involved in store-operated Ca2+ entry (SOCE) by interacting with calcium release-activated calcium channel protein 1 (Orai1) and stromal interaction molecule 1 (STIM1) (Ong et al. 2007), both of which were up-regulated during T-cell activation (Lioudyno et al. 2008). Furthermore, TRPC1 was shown to be expressed in a human T-cell line (HPB-ALL) and contributed to cannabinoid-induced Ca2+ influx in these cells independently of intracellular Ca2+ store depletion (Rao and Kaminski 2006).
Pattern recognition receptors (PRRs) are well known for their significant roles in immunity. Mannose receptor C-type 1 (MRC1) is a C-type lectin primarily present on macrophages and dendritic cells. MRC1 played significant roles in both innate and adaptive immune responses against various microorganisms (Taylor et al. 2005), and it was implicated in pathogen recognition, phagocytosis, cytokine production, antigen processing and presentation, cell adhesion and migration (Apostolopoulos and McKenzie 2001; East and Isacke 2002; Taylor et al. 2005; Gazi and Martinez-Pomares 2009). Due to the crucial roles in immune responses, much work on MRC1 has been done in human and mice, with a few studies in fish as well. In grass carp (Ctenopharyngodon idella), MRC1 was significantly up-regulated after Aeromonas hydrophila infection in liver, spleen, head kidney and intestine (Wang et al. 2014). Similarly, the expression of MRC1 was dramatically induced in the spleen of tilapia (Oreochromis niloticus) at all three studied timepoints (5 h, 50 h, and 7 days) following Streptococcus iniae infection (Zhu et al. 2015). In this study, the gene mrc1 like was within the QTL on LG23, and its expression in catfish was significantly up-regulated at 3 and 72 h after E. ictaluri infection in a previous study (Li et al. 2012).
The gene prkcq was located within the associated region on LG23, coding for protein kinase C theta (PKCθ). Predominantly expressed in T lymphocytes, PKCθ plays essential roles for T-cell activation, proliferation, differentiation, survival, and cytokine production (Anderson et al. 2006; Isakov and Altman 2002; Barouch-Bentov et al. 2005; Hayashi and Altman 2007). PKCθ is the only serine/threonine protein kinase C (PKC) recruited selectively to the center of the immunological synapse following stimulation with APCs, required for T-cell activation and downstream signaling (Monks et al. 1997; Bi et al. 2001). T lymphocytes in PKCθ-deficient mice displayed impaired activation of transcription factors including NF-κB (nuclear factor-kappa B), AP-1 (activator protein-1) and NFAT (Pfeifhofer et al. 2003; Sun et al. 2000). PKCθ was found to be significantly up-regulated in intestine of Pelodiscus sinensis after intragastric challenge with lipopolysaccharide (LPS) (Xu et al. 2016).
One interesting finding of our study was that the functionally related genes in immunity were in relatively close proximity on the chromosome. Of the seven immune-related genes within the significantly associated region on LG1, six genes were implicated in phagocytosis (nck1, vav3, abi1, actr3b, apbb1ip, and agtr1), and six genes were involved in T-cell activation (nck1, vav3, abi1, actr3b, apbb1ip, and trpc1). In plants, some resistance genes often cluster together in certain chromosome, sometimes so tightly that they can be considered as complex loci (Vale et al. 2001). The structural organization may facilitate coordinated expression and function (Michalak 2008). Further studies need to be conducted to confirm the involvement of these genes and their specific function in ESC disease resistance.
Our long-term goal is to enhance catfish stocks with a broad spectrum of disease resistance as well as other favorable traits, and support a sustainable and profitable aquaculture industry. To achieve this goal, the genetic basis underlying desirable traits should be understood, especially the accurate location of QTL affecting the traits. In this study, we identified two genomic regions associated with ESC resistance on LG1 and LG23. We previously reported one QTL in LG1 using the third-generation backcrosses (Zhou et al. 2017), which was produced by mating with channel catfish female in each generation. The QTL was validated here in the first-generation backcrosses, suggesting that this QTL was operating in various populations of a broad genetic background. SNPs which were associated with ESC resistance were identified in the present study. Individuals must be generated that are homozygous for these key SNPs, identified and selected as these SNPs are likely associated with the blue catfish resistance alleles that are incompletely dominant since blue catfish are almost totally resistant to ESC, but the F1 channel catfish female × blue catfish male (heterozygous) is not totally resistant though improved. Research should continue to identify the exact location and identity of the ESC resistance alleles using more families or populations. This would allow additional options to interspecific introgression to establish the ESC resistance alleles in a homozygous state in cultured catfish. These include gene transfer or even CRISPR/Cas9 technology to mutate the channel catfish alleles into their blue catfish ESC resistance counterparts (Dunham et al. 2014).
References
Abdelrahman H, ElHady M, Alcivar-Warren A, Allen S, Al-Tobasei R, Bao L, Beck B, Blackburn H, Bosworth B, Buchanan J, Chappell J, Daniels W, Dong S, Dunham R, Durland E, Elaswad A, Gomez-Chiarri M, Gosh K, Guo X, Hackett P, Hanson T, Hedgecock D, Howard T, Holland L, Jackson M, Jin Y, Khalil K, Kocher T, Leeds T, Li N, Lindsey L, Liu S, Liu Z, Martin K, Novriadi R, Odin R, Palti Y, Peatman E, Proestou D, Qin G, Reading B, Rexroad C, Roberts S, Salem M, Severin A, Shi H, Shoemaker C, Stiles S, Tan S, Tang KFJ, Thongda W, Tiersch T, Tomasso J, Prabowo WT, Vallejo R, Steen H, Vo K, Waldbieser G, Wang H, Wang X, Xiang J, Yang Y, Yant R, Yuan Z, Zeng Q, Zhou T (2017) Aquaculture genomics, genetics and breeding in the United States: current status, challenges, and priorities for future research. BMC Genom 18:191
Abecasis GR, Cardon LR, Cookson WOC (2000) A general test of association for quantitative traits in nuclear families. Am J Hum Genet 66:279–292
Andersen MH, Schrama D, thor Straten P, Becker JC (2006) Cytotoxic T cells. J Invest Dermatol 126:32–41
Anderson K, Fitzgerald M, DuPont M, Wang T, Paz N, Dorsch M, Healy A, Xu Y, Ocain T, Schopf L, Jaffee B, Picarella D (2006) Mice deficient in PKC theta demonstrate impaired in vivo T cell activation and protection from T cell-mediated inflammatory diseases. Autoimmunity 39:469–478
Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S (2009) Type 17 T helper cells—origins, features and possible roles in rheumatic disease. Nat Rev Rheumatol 5:325–331
Apostolopoulos V, McKenzie IFC (2001) Role of the mannose receptor in the immune response. Curr Mol Med 1:469–474
Areechon N, Plumb JA (1983) Pathogenesis of Edwardsiella ictaluri in channel catfish, Ictalurus punctatus. J World Aquac Soc 14:249–260
Baerwald MR, Petersen JL, Hedrick RP, Schisler GJ, May B (2011) A major effect quantitative trait locus for whirling disease resistance identified in rainbow trout (Oncorhynchus mykiss). Heredity 106:920
Baldwin TJ, Newton JC (1993) Pathogenesis of enteric septicemia of channel catfish, caused by Edwardsiella ictaluri: bacteriologic and light and electron microscopic findings. J Aquat Anim Health 5:189–198
Barouch-Bentov R, Lemmens EE, Hu J, Janssen EM, Droin NM, Song J, Schoenberger SP, Altman A (2005) Protein kinase C-θ is an early survival factor required for differentiation of effector CD8+ T cells. J Immunol 175:5126–5134
Bi K, Tanaka Y, Coudronniere N, Sugie K, Hong S, van Stipdonk MJB, Altman A (2001) Antigen-induced translocation of PKC-θ to membrane rafts is required for T cell activation. Nat Immunol 2:556
Bilodeau-Bourgeois L, Bosworth BG, Peterson BC (2008) Differences in mortality, growth, lysozyme, and toll-like receptor gene expression among genetic groups of catfish exposed to virulent Edwardsiella ictaluri. Fish Shellfish Immunol 24:82–89
Botelho RJ, Grinstein S (2011) Phagocytosis. Curr Biol 21:R533–R538
Burbach BJ, Medeiros RB, Mueller KL, Shimizu Y (2007) T-cell receptor signaling to integrins. Immunol Rev 218:65–81
Campbell NR, LaPatra SE, Overturf K, Towner R, Narum SR (2014) Association mapping of disease resistance traits in rainbow trout using restriction site associated DNA sequencing. G3 4:2473–2481
Cnaani A, Hallerman EM, Ron M, Weller JI, Indelman M, Kashi Y, Gall GAE, Hulata G (2003) Detection of a chromosomal region with two quantitative trait loci, affecting cold tolerance and fish size, in an F2 tilapia hybrid. Aquaculture 223:117–128
Coppolino MG, Krause M, Hagendorff P, Monner DA, Trimble W, Grinstein S, Wehland J, Sechi AS (2001) Evidence for a molecular complex consisting of Fyb/SLAP, SLP-76, Nck, VASP and WASP that links the actin cytoskeleton to Fcγ receptor signalling during phagocytosis. J Cell Sci 114:4307–4318
Correa K, Lhorente JP, López ME, Bassini L, Naswa S, Deeb N, Di Genova A, Maass A, Davidson WS, Yáñez JM (2015) Genome-wide association analysis reveals loci associated with resistance against Piscirickettsia salmonis in two Atlantic salmon (Salmo salar L.) chromosomes. BMC Genom 16:854
Darvasi A, Soller M (1992) Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor Appl Genet 85:353–359
Dong HT, Nguyen VV, Phiwsaiya K, Gangnonngiw W, Withyachumnarnkul B, Rodkhum C, Senapin S (2015) Concurrent infections of Flavobacterium columnare and Edwardsiella ictaluri in striped catfish, Pangasianodon hypophthalmus in Thailand. Aquaculture 448:142–150
Dunham RA, Argue BJ (2000) Reproduction among channel catfish, blue catfish, and their F1 and F2 hybrids. Trans Am Fish Soc 129:222–231
Dunham RA, Smitherman RO (1984) Ancestry and breeding of catfish in the United States. Circular 273, Ala Agric Exp Stat, Auburn University, Alabama
Dunham RA, Umali GM, Beam R, Kristanto AH, Trask M (2008) Comparison of production traits of NWAC103 channel catfish, NWAC103 channel catfish × blue catfish hybrids, Kansas Select 21 channel catfish, and blue catfish grown at commercial densities and exposed to natural bacterial epizootics. N Am J Aquac 70:98–106
Dunham RA, Taylor JF, Rise ML, Liu Z (2014) Development of strategies for integrated breeding, genetics and applied genomics for genetic improvement of aquatic organisms. Aquaculture 420–421:S121-123
East L, Isacke CM (2002) The mannose receptor family. BBA Gen Subj 1572:364–386
Fuji K, Kobayashi K, Hasegawa O, Coimbra MRM, Sakamoto T, Okamoto N (2006) Identification of a single major genetic locus controlling the resistance to lymphocystis disease in Japanese flounder (Paralichthys olivaceus). Aquaculture 254:203–210
Fuji K, Hasegawa O, Honda K, Kumasaka K, Sakamoto T, Okamoto N (2007) Marker-assisted breeding of a lymphocystis disease-resistant Japanese flounder (Paralichthys olivaceus). Aquaculture 272:291–295
Fulker DW, Cherny SS, Sham PC, Hewitt JK (1999) Combined linkage and association sib-pair analysis for quantitative traits. Am J Hum Genet 64:259–267
Gazi U, Martinez-Pomares L (2009) Influence of the mannose receptor in host immune responses. Immunobiology 214:554–561
Geng X, Sha J, Liu S, Bao L, Zhang J, Wang R, Yao J, Li C, Feng J, Sun F, Sun L, Jiang C, Zhang Y, Chen A, Dunham R, Zhi D, Liu Z (2015) A genome-wide association study in catfish reveals the presence of functional hubs of related genes within QTLs for columnaris disease resistance. BMC Genom 16:196
Geng X, Liu S, Yao J, Bao L, Zhang J, Li C, Wang R, Sha J, Zeng P, Zhi D, Liu Z (2016) A genome-wide association study identifies multiple regions associated with head size in catfish. G3 6:3389–3398
Geng X, Liu S, Yuan Z, Jiang Y, Zhi D, Liu Z (2017a) A genome-wide association study reveals that genes with functions for bone development are associated with body conformation in catfish. Mar Biotechnol 19:570–578
Geng X, Zhi D, Liu Z (2017b) Genome-wide association studies of performance traits. In: Liu Z (ed) Bioinformatics in aquaculture: principles and methods, 1st edn. Wiley, Hoboken, pp 415–433
Hall AB, Gakidis MAM, Glogauer M, Wilsbacher JL, Gao S, Swat W, Brugge JS (2006) Requirements for Vav guanine nucleotide exchange factors and Rho GTPases in FcγR-and complement-mediated phagocytosis. Immunity 24:305–316
Hanson LA (2006) Enteric septicaemia of catfish. In: Manual of diagnostic tests for aquatic animals, 5th edn. OIE, Paris, pp 228–235
Harburger DS, Calderwood DA (2009) Integrin signalling at a glance. J Cell Sci 122:159–163
Hawke JP (1979) A bacterium associated with disease of pond cultured channel catfish, Ictalurus punctatus. J Fish Board Canada 36:1508–1512
Hawke JP, McWhorter AC, Steigerwalt AG, Brenner DJ (1981) Edwardsiella ictaluri sp. nov., the causative agent of enteric septicemia of catfish. Int J Syst Bacteriol 31:396–400
Hawke JP, Durborow RM, Thune RL, Camus AC (1998) ESC: enteric septicemia of catfish. Southern Regional Aquaculture Center Stoneville, Mississippi
Hayashi K, Altman A (2007) Protein kinase C theta (PKCθ): a key player in T cell life and death. Pharmacol Res 55:537–544
Hornstein I, Alcover A, Katzav S (2004) Vav proteins, masters of the world of cytoskeleton organization. Cell Signal 16:1–11
Houston RD, Haley CS, Hamilton A, Guy DR, Tinch AE, Taggart JB, McAndrew BJ, Bishop SC (2008) Major quantitative trait loci affect resistance to infectious pancreatic necrosis in Atlantic salmon (Salmo salar). Genetics 178:1109–1115
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687
Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, Stradal TEB, Di Fiore PP, Carlier M-F, Scita G (2004) Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol 6:319
Isakov N, Altman A (2002) Protein kinase Cθ in T cell activation. Annu Rev Immunol 20:761–794
Jin Y, Zhou T, Geng X, Liu S, Chen A, Yao J, Jiang C, Tan S, Su B, Liu Z (2017) A genome-wide association study of heat stress-associated SNPs in catfish. Anim Genet 48:233–236
Kang HM, Sul JH, Zaitlen NA, Kong S, Freimer NB, Sabatti C, Eskin E (2010) Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42:348–354
Kasornchandra J, Rogers WA, Plumb JA (1987) Edwardsiella ictaluri from walking catfish, Clarias batrachus L., in Thailand. J Fish Dis 10:137–138
Katagiri K, Hattori M, Minato N, Irie S, Takatsu K, Kinashi T (2000) Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol Cell Biol 20:1956–1969
Kheir WA, Gevrey J-C, Yamaguchi H, Isaac B, Cox D (2005) A WAVE2-Abi1 complex mediates CSF-1-induced F-actin-rich membrane protrusions and migration in macrophages. J Cell Sci 118:5369–5379
Krause M, Sechi AS, Konradt M, Monner D, Gertler FB, Wehland J (2000) Fyn-binding protein (Fyb)/SLP-76-associated protein (SLAP), Ena/vasodilator-stimulated phosphoprotein (VASP) proteins and the Arp2/3 complex link T cell receptor (TCR) signaling to the actin cytoskeleton. J Cell Biol 149:181–194
Lettau M, Pieper J, Janssen O (2009) Nck adapter proteins: functional versatility in T cells. Cell Commun Signal 7:1
Li C, Zhang Y, Wang R, Lu J, Nandi S, Mohanty S, Terhune J, Liu Z, Peatman E (2012) RNA-seq analysis of mucosal immune responses reveals signatures of intestinal barrier disruption and pathogen entry following Edwardsiella ictaluri infection in channel catfish, Ictalurus punctatus. Fish Shellfish Immunol 32:816–827
Li Y, Geng X, Bao L, Elaswad A, Huggins KW, Dunham R, Liu Z (2017a) A deletion in the Hermansky–Pudlak syndrome 4 (Hps4) gene appears to be responsible for albinism in channel catfish. Mol Genet Genomics 292:663–670
Li N, Zhou T, Geng X, Jin Y, Wang X, Liu S, Xu X, Gao D, Li Q, Liu Z (2017b) Identification of novel genes significantly affecting growth in catfish through GWAS analysis. Mol Genet Genomics. https://doi.org/10.1007/s00438-017-1406-1
Lioudyno MI, Kozak JA, Penna A, Safrina O, Zhang SL, Sen D, Roos J, Stauderman KA, Cahalan MD (2008) Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc Natl Acad Sci 105:2011–2016
Liu S, Zhou Z, Lu J, Sun F, Wang S, Liu H, Jiang Y, Kucuktas H, Kaltenboeck L, Peatman E, Liu ZJ (2011) Generation of genome-scale gene-associated SNPs in catfish for the construction of a high-density SNP array. BMC Genom 12:53
Liu S, Sun L, Li Y, Sun F, Jiang Y, Zhang Y, Zhang J, Feng J, Kaltenboeck L, Kucuktas H, Liu Z (2014) Development of the catfish 250 K SNP array for genome-wide association studies. BMC Res Notes 7:135
Liu S, Vallejo RL, Palti Y, Gao G, Marancik DP, Hernandez AG, Wiens GD (2015) Identification of single nucleotide polymorphism markers associated with bacterial cold water disease resistance and spleen size in rainbow trout. Front Genet 6:298
Liu P, Wang L, Wan ZY, Ye BQ, Huang S, Wong S-M, Yue GH (2016a) Mapping QTL for resistance against viral nervous necrosis disease in Asian seabass. Mar Biotechnol 18:107–116
Liu Z, Liu S, Yao J, Bao L, Zhang J, Li Y, Jiang C, Sun L, Wang R, Zhang Y, Zhou T, Zeng Q, Fu Q, Gao S, Li N, Koren S, Jiang Y, Zimin A, Xu P, Phillippy AM, Geng X, Song L, Sun F, Li C, Wang X, Chen A, Jin Y, Yuan Z, Yang Y, Tan S, Peatman E, Lu J, Qin Z, Dunham R, Li Z, Sonstegard T, Feng J, Danzmann RG, Schroeder S, Scheffler B, Duke MV, Ballard L, Kucuktas H, Kaltenboeck L, Liu H, Armbruster J, Xie Y, Kirby ML, Tian Y, Flanagan ME, Mu W, Waldbieser G (2016b) The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat Commun 7:11757
Lucas JS, Southgate PC (2012) Aquaculture: farming aquatic animals and plants. Wiley, Oxford
Machesky LM, Gould KL (1999) The Arp2/3 complex: a multifunctional actin organizer. Curr Opin Cell Biol 11:117–121
Macian F (2005) NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 5:472
May RC, Caron E, Hall A, Machesky LM (2000) Involvement of the Arp2/3 complex in phagocytosis mediated by FcγR or CR3. Nat Cell Biol 2:246
Medzhitov R (2007) Recognition of microorganisms and activation of the immune response. Nature 449:819
Menanteau-Ledouble S (2009) Pathogen entrance and development of disease during infection of the American channel catfish Ictalurus punctatus by the enterobacterium Edwardsiella ictaluri. Dissertation, Mississippi State University
Menanteau-Ledouble S, Karsi A, Lawrence ML (2011) Importance of skin abrasion as a primary site of adhesion for Edwardsiella ictaluri and impact on invasion and systematic infection in channel catfish Ictalurus punctatus. Vet Microbiol 148:425–430
Michalak P (2008) Coexpression, coregulation, and cofunctionality of neighboring genes in eukaryotic genomes. Genomics 91:243–248
Miyazaki T, Plumb JA (1985) Histopathology of Edwardsiella ictaluri in channel catfish, Ictalurus punctatus (Rafinesque). J Fish Dis 8:389–392
Moen T, Baranski M, Sonesson AK, Kjøglum S (2009) Confirmation and fine-mapping of a major QTL for resistance to infectious pancreatic necrosis in Atlantic salmon (Salmo salar): population-level associations between markers and trait. BMC Genom 10:368
Monks CRF, Kupfer H, Tamir I, Barlow A, Kupfer A (1997) Selective modulation of protein kinase C-omega during T-cell activation. Nature 385:83
Newton JC, Wolfe LG, Grizzle JM, Plumb JA (1989) Pathology of experimental enteric septicaemia in channel catfish, Ictalurus punctatus (rafinesque), following immersion-exposure to Edwardsiella ictaluri. J Fish Dis 12:335–347
Nishimura S, Watanabe T, Mizoshita K, Tatsuda K, Fujita T, Watanabe N, Sugimoto Y, Takasuga A (2012) Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet 13:40
Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill D, Ambudkar IS (2007) Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem 282:9105–9116
Ortiz-Stern A, Rosales C (2003) Cross-talk between Fc receptors and integrins. Immunol Lett 90:137–143
Ozaki A, Khoo S-K, Yoshiura Y, Ototake M, Sakamoto T, Dijkstra JM, Okamoto N (2007) Identification of additional quantitative trait loci (QTL) responsible for susceptibility to infectious pancreatic necrosis virus in rainbow trout. Fish Pathol 42:131–140
Palti Y, Vallejo RL, Gao G, Liu S, Hernandez AG, Rexroad CE III, Wiens GD (2015) Detection and validation of QTL affecting bacterial cold water disease resistance in rainbow trout using restriction-site associated DNA sequencing. PLoS One 10:e0138435
Patel JC, Hall A, Caron E (2002) Vav regulates activation of Rac but not Cdc42 during FcγR-mediated phagocytosis. Mol Biol Cell 13:1215–1226
Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, Leitges M, Baier G (2003) Protein kinase C θ affects Ca2+ mobilization and NFAT activation in primary mouse T cells. J Exp Med 197:1525–1535
Plumb JA, Hanson LA (2011) Health maintenance and principal microbial diseases of cultured fishes. Wiley, Oxford
Poompuang S, Hallerman EM (1997) Toward detection of quantitative trait loci and marker-assisted selection in fish. Rev Fish Sci 5:253–277
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, De Bakker PIW, Daly MJ (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Rao GK, Kaminski NE (2006) Induction of intracellular calcium elevation by ∆9-tetrahydrocannabinol in T cells involves TRPC1 channels. J Leukoc Biol 79:202–213
Russo R, Shoemaker CA, Panangala VS, Klesius PH (2009) In vitro and in vivo interaction of macrophages from vaccinated and non-vaccinated channel catfish (Ictalurus punctatus) to Edwardsiella ictaluri. Fish Shellfish Immunol 26:543–552
Sebzda E, Bracke M, Tugal T, Hogg N, Cantrell DA (2002) Rap1A positively regulates T cells via integrin activation rather than inhibiting lymphocyte signaling. Nat Immunol 3:251
Shoemaker CA, Klesius PH, Evans JJ, Arias CR (2009) Use of modified live vaccines in aquaculture. J World Aquacult Soc 40:573–585
Shotts EB, Blazer VS, Waltman WD (1986) Pathogenesis of experimental Edwardsiella ictaluri infections in channel catfish (Ictalurus punctatus). Can J Fish Aquat Sci 43:36–42
Solovyev V, Kosarev P, Seledsov I, Vorobyev D (2006) Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol 7:S10
Sun Z, Arendt CW, Ellmeier W, Schaeffer EM (2000) PKCθ is required for TCR-induced NF-κB activation in mature but not immature T lymphocytes. Nature 404:402
Sun L, Liu S, Wang R, Jiang Y, Zhang Y, Zhang J, Bao L, Kaltenboeck L, Dunham R, Waldbieser G, Liu Z (2014) Identification and analysis of genome-wide SNPs provide insight into signatures of selection and domestication in channel catfish (Ictalurus punctatus). PLoS One 9:e109666
Taylor PR, Gordon S, Martinez-Pomares L (2005) The mannose receptor: linking homeostasis and immunity through sugar recognition. Trends Immunol 26:104–110
Tybulewicz VLJ (2005) Vav-family proteins in T-cell signalling. Curr Opin Immunol 17:267–274
Vale FXR, Parlevliet JE, Zambolim L (2001) Concepts in plant disease resistance. Fitopatol Bras 26:577–589
Villalba M, Coudronniere N, Deckert M, Teixeiro E, Mas P, Altman A (2000) A novel functional interaction between Vav and PKCθ is required for TCR-induced T cell activation. Immunity 12:151–160
Villalba M, Bi K, Hu J, Altman Y, Bushway P, Reits E, Neefjes J, Baier G, Abraham RT, Altman A (2002) Translocation of PKCθ in T cells is mediated by a nonconventional, PI3-K- and Vav-dependent pathway, but does not absolutely require phospholipase C. J Cell Biol 157:253–263
Wang R, Sun L, Bao L, Zhang J, Jiang Y, Yao J, Song L, Feng J, Liu S, Liu Z (2013) Bulk segregant RNA-seq reveals expression and positional candidate genes and allele-specific expression for disease resistance against enteric septicemia of catfish. BMC Genom 14:1
Wang L, Liu L, Zhou Y, Zhao X, Xi M, Wei S, Fang R, Ji W, Chen N, Gu Z, Liu X, Wang W, Asim M, Liu X, Lin L (2014) Molecular cloning and expression analysis of mannose receptor C type 1 in grass carp (Ctenopharyngodon idella). Dev Comp Immunol 43:54–58
Wang L, Bai B, Huang S, Liu P, Wan ZY, Ye B, Wu J, Yue GH (2017a) QTL mapping for resistance to iridovirus in Asian seabass using genotyping-by-sequencing. Mar Biotechnol 19:517–527
Wang X, Liu S, Jiang C, Geng X, Zhou T, Li N, Bao L, Li Y, Yao J, Yang Y, Zhong X, Jin Y, Dunham R, Liu Z (2017b) Multiple across-strain and within-strain QTLs suggest highly complex genetic architecture for hypoxia tolerance in channel catfish. Mol Genet Genomics 292:63–76
Wolters WR, Johnson MR (1994) Enteric septicemia resistance in blue catfish and three channel catfish strains. J Aquat Anim Health 6:329–334
Wolters WR, Wise DJ, Klesius PH (1996) Survival and antibody response of channel catfish, blue catfish, and channel catfish female × blue catfish male hybrids after exposure to Edwardsiella ictaluri. J Aquat Anim Health 8:249–254
Xu J, Zhao J, Li Y, Zou Y, Lu B, Chen Y, Ma Y, Xu H (2016) Evaluation of differentially expressed immune-related genes in intestine of Pelodiscus sinensis after intragastric challenge with lipopolysaccharide based on transcriptome analysis. Fish Shellfish Immunol 56:417–426
Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL (2014) Advantages and pitfalls in the application of mixed-model association methods. Nat Genet 46:100–106
Ye S, Li H, Qiao G, Li Z (2009) First case of Edwardsiella ictaluri infection in China farmed yellow catfish Pelteobagrus fulvidraco. Aquaculture 292:6–10
Zeng Q, Fu Q, Li Y, Waldbieser G, Bosworth B, Liu S, Yang Y, Bao L, Yuan Z, Li N, Liu Z (2017) Development of a 690 K SNP array in catfish and its application for genetic mapping and validation of the reference genome sequence. Sci Rep 7:40347
Zhong X, Wang X, Zhou T, Jin Y, Tan S, Jiang C, Geng X, Li N, Shi H, Zeng Q, Yang Y, Yuan Z, Bao L, Liu S, Tian C, Peatman E, Li Q, Liu Z (2017) Genome-wide association study reveals multiple novel QTL associated with low oxygen tolerance in hybrid catfish. Mar Biotechnol 19:1–12
Zhou T, Liu S, Geng X, Jin Y, Jiang C, Bao L, Yao J, Zhang Y, Zhang J, Sun L, Wang X, Li N, Tan S, Liu Z (2017) GWAS analysis of QTL for enteric septicemia of catfish and their involved genes suggest evolutionary conservation of a molecular mechanism of disease resistance. Mol Genet Genomics 292:231–242
Zhu J, Li C, Ao Q, Tan Y, Luo Y, Guo Y, Lan G, Jiang H, Gan X (2015) Trancriptomic profiling revealed the signatures of acute immune response in tilapia (Oreochromis niloticus) following Streptococcus iniae challenge. Fish Shellfish Immunol 46:346–353
Acknowledgements
This project was supported by Aquaculture and Food Research Initiative Competitive Grant no. 2015-67015-22975 from the USDA National Institute of Food and Agriculture (NIFA). Suxu Tan is supported by a scholarship from the China Scholarship Council (CSC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All the procedures involving the handling and treatment of fish were approved by the Institutional Animal Care and Use Committee (IACUC) at Auburn University. Blood samples were collected after euthanasia. All animal procedures were carried out according to the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act in the United States.
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tan, S., Zhou, T., Wang, W. et al. GWAS analysis using interspecific backcross progenies reveals superior blue catfish alleles responsible for strong resistance against enteric septicemia of catfish. Mol Genet Genomics 293, 1107–1120 (2018). https://doi.org/10.1007/s00438-018-1443-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-018-1443-4