
Abstract
Gene-based molecular markers are increasingly used in crop breeding programs for marker-assisted selection. However, identification of genetic variants associated with important agronomic traits has remained a difficult task in soybean. RNA-Seq provides an efficient way, other than assessing global expression variations of coding genes, to discover gene-based SNPs at the whole genome level. In this study, RNA isolated from four soybean accessions each with three replications was subjected to high-throughput sequencing and a range of 44.2–65.9 million paired-end reads were generated for each library. A total of 75,209 SNPs were identified among different genotypes after combination of replications, 89.1% of which were located in expressed regions and 27.0% resulted in amino acid changes. GO enrichment analysis revealed that most significant enriched genes with nonsynonymous SNPs were involved in ribonucleotide binding or catalytic activity. Of 22 SNPs subjected to PCR amplification and Sanger sequencing, all of them were validated. To test the utility of identified SNPs, these validated SNPs were also assessed by genotyping a relative large population with 393 wild and cultivated soybean accessions. These SNPs identified by RNA-Seq provide a useful resource for genetic and genomic studies of soybean. Moreover, the collection of nonsynonymous SNPs annotated with their predicted functional effects also provides a valuable asset for further discovery of genes, identification of gene variants, and development of functional markers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soybean (Glycine max) is the most important legume crops and one of the most widely grown crops all over the world. It is the world’s principal source of vegetable oil and provides high-quality plant proteins for animal feed (Hartman et al. 2011). Nutritional studies have also revealed that eating soybean-based food could reduce risks of multiple health problems including heart disease, cancer, and osteoporosis (Birt et al. 2004). In addition, soybean also benefits world agriculture by its capacity to fix atmospheric nitrogen through symbioses with microorganisms (Singh 2010). Similar to most crops, several agronomic traits of soybean including growth habit, morphology, and seed composition vary among different genotypes, which is caused by their genetic diversity (Palmer et al. 2004).
Advances in genetics and molecular biology have led to identification of several genes or molecular markers associated with important agronomic traits. There is ample evidence that sequence variations in coding regions, especially nonsynonymous variations, could alter gene function in soybean. For example, each of four nonsynonymous substitutions in the soybean Terminal Flower 1 (GmTfl1) gene and an A/G nonsynonymous substitution in the Dt2 (Glyma18g50910) gene all have major effects on the formation of stem growth habit, which is a key adaptive and agronomic trait affecting soybean yield (Tian et al. 2010; Ping et al. 2014). Amino acid variations (R130P and Y358N) in a serine hydroxymethyltransferase (SHMT) resulting from changes of two nucleotides affect the role of this gene in soybean cyst nematode resistance (Liu et al. 2012). In addition, the presence of a nonsynonymous substitution (G/C) in the EAR motif region of the Gm-JAG1(Ln) locus indicated that this gene is a key regulator of leaflet shape and number of seeds per pod (Jeong et al. 2012). Moreover, identification of single-nucleotide substitutions in E1, E2, E3, E4 and FAD genes also revealed their critical functions in regulation of flowering, maturity and fatty acid composition in soybean (Liu et al. 2008; Watanabe et al. 2009, 2011; Pham et al. 2010; Xia et al. 2012). Based on these sequence variations, several functional molecular markers have been successfully developed and used for marker-assisted selection in breeding programs (Xu et al. 2013; Liu et al. 2015; Shi et al. 2015a, b).
Due to the global importance, soybean was the first legume species to have its whole genome sequenced (Schmutz et al. 2010). Since the release of soybean reference genome, interest in sequence variations among different genotypes at the genomic level has grown and millions of SNPs have been detected (Kim et al. 2010; Li et al. 2014). With the increasing of soybean accessions employed in different resequencing studies, the number of SNPs identified has grown from a few to nearly ten million (Lam et al. 2010; Li et al. 2013; Chung et al. 2014; Zhou et al. 2015; dos Santos et al. 2016). These SNPs have been widely used in developing SNP genotyping arrays with various densities for QTL mapping or genome-wide association studies (Akond et al. 2013; Song et al. 2013; Lee et al. 2015). However, whole genome sequencing is still somewhat expensive and time-consuming for species with large genomes such as soybean. Most importantly, given that most SNPs commonly used in genotyping arrays are not located in coding regions of genes, SNPs associated with phenotypes of interest could not be directly used for isolating functional genes. Therefore, identification of genetic variants influencing important agronomic traits remains a difficult task in soybean now.
With the development of next generation sequencing, transcriptome sequencing (RNA-Seq) has recently been served as a cheaper and time-saving alternative method for identifying variations in transcribed regions in species from animals to plants (Bellucci et al. 2014; Geraldes et al. 2011; Djari et al. 2013; Chopra et al. 2015; Yang et al. 2015). Generally, RNA-Seq can generate large-scale sequences with high read depth by reducing the effective genome size to about 10% of the whole genome. For example, the transcribed sequences of soybean only account for no more than 100 Mb, whereas the entire genome is about 1.1 Gb (Schmutz et al. 2010). However, RNA-Seq technology has still been applied mainly for identifying expression variations of coding genes as usual in soybean (Libault et al. 2010; Jones and Vodkin 2013; Wan et al. 2015). Only a few studies have focused on discovery of SNPs associated with drought (Vidal et al. 2012) or oil composition and content (Goettel et al. 2014) by transcriptome analysis of soybean accessions.
To identify gene-based SNPs among different soybean genotypes, a transcriptome sequencing approach was performed in different soybean accessions with replications. About 75K putative SNPs were identified from four accessions after combination of replications. The distribution and predicted function of all these SNPs were analyzed in detail. To evaluate this SNP dataset, 22 SNPs were amplified and sequenced using the Sanger method. All of them were validated and these SNPs were then used for genotyping a panel of accessions including wild and cultivated soybeans. These sequence variations in expressed regions of the genome, especially nonsynonymous SNPs, will provide a useful resource for genetic and genomic research in soybean.
Materials and methods
Plant materials
Four soybean accessions, Zhongpin 661, Lincoln, Jidou 12, and Dongnong 42, were subjected to RNA-Seq. These four accessions and the reference accession Williams 82 all originate from different eco-regions and have high diversity in phenotypes including stem growth habit, plant morphology, and seed characteristics (Online Resource 1). Among them, Lincoln is a soybean cultivar cultivated in North America while Jidou 12 and Dongnong 42 are accessions from Huanghuaihai and Northeast eco-regions, respectively, of China. Although Zhongpin 661 is a Chinese cultivar, both of its parental lines (Williams 82 and Buffalo) are from North America. Leaves of 2-week-old seedlings from these four accessions were collected, each with three replications. All 12 samples were immediately frozen in liquid nitrogen and stored at − 80 °C until RNA extraction.
RNA isolation and sequencing
Total RNA was extracted using the TRIzol reagent (Invitrogen, USA) following the manufacturer’s instructions. The RNA was then treated with RNase-free DNase I (Takara, Japan) to remove genomic DNA. The quality and quantity of RNA samples were assessed with a Nanodrop spectrophotometer (Thermo Scientific, USA). Twelve RNA-Seq libraries were constructed using RNA-Seq Sample Preparation Kit (Illumina, USA). Finally, 11 libraries were subjected for sequencing with one replication of Jidou 12 discarded owing to the low quality of the library, and 100-base paired-end reads were generated on the Illumina HiSeq 2000 platform.
Data filtering and RNA-Seq read mapping
Sequencing data originated from the sequencer were processed for quality control. All short reads were submitted to the NCBI website with SRA accession # SRP082550. Raw reads were filtered by removal of adapter sequences, reads containing poly-Ns, and low-quality sequences (Q < 20). Clean reads of each sample were then aligned to the Williams 82 reference genome (Glyma1.01) using TopHat (Trapnell et al. 2009). Sequence reads mapped to the reference genome with no more than two mismatches were used for further analysis.
SNP calling and annotation
The SAMtools software package was used to call SNPs across all samples simultaneously (Li et al. 2009). The results were filtered to discard SNPs with quality scores less than 70. To ensure reliability of these SNPs, those identified in at least two replications for a cultivar and having total read depth of at least six were assigned as SNPs for that cultivar. The functional effects of these identified SNPs on known transcripts were analyzed using the program SnpEff (Cingolani et al. 2012).
Gene Ontology enrichment analysis
Gene Ontology (GO) analysis was performed to annotate genes containing nonsynonymous SNPs. Enriched GO terms were identified with the AgriGO web tool using annotated genes of Williams 82 reference genome (Glyma1.01) as background reference (Du et al. 2010). GO terms with corrected P value less than 0.05 were considered significantly enriched.
Validation of SNPs
To validate SNPs identified from RNA-Seq, 22 of them were selected for PCR amplification and Sanger sequencing. Flanking sequences of selected SNPs were extracted from the reference genome and PCR primers were designed with Premier Primer 5 (Singh et al. 1998). All primers were listed in Online Resource 2. DNA samples of four soybean accessions and Williams 82 were used as templates for PCR amplification. The amplified PCR products were sequenced by the Sanger method and sequencing results were analyzed with BioEdit (Hall 1999).
SNP genotyping
Genomic DNA of 393 soybean accessions used for genotyping (Online Resource 3) was isolated from leaves of each accession using a modified CTAB method (Porebski et al. 1997). Twenty-one of the 22 validated SNPs were used for genotyping while the other one was failed in designing primers for genotyping. The genotype of each allele was analyzed using the Sequenom MassARRAY iPLEX platform (Gabriel et al. 2009). The resulting data were analyzed with the MassARRAY Typer 4.0 Analyzer software. Minor allele frequency (MAF), gene diversity, and PIC value of these SNPs were calculated with PowerMarker V3.25 (Liu and Muse 2005).
Results
RNA isolation and sequencing
To acquire a global view of the transcriptome at single-nucleotide resolution in soybean, RNA samples from four cultivars, each with three biological replications, were isolated and subjected to high-throughput sequencing. After quality control and filtering of raw reads, 44.2–65.9 million high-quality paired-end reads with average length of 94.7 bases were generated for the remaining 11 libraries after removal of one replication of Jidou 12 (Table 1). Clean reads of each sample were then mapped to the Williams 82 reference genome (Glyma1.01). The mapping results showed that 90.3–94.2% of short reads in all these samples could be mapped to the reference genome (Table 1), indicating the high quality of the sequence data.
SNP discovery
Putative SNPs were identified in 11 samples based on quality scores of alignments. SNP loci from replications of each accession were combined using the following criteria: (1) SNPs appearing in at least two replications and (2) read depth ≥ 6. After combination of the data, a total of 5478, 21,052, 36,930, and 40,144 SNPs were detected in Zhongpin 661, Lincoln, Jidou 12, and Dongnong 42 compared to reference genome, respectively. Polymorphism was much lower in Zhongpin 661 than other accessions because it is the progeny of the reference accession Williams 82. When SNPs of all four soybean accessions were combined together, a total of 75,209 putative SNP loci were included across all libraries (Fig. 1a, Online Resource 4). Among all these variations, 1984, 9818, 19,529, and 21,460 were specific to respective accessions and 22,418 were common to at least two cultivars (Fig. 1b).

Number and type of SNPs identified in soybean transcriptome sequencing. a Physical positions of all SNPs identified in soybean transcriptome sequencing. b Venn diagram of SNPs among soybean accessions and c summary of SNP types identified in soybean transcriptome sequencing
In this SNP dataset, transitions (A/G and C/T) were the two most-abundant substitutions, each accounting for about 30% of all SNPs (Fig. 1c). Transversion substitutions accounted for only 8.2–13.4% of total SNPs (A/T, 13.4%; G/T, 9.6%; A/C, 9.5%; and C/G, 8.2%). The observed transition: transversion ratio was thus 1.46:1, close to the ratio in previous study using the resequencing of sequence-tagged sites (STSs) developed from expressed sequence tag (EST) sequences, in which 55.7% were transitions and 44.3% transversions were identified (Choi et al. 2007).
The distribution of SNPs
The distribution of SNPs across chromosomes and genes is of particular importance for evaluating their genome coverage and marker density. Therefore, we analyzed the SNP distribution among all soybean chromosomes and expressed genes (Fig. 2). The results showed that 75,209 SNPs identified in our RNA-Seq data were distributed across all 20 chromosomes and 50 scaffolds, with an average of one SNP per 12.9 kb. Chromosome 18 contained the most SNPs (5340) and chromosome 11 contained the fewest (2022). The SNP distribution was in close agreement with the distribution of annotated genes across chromosomes (Fig. 2a).

SNP distribution among chromosomes and genes. a Distribution of SNPs and annotated genes on the 20 soybean chromosomes and scaffolds and b distribution of SNPs in genes
Further analysis revealed that all these SNPs were distributed in 19,976 annotated genes. The number of SNPs per gene ranged from 1 (6000 genes) to 75 (in Glyma07g07100), with an average of 3.8 SNPs for genes harboring at least one SNP. Among these genes, those containing fewer than ten SNPs accounted for 95.7% of total genes with SNPs (Fig. 2b).
Functional annotation of SNPs
According to their locations in the genome, all these SNPs were classified into several categories. As shown in Table 2, of the putative SNPs, 95.5% (71,806) were identified in genic regions while the other 4.5% (3403) in intergenic regions according to Glyma1.01 annotation of the soybean genome. A total of 66,957 (89.1%) SNPs were identified in expressed regions or splicing region of introns (which could result in splicing variants) and 4849 SNPs (6.4%) were annotated in introns. In particular, 18,362 (24.4%) SNPs were classified as nonsynonymous SNPs resulting in amino acid changes, and 1907 (2.5%) other SNPs gave rise to variants in start or stop codons or splicing regions, also altering amino acid sequences of coding genes. Interestingly, Some SNPs associated with important traits were also identified. For example, the G/C substitution at position 34,688,649 of chromosome 20 was associated with the leaflet shape function of Ln (Glyma20g25000), which was identified in functional characterization study (Jeong et al. 2012). The A/T substitution at position 44,732,850 of chromosome 10 resulted in a premature stop codon in the E2 gene (Glyma10g36600) regulating the flowering and maturity of soybean (Watanabe et al. 2011).
GO enrichment analysis was performed for 10,758 genes with nonsynonymous SNPs and other SNPs altering amino acid sequence using all Glyma1.01-annotated genes as the gene enrichment reference. A total of 6011 genes were assigned one or more GO terms. For “molecular function”, genes involved in nucleotide or ribonucleotide binding showed significant enrichment. In addition, several GO terms related to catalytic activity and protein kinase activity were also statistically significantly enriched (Fig. 3).

GO enrichment of genes with nonsynonymous SNPs in soybean. Enriched GO terms were identified with the AgriGO web tool (http://bioinfo.cau.edu.cn/agriGO/) and GO terms with corrected P value less than 0.05 were considered significantly enriched
Sanger validation and genotyping of putative SNPs
To assess the validity of these SNPs identified by RNA-Seq, a set of SNPs were selected from the whole SNP dataset for validation by Sanger sequencing. A total of 22 SNP loci were amplified and sequenced from the four original soybean accessions and Williams 82. The results showed that Sanger sequencing results of all these 22 SNPs were actually the “true” SNPs, indicating the accuracy of the SNP dataset (Table 3). Meanwhile, five other SNPs with relatively low quality scores (score < 70 and this kind of SNP was not in the final SNP set) were also sequenced and the results showed that these SNPs may be false positive results of bioinformatics analysis. Therefore, increasing the threshold of quality scores to a reasonable level could reduce false positive results of SNPs identified.
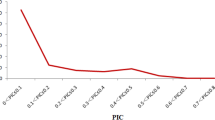
To further test the utility of identified SNPs, 21 SNPs (all except SNP09) were used to genotype 393 soybean accessions including 238 from the mini-core collection of Chinese cultivated soybeans and 155 wild soybeans, using the Sequenom MassARRAY iPLEX platform. All but two SNPs (SNP11 and SNP14) had SNP call rates above 90.0% (Table 4). Each of the 19 SNPs showed a minor allele frequency (MAF) ≥ 0.08 in this population and the highest MAF observed was 0.48. The genetic diversity and polymorphism information content (PIC) values were in the ranges of 0.15–0.50 and 0.14–0.37, respectively. All these results indicated the high diversity of these validated SNP markers among different soybean genotypes.
Discussion
SNPs are the most abundant DNA markers in plant genomes and have been widely used in genetic studies and breeding programs. We used RNA-Seq technology to sequence transcriptomes of four soybean accessions with high diversity in phenotype and finally identified 75,209 high-quality SNPs distributed in 19,976 annotated genes. Of these, 24.4% (18,362) were nonsynonymous SNPs and 1907 SNPs showed variations in start or stop codons or splicing regions, all resulting in amino acid changes of encoded genes. Selected SNPs were validated by Sanger sequencing and used for genotyping a population of wild and cultivated soybeans. These gene-based SNPs will expand the genomic resources available for soybean and can be used to develop genotyping platforms to perform marker-trait association studies.
SNPs among wild and cultivated soybeans have previously been discovered via next-generation whole genome sequencing (Lam et al. 2010; Li et al. 2013; Zhou et al. 2015). Compared with whole genome sequencing, transcriptome sequencing could reduce genome complexity and is an attractive strategy for organisms with large genomes. Given that most regions of soybean genome are composed of highly repetitive transposable elements (Schmutz et al. 2010), protein-coding genes only account for about 10% of the genome. Since RNA-Seq only focuses on functional components of the genome, it provides a much more efficient method for identifying sequence variations in expressed genes and was proven to be a better approach to find a large number of SNPs to efficiently tag most variants in genes that might influence agronomic traits. Just like previous studies identified 48,792 SNPs from nine soybean lines and 6698 SNPs from two cultivars (Vidal et al. 2012; Goettel et al. 2014), 75,209 high-quality SNPs were identified in our study using four accessions with high genetic and phenotypic diversity. Moreover, compared with the 5.1M SNPs identified by DNA resequencing of wild soybean, landrace and breeding lines (Li et al. 2013), about 1/4 (18,409 out of 75,209) high-quality SNPs were new identified ones.
Sequence variants that affect gene functions and cause phenotypic variations could be used for development of functional molecular markers (Andersen and Lubberstedt 2003). Owing to their complete linkage with trait phenotypes, functional markers are ideal for marker-assisted breeding (Poczai et al. 2013). Our study identified a large collection of SNPs that could lead to protein sequence changes and potentially to biological functional changes in genes. The sequence variants of some important agronomic genes including Ln and E2 genes were all observed in our dataset. As to Ln gene, the C allele at position 34,688,649 of chromosome 20 was identified in Dongnong 42 while the G allele was identified in other three cultivars, which is in agreement with phenotypes of narrow leaflet in Donong42 and broad leaflet in other cultivars (Jeong et al. 2012). Therefore, these identified transcript sequence polymorphisms could be further used for developing functional markers. However, 6.4% of these SNPs were annotated as in introns and 4.5% in intergenic regions. Previous study also indicated that about 15.3% (7488/48,792) SNPs were located in annotated introns (Goettel et al. 2014). There are at least two possible explanations for this result: novel transcripts and false-positive SNPs. RNA-Seq has the potential to improve genome annotation by detecting expression of regions previously thought to be intergenic and alternatively spliced variants of previously annotated genes. Previous studies suggested that approximately 90% of genes in the human genome and 42% in the Arabidopsis thaliana genome underwent some form of alternative splicing (Wang et al. 2008; Filichkin et al. 2010). A study in soybean also revealed that more than 23,764 (about 63%) multiexonic-expressed genes underwent alternative splicing detected by high-throughput RNA sequencing (Shen et al. 2014). These results indicated that many of the putative intronic SNPs found in our study are in fact intronic only in the assembly of soybean transcripts that we used.
Our Sanger sequencing results pointed to false SNP discovery as a further alternative explanation. Of 22 high-quality SNPs selected for Sanger sequencing, all were proven to be “true” variations. However, five other SNPs with low quality could not be confirmed as polymorphic among different soybean accessions. The quality scores of these five SNPs were all less than 70, suggesting the need of a higher SNP quality threshold for selection of high-confidence SNPs. We increased the threshold of quality score to 70 and discarded all the low-quality SNPs in the final SNP set. In addition, the advantage of using biological replication for SNP discovery is that we can further filter putative SNPs based not only on mapping quality and depth in individual library but also on the replications of each locus. Biological replications increase the accuracy of SNPs and a SNP found in two or three replications is more likely to be a reliable SNP than one found in only a single replication.
References
Akond M, Liu S, Schoener L, Anderson JA, Kantartzi SK, Meksem K, Song Q, Wang D, Wen Z, Lightfoot DA, Kassem MA (2013) A SNP-based genetic linkage map of soybean using the SoySNP6K Illumina Infinium BeadChip genotyping array. J Plant Genome Sci 1:80–89
Andersen JR, Lubberstedt T (2003) Functional markers in plants. Trends Plant Sci 8:554–560
Bellucci E, Bitocchi E, Ferrarini A, Benazzo A, Biagetti E, Klie S, Minio A, Rau D, Rodriguez M, Panziera A, Venturini L, Attene G, Albertini E, Jackson SA, Nanni L, Fernie AR, Nikoloski Z, Bertorelle G, Delledonne M, Papa R (2014) Decreased nucleotide and expression diversity and modified coexpression patterns characterize domestication in the common bean. Plant Cell 26(5):1901–1912
Birt DF, Hendrich S, Alekel DL, Anthony M (2004) Soybean and the prevention of chronic human disease. In: Boerma HR, Specht JE (eds) Soybeans: improvement, production, and uses. American Society of Agronomy, Madison, pp 1047–1117
Choi IY, Hyten DL, Matukumalli LK, Song Q, Chaky JM, Quigley CV, Chase K, Lark KG, Reiter RS, Yoon MS, Hwang EY, Yi SI, Young ND, Shoemaker RC, van Tassell CP, Specht JE, Cregan PB (2007) A soybean transcript map: gene distribution, haplotype and single-nucleotide polymorphism analysis. Genetics 176(1):685–696
Chopra R, Burow G, Farmer A, Mudge J, Simpson CE, Wilkins TA, Baring MR, Puppala N, Chamberlin KD, Burow MD (2015) Next-generation transcriptome sequencing, SNP discovery and validation in four market classes of peanut, Arachis hypogaea L. Mol Genet Genomics 290:1169–1180
Chung WH, Jeong N, Kim J, Lee WK, Lee YG, Lee SH, Yoon W, Kim JH, Choi IY, Choi HK, Moon JK, Kim N, Jeong SC (2014) Population structure and domestication revealed by high-depth resequencing of Korean cultivated and wild soybean genomes. DNA Res 21:153–167
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu XY, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6:80–92
Djari A, Esquerre D, Weiss B, Martins F, Meersseman C, Boussaha M, Klopp C, Rocha D (2013) Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC Genom 14:307
dos Santos JVM, Valliyodan B, Joshi T, Khan SM, Liu Y, Wang JX, Vuong TD, de Oliveira MF, Marcelino-Guimaraes FC, Xu D, Nguyen HT, Abdelnoor RV (2016) Evaluation of genetic variation among Brazilian soybean cultivars through genome resequencing. BMC Genom 17:110
Du Z, Zhou X, Ling Y, Zhang ZH, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38:W64-W70
Filichkin SA, Priest HD, Givan SA, Shen RK, Bryant DW, Fox SE, Wong WK, Mockler TC (2010) Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res 20:45–58
Gabriel S, Ziaugra L, Tabbaa D (2009) SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet Chapter 2:Unit 2.12
Geraldes A, Pang J, Thiessen N, Cezard T, Moore R, Zhao YJ, Tam A, Wang SC, Friedmann M, Birol I, Jones SJM, Cronk QCB, Douglas CJ (2011) SNP discovery in black cottonwood (Populus trichocarpa) by population transcriptome resequencing. Mol Ecol Resour 11:81–92
Goettel W, Xia E, Upchurch R, Wang ML, Chen PY, An YQC (2014) Identification and characterization of transcript polymorphisms in soybean lines varying in oil composition and content. BMC Genom 15:299
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Hartman GL, West ED, Herman TK (2011) Crops that feed the World 2. Soybean-worldwide production, use, and constraints caused by pathogens and pests. Food Secur 3:5–17
Jeong N, Suh SJ, Kim MH, Lee S, Moon JK, Kim HS, Jeong SC (2012) Ln is a key regulator of leaflet shape and number of seeds per pod in soybean. Plant Cell 24:4807–4818
Jones SI, Vodkin LO (2013) Using RNA-Seq to profile soybean seed development from fertilization to maturity. PLoS ONE 8:e59270
Kim MY, Lee S, Van K, Kim TH, Jeong SC, Choi IY, Kim DS, Lee YS, Park D, Ma J, Kim WY, Kim BC, Park S, Lee KA, Kim DH, Kim KH, Shin JH, Jang YE, Do Kim K, Liu WX, Chaisan T, Kang YJ, Lee YH, Kim KH, Moon JK, Schmutz J, Jackson SA, Bhak J, Lee SH (2010) Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proc Natl Acad Sci USA 107:22032–22037
Lam HM, Xu X, Liu X, Chen WB, Yang GH, Wong FL, Li MW, He WM, Qin N, Wang B, Li J, Jian M, Wang JA, Shao GH, Wang J, Sun SSM, Zhang GY (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42:1053–1059
Lee YG, Jeong N, Kim JH, Lee K, Kim KH, Pirani A, Ha BK, Kang ST, Park BS, Moon JK, Kim N, Jeong SC (2015) Development, validation and genetic analysis of a large soybean SNP genotyping array. Plant J 81:625–636
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Li YH, Zhao SC, Ma JX, Li D, Yan L, Li J, Qi XT, Guo XS, Zhang L, He WM, Chang RZ, Liang QS, Guo Y, Ye C, Wang XB, Tao Y, Guan RX, Wang JY, Liu YL, Jin LG, Zhang XQ, Liu ZX, Zhang LJ, Chen J, Wang KJ, Nielsen R, Li RQ, Chen PY, Li WB, Reif JC, Purugganan M, Wang J, Zhang MC, Wang J, Qiu LJ (2013) Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing. BMC Genom 14:579
Li YH, Zhou GY, Ma JX, Jiang WK, Jin LG, Zhang ZH, Guo Y, Zhang JB, Sui Y, Zheng LT, Zhang SS, Zuo QY, Shi XH, Li YF, Zhang WK, Hu YY, Kong GY, Hong HL, Tan B, Song J, Liu ZX, Wang YS, Ruan H, Yeung CKL, Liu J, Wang HL, Zhang LJ, Guan RX, Wang KJ, Li WB, Chen SY, Chang RZ, Jiang Z, Jackson SA, Li RQ, Qiu LJ (2014) De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat Biotechnol 32:1045–1052
Libault M, Farmer A, Joshi T, Takahashi K, Langley RJ, Franklin LD, He J, Xu D, May G, Stacey G (2010) An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J 63:86–99
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21(9):2128–2119
Liu B, Kanazawa A, Matsumura H, Takahashi R, Harada K, Abe J (2008) Genetic redundancy in soybean photoresponses associated with duplication of the Phytochrome A gene. Genetics 180:995–1007
Liu SM, Kandoth PK, Warren SD, Yeckel G, Heinz R, Alden J, Yang CL, Jamai A, El-Mellouki T, Juvale PS, Hill J, Baum TJ, Cianzio S, Whitham SA, Korkin D, Mitchum MG, Meksem K (2012) A soybean cyst nematode resistance gene points to a new mechanism of plant resistance to pathogens. Nature 492:256–260
Liu G, Zhao L, Averitt BJ, Liu Y, Zhang B, Chang R, Ma Y, Luan X, Guan R, Qiu L (2015) Geographical distribution of GmTfl1 alleles in Chinese soybean varieties. Crop J 3:371–378
Palmer RG, Pfeiffer TW, Buss GR, Kilen TC (2004) Qualitative genetics In: Soybeans: improvement, production, and uses, 3rd edn. ASA, CSSA, and SSSA, Madison (WI), pp 137–214
Pham AT, Lee JD, Shannon JG, Bilyeu KD (2010) Mutant alleles of FAD2-1A and FAD2-1B combine to produce soybeans with the high oleic acid seed oil trait. BMC Plant Biol 10:195
Ping JQ, Liu YF, Sun LJ, Zhao MX, Li YH, She MY, Sui Y, Lin F, Liu XD, Tang ZX, Nguyen H, Tian ZX, Qiu LJ, Nelson RL, Clemente TE, Specht JE, Ma JX (2014) Dt2 is a gain-of-function MADS-domain factor gene that specifies semideterminacy in soybean. Plant Cell 26:2831–2842
Poczai P, Varga I, Laos M, Cseh A, Bell N, Valkonen JPT, Hyvonen J (2013) Advances in plant gene-targeted and functional markers: a review. Plant Methods 9(1):6
Porebski S, Bailey LG, Baum BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep 15:8–15
Schmutz J, Cannon SB, Schlueter J, Ma JX, Mitros T, Nelson W, Hyten DL, Song QJ, Thelen JJ, Cheng JL, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu SQ, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du JC, Tian ZX, Zhu LC, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Shen YT, Zhou ZK, Wang Z, Li WY, Fang C, Wu M, Ma YM, Liu TF, Kong LA, Peng DL, Tian ZX (2014) Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 26:996–1008
Shi Z, Bachleda N, Pham AT, Bilyeu K, Shannon G, Nguyen H, Li ZL (2015a) High-throughput and functional SNP detection assays for oleic and linolenic acids in soybean. Mol Breeding 35:1–10
Shi Z, Liu SM, Noe J, Arelli P, Meksem K, Li ZL (2015b) SNP identification and marker assay development for high-throughput selection of soybean cyst nematode resistance. BMC Genom 16:314
Singh G (2010) The soybean: botany, production and uses. CABI Publishing, Wallingford
Singh VK, Mangalam AK, Dwivedi S, Naik S (1998) Primer premier: program for design of degenerate primers from a protein sequence. Biotechniques 24:318–319
Song QJ, Hyten DL, Jia GF, Quigley CV, Fickus EW, Nelson RL, Cregan PB (2013) Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS ONE 8:e54985
Tian ZX, Wang XB, Lee R, Li YH, Specht JE, Nelson RL, McClean PE, Qiu LJ, Ma JX (2010) Artificial selection for determinate growth habit in soybean. Proc Natl Acad Sci USA 107:8563–8568
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-SEq. Bioinformatics 25:1105–1111
Vidal RO, do Nascimento LC, Mondego JMC, Pereira GAG, Carazzolle MF (2012) Identification of SNPs in RNA-seq data of two cultivars of Glycine max (soybean) differing in drought resistance. Genet Mol Biol 35:331–334
Wan JR, Vuong T, Jiao YQ, Joshi T, Zhang HX, Xu D, Nguyen HT (2015) Whole-genome gene expression profiling revealed genes and pathways potentially involved in regulating interactions of soybean with cyst nematode (Heterodera glycines Ichinohe). BMC Genom 16:148
Wang ET, Sandberg R, Luo SJ, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456:470–476
Watanabe S, Hideshima R, Xia ZJ, Tsubokura Y, Sato S, Nakamoto Y, Yamanaka N, Takahashi R, Ishimoto M, Anai T, Tabata S, Harada K (2009) Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics 182:1251–1262
Watanabe S, Xia ZJ, Hideshima R, Tsubokura Y, Sato S, Yamanaka N, Takahashi R, Anai T, Tabata S, Kitamura K, Harada K (2011) A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics 188:395–407
Xia ZJ, Watanabe S, Yamada T, Tsubokura Y, Nakashima H, Zhai H, Anai T, Sato S, Yamazaki T, Lu SX, Wu HY, Tabata S, Harada K (2012) Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci USA 109:E2155–E2164
Xu ML, Xu ZH, Liu BH, Kong FJ, Tsubokura Y, Watanabe S, Xia ZJ, Harada K, Kanazawa A, Yamada T, Abe J (2013) Genetic variation in four maturity genes affects photoperiod insensitivity and PHYA-regulated post-flowering responses of soybean. BMC Plant Biol 13:91
Yang M, Xu LM, Liu YL, Yang PF (2015) RNA-Seq uncovers SNPs and alternative splicing events in Asian lotus (Nelumbo nucifera). PLoS ONE 10:e0125702
Zhou ZK, Jiang Y, Wang Z, Gou ZH, Lyu J, Li WY, Yu YJ, Shu LP, Zhao YJ, Ma YM, Fang C, Shen YT, Liu TF, Li CC, Li Q, Wu M, Wang M, Wu YS, Dong Y, Wan WT, Wang X, Ding ZL, Gao YD, Xiang H, Zhu BG, Lee SH, Wang W, Tian ZX (2015) Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol 33:408–414
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31471520), 13th Five-Year Plan for Precise Identification and Germplasm Enhancement of Economic Crops, and the Agricultural Science and Technology Innovation Program (ASTIP) of Chinese Academy of Agricultural Sciences.
Author information
Authors and Affiliations
Contributions
YG and LJQ conceived and designed the experiments. YG, BS, JT and FZ performed the experiments. YG and LJQ analyzed data and wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Research involving human and animal participants
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Guo, Y., Su, B., Tang, J. et al. Gene-based SNP identification and validation in soybean using next-generation transcriptome sequencing. Mol Genet Genomics 293, 623–633 (2018). https://doi.org/10.1007/s00438-017-1410-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-017-1410-5