
Abstract
Flower color in Brassica spp. is an important trait and considered a major visual signal for insect-pollinated plants. In previous study, we isolated and identified two genes (BjPC1 and BjPC2) that control the flower color in B. juncea, and mapped BjPC1 to a 0.13-cM region. In this study, we report the fine-mapping and candidate analysis of BjPC2. We conducted whole-genome resequencing, using bulked segregant analysis (BSA) to determine the BjPC2 candidate intervals. Crossing, allelism testing, and repeated full-sib mating were used to generate XG3, a near isogenic line (NIL) population that segregated on the BjPC2 locus. Through a genome-wide comparison of single nucleotide polymorphism (SNP) profiles between the yellow- and white-flowered bulks, a candidate interval for BjPC2 was identified on chromosome B04 (2.45 Mb). The BjPC2 linkage map was constructed with the newly developed simple sequence repeat (SSR) markers in the candidate interval to narrow the candidate BjPC2 region to 31-kb. Expression profiling and RNA-seq analysis partially confirmed that the AtPES2 homolog, BjuB027334 is the most promising candidate gene for BjPC2. Furthermore, analyses with high pressure liquid chromatography and transmission electron microscopy demonstrated that BjPC2 might be important in xanthophyll esterification, a process that limits xanthophyll degradation and increases sequestration. Overall, we mapped the BjPC2 to a 31-kb region on the B04 in B. juncea and identified BjuB027334 as a valuable candidate gene. Our results provide a basis for understanding the molecular mechanisms underlying the white-flowered trait and for molecular marker-assisted selection of flower color in B. juncea breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brassica juncea (AABB, 2n = 36) is an allopolyploid species that originated by the hybridization of B. rapa (AA, 2n = 20) and B. nigra (BB, 2n = 16). As one of the major oilseed crops in Brassica, Brassica juncea has several valuable agronomic characteristics, including early maturity, drought tolerance, disease resistance, tolerance to poor soil, shattering resistance, and yellow seeds (Downey 1990; Woods et al. 1991), making it highly suitable for planting in drought-prone regions. Recently, B. juncea has begun to gain importance as an alternative to B. napus in Canada and the USA (Negi et al. 2000). However, despite its broad distribution in western plateau region of China and the abundance of its genetic resources (Xiao et al. 2013), B. juncea research is still in its early stages. Therefore, investigation of genetic patterns and development of molecular markers and identification of candidate genes provide a basis for using the genetic resources and molecular marker-assisted selection (MAS) for valuable agronomic characteristics in B. juncea. In this study, we aimed to identify a candidate gene for the flower color trait in B. juncea.
The flower color is among the traits of greatest interest in Brassica spp. (Pearson 1929); it is an environmentally independent character that is particularly useful for ornamental and landscaping purposes. The phenotype is either yellow or white, with the clearly visible white coloration being valuable for identifying the true/false hybrids or for evaluating the seed purity in hybrid production. The inheritance mechanism of Brassica flower color varies across species. In B. rapa, a single, fully dominant gene controls the white flower coloration and no cytoplasmic effects are apparent (Lee et al. 2014; Rahman 2001), whereas in B. carinata, the single gene has incomplete dominance over the yellow flower trait (Jambhulkar and Raut 1995). Although also controlled by a single nuclear gene (carotenoid cleavage dioxygenase 4, BnaC3.CCD), the white coloration is dominant over yellow in B. napus (Huang et al. 2014; Liu et al. 2004; Zhang et al. 2015). Currently, we know that white flower pigmentation in B. juncea is influenced by the interaction of two gene pairs (Alam and Aziz 1954; Bhuiyan 1986; Rawat and Anand 1986; Singh et al. 1964; Singh and Chauhan 2011), but the exact molecular basis remains poorly understood.
In nature, carotenoids, flavonoids, and betalains are the three pigments responsible for flower color, with the former generating yellows, oranges, and reds (Grotewold 2006). The most common carotenoids in the yellow flower organs are the xanthophylls. Carotenoids are present in both chloroplasts and chromoplasts. In the former, carotenoids are essential to the antenna complex of photosynthesis and to photoprotective mechanisms in plant cells (Grotewold 2006; Walter and Strack 2011). In the latter, carotenoids are the primary pigments (Bramley 2002; Camara et al. 1995) and the source of distinct pigmentation that attracts pollinators or seed dispersers (And and Baker 1983). Carotenoid levels are modulated by sequestration within specific lipoproteins or plastoglobuli of chromoplasts (Ariizumi et al. 2014; Deruère et al. 1994), as well as enzymatic degradation (Auldridge et al. 2006; Zhang et al. 2015). Two groups of carotenoid oxygenases have been identified to work in different plant processes: carotenoid cleavage dioxygenases (CCDs) and the cis 9-carotenoid cleavage dioxygenases (NCEDs). The latter are involved in the synthesis of major plant apocarotenoids, including phytohormones strigolactone and abscisic acid (ABA) (Walter and Strack 2011). Specifically, the cleavage reaction of 9-cis-violaxanthin and 9-cis-neoxanthin is the rate-limiting step in ABA biosynthesis (Cutler and Krochko 1999).
Bulked segregant analysis (BSA) is a simple and effective technique used to identify molecular markers linked to target genes. The method genotypes bulked DNA samples from two groups of individuals with extremely different phenotypes in a given trait of interest (Michelmore et al. 1991). Newly available next-generation sequencing (NGS) techniques, including whole-genome resequencing, can make use full of BSA and high-throughput genotyping to clarify genetic architecture, identifying quantitative trait loci (QTLs) and candidate genes. For example, BSA and whole-genome resequencing have successfully identified the candidate gene, BrTT1, for the seed coat color trait in B. rapa (Wang et al. 2016), while genome resequencing found candidate genes for an early-maturing soybean mutant (Lee et al. 2016). Detailed genomic studies on non-model organisms are even possible now, thanks to the NGS technique of transcriptome analysis (RNA-seq), a rapid and cost-effective approach for obtaining a massive database of expressed genes without requiring a full sequenced genome. The resultant information is strongly applicable for research in ecological, comparative, structural, regulatory, and evolutionary genomics (Khan et al. 2017; Li et al. 2017; Shi et al. 2015; Xiong et al. 2017; Zhang et al. 2017).
Previously, we reported a white-flowered trait in B. juncea that exhibited reduced yellow flower pigmentation. In this study, we aimed to identify a single-gene locus for this trait, performing whole-genome resequencing on the parents and on two bulked samples of yellow-flowered and white-flowered offspring. Expression profiling and RNA-seq results were then confirmed using a combination of fine-mapping and whole-genome resequencing. Finally, high-performance liquid chromatography (HPLC) and transcription electron microscopy (TEM) demonstrated that BjPC2 might be important in xanthophyll esterification, a process that limits degradation and increases sequestration of this major carotenoid group. Our results provide insight into the genetic mechanisms of flower color in B. juncea and act as a solid foundation for further functional validation of candidate genes and marker-based breeding of B. juncea varieties.
Materials and methods
Plant materials and population construction
The white-flowered JG800-1(genotype: Bjpc1Bjpc1Bjpc2Bjpc2) and yellow-flowered L12-5 (genotype: BjPC1BjPC1BjPC2BjPC2) lines of B. juncea were self-pollinated for over five generations to yield stable petal coloration. In our previous study, we successfully generated nine BC3 lines (designated XG1-9) with 1:1 segregation at the white locus. Line XG1 was selected for mapping BjPC1. After applying the developed BjPC1 linkage markers to identify the remaining eight lines (XG2–9), we confirmed that XG2, XG7, XG8 and XG9 had the same genotype as XG1. However XG3, XG4, XG5 and XG6 segregated in the BjPC2/Bjpc2 locus. Therefore, line XG3 was used to map BjPC2. A BC4 population (donor parent: L12-5; recurrent parent: JG800-1) comprising 2,016 individuals was used for the molecular mapping of BjPC2 (for pedigree and genesis, see supplementary Fig. S1). All the materials were provided by the Department of Rapeseed Research at Huazhong Agricultural University.
Allelism analysis
To determine allelism between XG3 and XG1, the yellow-flowered XG1 (expected genotype: BjPC1Bjpc1Bjpc2Bjpc2) and XG3 (expected genotype: Bjpc1Bjpc1BjPC2Bjpc2) were self-fertilized and the crossed. All resultant progeny (15 individuals; expected phenotype: yellow flowers; expected genotypes: BjPC1Bjpc1BjPC2Bjpc2, BjPC1Bjpc1Bjpc2Bjpc2 or Bjpc1Bjpc1BjPC2Bjpc2) were self-fertilized and simultaneously test-crossed with JG800-1 (genotype: Bjpc1Bjpc1Bjpc2Bjpc2) to yield 19 populations. The allelic relationship between XG3 and XG1 was then identified through an assay of flower-color segregation ratio. If XG3 was not allelic to XG1, selfed populations would have a segregation ratio of 15:1 or 3:1 (yellow: white); ratios for test-crossed populations would be 3:1 or 1:1 (Supplementary Fig. S2a). However, if XG3 was allelic to XG1, the two genotypes would be identical and selfing would result in two segregating populations: one with a 3:1ratio and the other comprised entirely of yellow-flowered plants. Test-crossed populations would exhibit all yellow flowers and 1:1 segregating populations (Supplementary Fig. S2b).
DNA library construction and whole-genome resequencing
Using CTAB, total genomic DNA was isolated from young leaves of parents and BC4 plants (Doyle 1990). The DNA was quantified using the NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). To generate the bulked samples, 30 yellow-flowered and 30 white-flowered plants were chosen from the BC4 population; an equal amount of DNA from each plant per group was mixed to form the yellow-flowered (Y-pool) and white-flowered (W-pool) set at a final concentration of 40 ng/μL. Isolated DNA was used to generate sequencing libraries. DNA samples were sonicated to produce 350-bp fragments. Sheared DNA was end-repaired, a single nucleotide (A) overhang was added subsequently, and then sequencing adapters were ligated using T4 DNA ligase. Polymerase chain reaction was performed and the products were then purified and sequenced on an Illumina HiSeq system using the standard protocol. The two parental lines had a sequencing depth of ~ 20×; each bulk, ~ 30×. The low-quality reads (quality score < 20e) were filtered out, and raw reads were sorted to each progeny based on barcode sequences. After barcodes were trimmed, clean, high-quality reads from the same sample were mapped onto the B. juncea genome sequence (955.08 Mb; Yang et al. 2016) in Burrows-Wheeler Aligner (Li et al. 2009). Duplicates were marked in Samtools (Li and Durbin 2009), while local realignment and base recalibration were performed in GATK (Mckenna et al. 2010). Both programs were combined for a SNP-calling analysis using default parameters, generating a SNP set. All identified SNPs shared across the bulk were considered polymorphic in association studies. Next, Δ (SNP-index) was calculated; this is an association analysis method that finds significant differences in genotype frequency between two pools (Abe et al. 2012). The upper limit of Δ (SNP-index) is expected to be 0.5 for a BC4 population. To determine and obtain the association threshold, we performed loess regression fitting (Abe et al. 2012). Candidate BjPC2 regions over the threshold (99.7th -percrntile) were extracted from Linkage group by BMK (Beijing, China).
Fine mapping of the white-flowered gene Bjpc2
The BC4 population (1:1 yellow: white ratio) was used to fine-map Bjpc2. Equivalent amounts of DNA from 12 yellow-flowered BC4 plants were randomly selected to construct two yellow-flowered gene pools. Two white-flowered gene pools were built in the same way, and all four were used to screen molecular markers. New simple-sequence repeat (SSR) markers were developed in candidate intervals according to the B. juncea reference genome sequences. The SSR primers were developed using WebSat (http://wsmartins.net/websat/) (see Supplementary Table S1 for full list). Recombinant individuals in BC4 were screened using markers with tight linkage to BjPC2, and their genetic distances were calculated using the Kosambi function (Kowalski et al. 1994). Synteny analysis was performed between the linked markers and the B. juncea genome, and then a physical map of the region encompassing the BjPC2 was constructed. To further fine map the BjPC2 and to identify the candidate gene, total RNA was extracted from the blooming petals of three yellow and white flowers in individuals from the BC4 population and was separately pooled to create two independent samples that were used to identify the differentially expressed genes (DEGs) between the yellow- and white-flowered B. juncea plants. For DEG screening, we used a P value < 0.005 and |log2 (fold change)| ≥ 1 as the threshold for determining the significance of differences in the gene expression. FC is the ratio of FPKM (fragments per kilobase per million) between yellow (Y) and white (W) petals.
Carotenoid analysis
Fresh petals were used for HPLC analysis coupled with photo-diode array detection analysis, following previously published methods (Cao et al. 2012). Non-esterified carotenoids were identified and quantified using HPLC with saponification, while esterified carotenoids were subjected to HPLC without saponification (Yamamizo et al. 2010). Carotenoid compounds were identified based on characteristic absorption spectra and typical retention time, taken from existing literature and standards of CaroNature Co. (Bern, Switzerland). Individual carotenoid content was calculated following previous methods (Morris et al. 2004). All analyses were conducted using at least three biological replicates. Means and standard errors were also calculated.
Transmission electron microscopy analysis
Yellow and white flower petals from three developmental stages (Fig. 1) were cut into 1 × 1 cm sections, fixed in a 2.5% (w/v) glutaraldehyde with 0.1 M phosphate buffer (pH 7.4), then fixed again in 1% OsO4 made using the same buffer (Zhu et al. 2014). Transmission electron microscopy of the petal samples was performed as previously described (Yi et al. 2010).

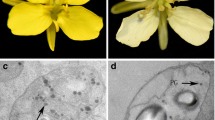
Petal coloration in the yellow- and white-flowered plants from the Brassica juncea BC4 population. Petals in three different developmental stages are shown. Stage 1 (a, e) is equivalent to 5 days before the flowering period, stage 2 (b, f) is 1 day before the flowering period, stage 3 (c, g) is the flowering period, bar 0.5 cm
Real-time quantitative PCR (qRT-PCR) analysis
Total RNA was isolated using Trizol (Invitrogen). One microgram of total RNA from stems, leaves, sepals, stamens, pistils, and petals of yellow- and white-flowered plants were processed by RNase-free DNaseI to remove contamination. Samples were then reverse-tran-scribed into first-strand cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, USA). Gene-specific primers were used in reactions performed with the SYBR Green Real-time PCR Master Mix (TOYOBO, Japan) in a Bio-Rad CFX96 in strument. Each qRT-PCR experiment was repeated three times. Relative expression levels were calculated using the 2-ΔΔCt method with Actin as an internal control.
Results
Allelism analysis between XG3 and XG1
The flower-color segregation ratios of 13 self-fertilized populations (F1 progeny of XG1 × XG3) were 15:1 (yellow:white) or 3:1 (X 2 0.05 = 3.84; P > 0.05). We did not observe populations with only yellow flowers. Furthermore, six corresponding test-crossed populations (F1 crossed with JG800-1) showed a flower color ratio of 3:1 or 1:1 (X 2 0.05 = 3.84; P > 0.05), with no population consisting entirely of yellow-flowered plants (Supplementary Tables S2, S3). These data clearly indicate that the segregating loci of XG3 and XG1 were not allelic. Thus, the genotypes of yellow-flowered plants in XG3 and XG1 were Bjpc1Bjpc1BjPC2Bjpc2 and BjPC1Bjpc1Bjpc2Bjpc2, respectively.
Whole-genome resequencing analysis
After filtering 149.09 G of raw data, 148.53 G of clean data was obtained for further analysis. The Q30 ratio was 88.76% and the GC content was 41.67%. The genome resequencing depth was 110 for four samples with an average > 37× and genome coverage > 88% (Supplementary Tables S3). Compared with the reference genome, 2,597,605 SNPs and 1,004,480 insertion–deletions (indels) were identified between the yellow- and white-flowered parents (Supplementary Tables S3). Additionally, 2,700,072 SNPs and 1,068,848 indels were found in the two BC4 bulk segregant populations. Association analysis between the two bulks was performed on 1,193,969 SNPs and a Δ (SNP-index) was indicated by calculating SNP-index (Fig. 2). Two candidate intervals (0.06–1.66 and 2.12–2.97 Mb) exceeded the threshold value for BjPC2 on J16 (chromosome B04 of B. juncea) (Fig. 3a). Together, the candidate regions were around 2.45 Mb with approximately 371 annotated genes. We found 431 SNPs with non-synonymous coding between the parents, and 291 SNPs with non-synonymous coding across the 110 genes between the two bulks. These SNPs are likely to be directly associated with flower color. In summary, we showed that the candidate genes for flower color were all located on chromosome B04 of B. juncea.

The layout of SNP index and Δ(SNP Index). a The distribution of SNP index of the white-flowered bulk from the BC4 population. b The distribution of SNP index of the yellow-flowered bulk from the BC4 population. c The layout of Δ(SNP Index) value. The red dashed line represents the threshold line. The x-axis (J01–J18) represents chromosome number in B. juncea chromosome V1. (Color figure online)

Fine-mapping of the BjPC2 in Brassica. juncea. a The candidate interval for BjPC2 on chromosome B04. b The genetic linkage map of the BjPC2 and associated molecular markers in BC4 population. c A partial physical map of linkage markers for BjPC2 on chromosome B04 of B. juncea. The black section shows the candidate region for BjPC2. d A partial physical map of Arabidopsis thaliana showing regions homologous to chromosome B04 of B. juncea
Fine mapping of BjPC2
For whole-genome resequencing, we developed 138 SSR primer pairs based on the sequence of the 2.45 Mb candidate regions. 16 SSR primer pairs detected polymorphisms between the two flower-color bulks were used to genomic DNA markers in 96 individuals, and nine polymorphic markers were identified as being linked to BjPC2. Meanwhile, the BjPC1 linkage markers were applied to screen the above 96 individuals, and the result revealed one SSR marker Z6SSR26 also linked with the BjPC2. To more precisely map BjPC2, all individuals in the BC4 population were screened by the linkage markers to evaluate their genetic distance. 81 recombinants between Z6SSR26 and, and 17 between SSR104 and BjPC2 were identified. On the side of SSR104, one recombinant was detected between the closest marker SSR111 and BjPC2, while four recombinants were detected between the closest marker SSR78 on the other side and BjPC2. By screening, we found that SSR80 and SS110 co-segregated with BjPC2 were no recombinant. Finally, we constructed a genetic linkage map surrounding the BjPC2 (Fig. 3), and the BjPC2 was located in an interval between the markers SSR78 and SSR111, corresponding to a genetic distance of 0.25-cM.
Candidate gene identification
Based on the linkage map (Fig. 3), BjPC2 was located on chromosome B04 of B. juncea. The target region for BjPC2 was narrowed to a 31-kb interval containing six predicted genes in the B. juncea reference genome (Table 1). The functional genes predicted in this region included two genes encoding phytyl ester synthase 2, one encoding an esterase/lipase/thioesterase family protein, one encoding a hydroxyproline-rich glycoprotein family protein, one encoding a DHHC-type zinc finger family protein and one encoding dsRNA-binding protein 3.
To study the differential expression of these six annotated genes and identify the candidate gene for BjPC2, we examined their transcripts in yellow and white petals. Five of the six genes contained non-synonymous mutations between the yellow- and white-flowered traits. Among these five genes, BjuB027331 and BjuB027333had extremely low transcript levels and were barely detected in petals. BjuB027330 and BjuB027332 expression were also low, but the latter increased in yellow petals compared to white petals. BjuB027334 is highly homologous to AT3G26840 (PES2) in Arabidopsis, which encodes a protein with phytyl-ester synthesis and diacylglycerol acyltransferase activities. BjuB027334 expression decreased approximately sevenfold in white petals than in yellow petals (Fig. 4a).Therefore, we hypothesized that the BjuB027334 gene was the most likely candidate gene for BjPC2. Subsequently, the expression analysis in different organizations from BC4 population showed that BjuB027334 was predominantly expressed in petals (Fig. 4b). Our RNA-seq analysis identified 164 DEGs, including 50 down-regulated and 114 up-regulated DEGs in the white petals. The BjuB027334 gene was down-regulated five-fold in the white petals than in the yellow petals. Nevertheless, there were no DEGs annotated to the carotenoid biosynthesis pathway. Together, these data further suggest that BjuB027334 is a candidate gene for the flower-color locus in B. juncea.

Gene expression data analysis. a Relative expression of genes in the candidate region of BjPC2 from differently colored BC4 plants at anthesis. “Yellow” and “white” represent individuals from yellow- and white-flowered plants, respectively. b Relative expression of BjuB027334 in different tissues of BC4 plants. (Color figure online)
Carotenoid analysis in yellow and white petals using HPLC
We analyzed carotenoid profiles in yellow and white petals to investigate whether low pigmentation was due to decreased carotenoid accumulation. Esterified carotenoids were abundant in yellow petals (Fig. 5). Furthermore, HPLC analysis without saponification revealed more carotenoid peaks in yellow flowers than white flowers (Fig. 5). The peaks that appeared after 20 min were more likely to be esterified carotenoids, saponified extracts rarely exhibited later peaks (Fig. 5). We then compared the retention times and absorbance spectra of carotenoid peaks with those of standards to identify six carotenoids: violaxanthin, 9-cis-violaxanthin, cis-neoxanthin, luteoxanthin, lutein and β-cryptoxanthin (Table 2). The six peaks exhibited some minor differences under HPLC without saponification; however, HPLC with saponification showed that violaxanthin, 9-cis-violaxanthin and cis-neoxanthin accounted for 91.6% of the total carotenoids in yellow petals, approximately eight times higher than their percentage in white petals (a significant difference; Table 2). These results indicate that white petals do not accumulate carotenoid esters, thus leading to a decrease in carotenoid accumulation. Moreover, yellow-flowered petals in B. juncea are the result of heavy carotenoid (particularly violaxanthin) accumulation.

Carotenoids profiles in yellow and white petals. Carotenoid extracts from mature petals were subjected to high-performance liquid chromatography without saponification (left column) or with saponification (right column). Carotenoid esters were detected in non-saponified yellow mature petals, while no such esters were detected in the saponified yellow mature petals due to the hydrolysis of ester compounds. The x-axis represents retention time and the y-axis represents milli-absorbance units (AU). (Color figure online)
Ultrastructure analysis of chromoplasts in yellow and white petals
We used TEM to determine whether changes in chromoplast morphology contributed to reduced petal pigmentation in white B. juncea flowers. At stage 1 in both yellow- and white-flowered plants, petal plastids exhibited a chloroplastic structure with granal stacking. In yellow flowers only, a few plastoglobules (PGs) appeared as electron-dense granules in plastids (Fig. 6a, d). In stage 2, plastids of yellow petals began differentiating into chromoplasts, and PGs were less electron-dense than stage-1 PGs (Fig. 6b). However, plastids in white petals were barely visible, and only a few, electron-dense PGs were present (Fig. 6e). During stage 3, complete chromoplasts were present in yellow petals, filled with numerous, fully developed PGs (as suggested by a low electron density inside and high density on the surface) (Fig. 6c). In contrast, stage-3 white petals did not exhibit further chromoplast development; the plastids appeared flattened, with only a few small PGs surrounded by an incomplete envelope membrane (Fig. 6f).

Transmission electron microscopy was used to analyze plastid morphology of petal three developmental stages (stages 1, 2, and 3) in the yellow (a–c) and white flowers (d–f). plastoglobules (PG), granal stacks (GS), and starch grain (S). Bar 0.5 µm
Discussion
This study characterized a B.juncea mutant with low petal pigmentation (Fig. 1). Using HPLC with saponification, we observed that violaxanthin and 9-cis-violaxanthin, which accounted for approximately 93% of the total carotenoids present and significantly greater than concentrations in white petals. Our results are consistent with previous work showing that yellow petal is due to the accumulation of these xanthophylls (Ariizumi et al. 2014; Neuman et al. 2014; Zhang et al. 2015). Moreover, HPLC without saponification showed that yellow-petals contained abundant carotenoid esters, mostly with backbones derived from violaxanthin, 9-cis-violaxanthin and cis-neoxanthin Thus, the three xanthophylls exist as a mixture of free (non-esterified) and esterified forms, with the latter being more prominent in yellow petals. Together, these results show that BjPC2 plays critical role in the production of xanthophyll esters required for yellow pigmentation in B. juncea petals.
In this study, we successfully combined BSA with whole-genome resequencing to fine-map the target gene BjPC2. Aided by the newly released B. juncea genome assemblies (Yang et al. 2016), the rapid development of a Brassica genomic infrastructure (including molecular marker techniques such SNP chips), and high-throughput genotyping by sequencing (GBS) using SNPs (Geng et al. 2016; Lee et al. 2016; Wang et al. 2016), we were able to detect two significant genomic regions for BjPC2, covering 2.45 Mb on chromosome B04. New SSR markers designed from sequences in these two regions allowed us to fine-map BjPC2, then localize it on to a 31-kb region on chromosome B04, where six genes were annotated. Our research clearly shows that combining BSA with whole genome resequencing is an effective and rapid method to locate genes for important traits in diploid plants. These two techniques can verify molecular marker-based mapping results while improving their accuracy (Wang et al. 2016).
Our qRT-PCR analyses found that most of the six annotated genes within the 31-kb region were lowly expressed or not detected in both yellow and white petals. Only BjuB027334 was significantly and differentially expressed, being far higher in yellow B. juncea petals. Additionally, whole-genome resequencing revealed that non-synonymous mutations between the yellow- and the white-flowered traits are present in BjuB027334. The loss of BjuB027334 expression is likely responsible for pigment absence in white B. juncea petals. Further expression profiling and RNA-seq analysis have partially confirmed this hypothesis. Moreover, RNA-seq results showed that white and yellow flowers did not have significantly different carotenogenic genes, suggesting that the former plants do not influence their transcription, despite a known correlation between carotenogenic gene expression with carotenoid content(Fraser et al. 2007; Ruiz-Sola and Rodríguez-Concepción 2012). In addition, the biosynthesis of xanthophyll was likely unaffected in white flowers, as HPLC without saponification showed that white petals had nearly the same xanthophyll content as yellow petals. Degradation pathways for modulated carotenoid levels yielded the same result. Further, none of the DEGs were annotated as either CCDs or NCEDs. Taken together, these results suggest that changes in the carotenoid biosynthesis or degradation are not the cause of any carotenoid reduction in white petals. However, we found evidence that variation in carotenoid accumulation has more to do with plastid morphology. Chloroplast–chromoplast conversion and chromoplast structure differed between yellow- and white-flowered plants. In the latter, PGs failed to fully develop, with far fewer PGs in the abnormal chromoplasts. Corroborating our results, several evidences proved that carotenoid sequestration by lipoproteins (Deruère et al. 1994) is associated with the plastid morphology, such as the chromoplast size and PG (or fibril) number within the chromoplast (Deruère et al. 1994; Fraser et al. 2007; Egea et al. 2010; Nogueira et al. 2013; Ariizumi et al. 2014). These findings suggest that the loss of xanthophyll esterification in white petals alters carotenoid sequestration, thereby reducing total carotenoid content and changing petal color.
Our HPLC analysis strongly suggests that BjPC2 primarily functions accumulate xanthophylls in the chromoplast PGs of yellow flowers by esterify. However, to date, research has isolated only one enzyme known to influence xanthophyll esterification in plants: the tomato PYP1 (Arabidopsis PES1 homolog) (Ariizumi et al. 2014). BjuB027334 is a down-regulated DEG annotated as PES2 and is a member of an acyltransferase family. In Arabidopsis, PES2 encodes a protein with phytyl ester synthesis and diacylglycerol acyltransferase activities during chlorosis, similar to PES1 (Lippold et al. 2012), although PES2 may contribute more to phytyl-ester synthesis. Both proteins are also acyltransferase restricted to chlorophyll-containing organisms. To date, no studies have directly reported a relationship between PES2 and yellow-pigment accumulation. In Arabidopsis, PES2 is also a prime candidate for wax-synthase activity involved in the production of medium-chain wax esters (Aslan et al. 2014). However, in tomato, loss of PYP1 function produces pale yellow petals and anthers (Ariizumi et al. 2014). We, therefore, speculate that BjuB027334 is the candidate gene involved in xanthophyll-ester production and thus the accumulation of more yellow pigment in B. juncea petals. These findings are consistent with results showing that esterification prevents xanthophyll degradation and increases xanthophyll sequestration efficiency. This mechanism facilitates the yellow flower organs to accumulate more esterified xanthophylls than free-form xanthophylls (Ariizumi et al. 2014).
In conclusion, our study demonstrates that BjPC2 is necessary for the production of xanthophyll esters in yellow B. juncea flowers. This gene may encode a protein that catalyzes xanthophyll-ester formation and leads to more carotenoid accumulation in chromoplasts of yellow flowers. We identified two candidate regions (totaling 2.45 Mb) on chromosome B04 with 271 candidate genes that are tightly associated with the white-flowered trait. Subsequently, the BjPC2 gene was fine-mapped on B04 and was physically localized to a 31-kb region. Expression profiling and RNA-seq analysis then revealed that BjuB027334, a gene with non-synonymous mutations between white- and yellow-petal genotypes, seems to be a candidate for the flower color loci in B. juncea. To conclude, this study successfully combined whole-genome resequencing technology with BSA to identify candidate genes required for B. juncea flower pigmentation, demonstrating the utility of these techniques. Our results lay both technical and empirical groundwork for further functional studies of white flower-related genes that will ultimately decipher the mechanisms responsible for the carotenoid content in yellow flowers. Such knowledge will also be valuable for research on other polychrome horticultural crops.
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178
Alam Z, Aziz M (1954) Inheritance of flower colour in some self-fertile oleiferous Brassicae. Pakistan J Sci Res 6:27–36
And PGK, Baker HG (1983) Insects as flower visitors and pollinators. Entomology 28:407–453
Ariizumi T, Kishimoto S, Kakami R, Maoka T, Hirakawa H, Suzuki Y, Ozeki Y, Shirasawa K, Bernillon S, Okabe Y, Moing A, Asamizu E, Rothan C, Ohmiya A, Ezura H (2014) Identification of the carotenoid modifying gene PALE YELLOW PETAL 1 as an essential factor in xanthophyll esterification and yellow flower pigmentation in tomato (Solanum lycopersicum). Plant J 79:453–465
Aslan S, Sun C, Leonova S, Dutta P, Dormann P, Domergue F, Stymne S, Hofvander P (2014) Wax esters of different compositions produced via engineering of leaf chloroplast metabolism in Nicotiana benthamiana. Metab Eng 25:103–112
Auldridge ME, Mccarty DR, Klee HJ (2006) Plant carotenoid cleavage oxygenases and their apocarotenoid products. Curr Opin Plant Biol 9:315–321
Bhuiyan M (1986) Inheritance of flower colour in Brassica juncea. Indian J Genet Plant Breed 46:563
Bramley PM (2002) Regulation of carotenoid formation during tomato fruit ripening and development. J Exp Bot 53:2107–2111
Camara B, Hugueney P, Bouvier F, Kuntz M, Monéger R (1995) Biochemistry and molecular biology of chromoplast development. Int Rev Cytol 163:175–247
Cao H, Zhang J, Xu J, Ye J, Yun Z, Qiang X, Xu J, Deng X (2012) Comprehending crystalline β-carotene accumulation by comparing engineered cell models and the natural carotenoid-rich system of citrus. J Exp Bot 63:4403–4417
Cutler AJ, Krochko JE (1999) Formation and breakdown of ABA. Trends Plant Sci 4:472–478
Deruère J, Römer S, D’Harlingue A, Backhaus RA, Kuntz M, Camara B (1994) Fibril assembly and carotenoid overaccumulation in chromoplasts: a model for supramolecular lipoprotein structures. Plant Cell 6:119–133
Downey RK (1990) Brassica oilseed breeding - achievements and opportunities. Plant Breed Abstr 60:1165–1170
Doyle J (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Egea I, Barsan C, Bian W, Purgatto E, Latché A, Chervin C, Bouzayen M, Pech JC (2010) Chromoplast differentiation: current status and perspectives. Plant Cell Physiol 51:1601–1611
Fraser PD, Enfissi EMA, Halket JM, Truesdale MR, Dongmei Y, Christopher G, Bramley PM (2007) Correction: manipulation of phytoene levels in tomato fruit: effects on isoprenoids, plastids, and intermediary metabolism. Plant Cell 19:3194–3211
Geng X, Jiang C, Yang J, Wang L, Wu X, Wei W (2016) Rapid Identification of candidate genes for seed weight using the SLAF-seq method in Brassica napus. PloS One 11:e0147580
Grotewold E (2006) The genetics and biochemistry of floral pigments. Annu Rev Plant Biol 57:761–780
Huang Z, Ban YY, Bao R, Zhang XX, Xu AX, Ding J (2014) Inheritance and gene mapping of the white flower in Brassica napus L. New Zeal J Crop Hort 42:111–117
Jambhulkar S, Raut R (1995) Inheritance of flower colour and leaf waxiness in Brassica carinata A. Br. Cruciferae Newsl 17:66–67
Khan F, Chai HH, Ajmera I, Hodgman C, Mayes S, Lu C (2017) A transcriptomic comparison of two bambara groundnut landraces under dehydration stress. Genes 8:121
Kowalski SP, Lan TH, Feldmann KA, Paterson AH (1994) Comparative mapping of Arabidopsis thaliana and Brassica oleracea chromosomes reveals islands of conserved organization. Genetics 138:499–510
Lee S, Lee SC, Byun DH, Lee DY, Park JY, Lee JH, Lee HO, Sung SH, Yang TJ (2014) Association of molecular markers derived from the BrCRTISO1 gene with prolycopene-enriched orange-colored leaves in Brassica rapa [corrected]. Theor Appl Genet 127:179–191
Lee KJ, Kim DS, Kim JB, Jo SH, Kang SY, Choi HI, Ha BK (2016) Identification of candidate genes for an early-maturing soybean mutant by genome resequencing analysis. Mol Genet Genom 291:1561–1571
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Li H, Huang W, Wang GL, Wang WL, Cui X, Zhuang J (2017) Transcriptomic analysis of the biosynthesis, recycling, and distribution of ascorbic acid during leaf development in tea plant (Camellia sinensis (L.) O. Kuntze). Sci Rep 7:46212
Lippold F, Dorp KV, Abraham M, Hölzl G, Wewer V, Yilmaz JL, Lager I, Montandon C, Besagni C, Kessler F (2012) Fatty acid phytyl ester synthesis in chloroplasts of Arabidopsis. Plant Cell 24:2001–2014
Liu XP, Tu JX, Chen BY, Fu TD (2004) Identification of the linkage relationship between the flower colour and the content of erucic acid in the resynthesized Brassica napus L. Yi Chuan Xue Bao 31:357–362
Mckenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA 88:9828–9832
Morris WL, Ducreux L, Griffiths DW, Stewart D, Davies HV, Taylor MA (2004) Carotenogenesis during tuber development and storage in potato. J Exp Bot 55:975–982
Negi MS, Devic M, Delseny M, Lakshmikumaran M (2000) Identification of AFLP fragments linked to seed coat colour in Brassica juncea and conversion to a SCAR marker for rapid selection. Theor Appl Genet 101:146–152
Neuman H, Galpaz N, Jr CF, Zamir D, Hirschberg J (2014) The tomato mutation nxd1 reveals a gene necessary for neoxanthin biosynthesis and demonstrates that violaxanthin is a sufficient precursor for abscisic acid biosynthesis. Plant J 78:80–93
Nogueira M, Mora L, Enfissi EMA, Bramley PM, Fraser PD (2013) Subchromoplast sequestration of carotenoids affects regulatory mechanisms in tomato lines expressing different carotenoid gene combinations. Plant Cell 25:4560–4579
Pearson OH (1929) A Dominant white flower color in Brassica oleracea L. Am Nat 63:561–565
Rahman MH (2001) Inheritance of petal colour and its independent segregation from seed colour in Brassica rapa. Plant Breeding 120:197–200
Rawat DS, Anand IJ (1986) Inheritance of flower colour in mustard mutant. Indian J Agric Sci 56:206–208
Ruiz-Sola MÁ, Rodríguez-Concepción M (2012) Carotenoid biosynthesis in Arabidopsis: a colorful pathway. Arabidopsis Book 10:e0158. https://doi.org/10.1199/tab.0158
Shi Q, Zhou L, Wang Y, Li K, Zheng B, Miao K (2015) Transcriptomic analysis of Paeonia delavayi wild population flowers to identify differentially expressed genes involved in purple-red and yellow petal pigmentation. PloS One 10:e0135038
Singh KH, Chauhan JS (2011) Genetics of flower colour in Indian mustard (Brassica juncea L. Czern & Coss). Indian J Genet Plant Breed 71:377–378
Singh D, Pokhriyal S, Mangath K (1964) Inheritance of some characters in Brassica juncea. Indian J Genet Plant Breed 24:288–290
Walter MH, Strack D (2011) Carotenoids and their cleavage products: biosynthesis and functions. ChemInform 42:663–692
Wang Y, Xiao L, Guo S, An F, Du D (2016) Fine mapping and whole-genome resequencing identify the seed coat color gene in Brassica rapa. PloS One 11:e0166464
Woods DL, Capcara JJ, Downey RK (1991) The potential of mustard (Brassica juncea (L.) Coss) as an edible oil crop on the Canadian Prairies. Can J Plant Sci 71:195–198
Xiao L, Zhao H, Zhao Z, Du D, Xu L, Yao Y, Zhao Z, Xing X, Shang G, Zhao H (2013) Genetic and physical fine mapping of a multilocular gene Bjln1 in Brassica juncea to a 208-kb region. Mol Breed 32:373–383
Xiong B, Ye S, Qiu X, Liao L, Sun G, Luo J, Dai L, Rong Y, Wang Z (2017) Transcriptome analyses of two citrus cultivars (Shiranuhi and Huangguogan) in seedling etiolation. Sci Rep 7:46245
Yamamizo C, Kishimoto S, Ohmiya A (2010) Carotenoid composition and carotenogenic gene expression during Ipomoea petal development. J Exp Bot 61:709–719
Yang J, Liu D, Wang X, Ji C, Cheng F, Liu B, Hu Z, Chen S, Pental D, Ju Y, Yao P, Li X, Xie K, Zhang J, Wang J, Liu F, Ma W, Shopan J, Zheng H, Mackenzie SA, Zhang M (2016) The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat Genet 48:1225–1232
Yi B, Zeng F, Lei S, Chen Y, Yao X, Zhu Y, Wen J, Shen J, Ma C, Tu J (2010) Two duplicate CYP704B1-homologous genes BnMs1 and BnMs2 are required for pollen exine formation and tapetal development in Brassica napus. Plant J 63:925–938
Zhang B, Liu C, Wang Y, Yao X, Wang F, Wu J, King GJ, Liu K (2015) Disruption of a CAROTENOID CLEAVAGE DIOXYGENASE 4 gene converts flower colour from white to yellow in Brassica species. New Phytol 206:1513–1526
Zhang L, Xu B, Wu T, Yang Y, Fan L, Wen M, Sui J (2017) Transcriptomic profiling of two Pak Choi varieties with contrasting anthocyanin contents provides an insight into structural and regulatory genes in anthocyanin biosynthetic pathway. BMC Genom 18:288
Zhu L, Zeng X, Chen Y, Yang Z, Qi L, Pu Y, Yi B, Wen J, Ma C, Shen J, Tu J, Fu T (2014) Genetic characterisation and fine mapping of a chlorophyll-deficient mutant (BnaC.ygl) in Brassica napus. Mol Breed 34:603–614
Acknowledgements
This research was funded by the National Key Research and Development Program of China (Grant number 2016YFD0101300), the National Science Foundation of China (NSFC, Grant number 31571698) and the Program for Modern Agricultural Industrial Technology System (Grant number nycytx-00501).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Ethical standards
The authors declare that the experiments complied with the current laws of China.
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, X., Li, R., Chen, L. et al. Fine-mapping and candidate gene analysis of the Brassica juncea white-flowered mutant Bjpc2 using the whole-genome resequencing. Mol Genet Genomics 293, 359–370 (2018). https://doi.org/10.1007/s00438-017-1390-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-017-1390-5