
Abstract
Spinach is a popular leafy green vegetable due to its nutritional composition. It contains high concentrations of vitamins A, E, C, and K, and folic acid. Development of genetic markers for spinach is important for diversity and breeding studies. In this work, Next Generation Sequencing (NGS) technology was used to develop genomic simple sequence repeat (SSR) markers. After cleaning and contig assembly, the sequence encompassed 2.5% of the 980 Mb spinach genome. The contigs were mined for SSRs. A total of 3852 SSRs were detected. Of these, 100 primer pairs were tested and 85% were found to yield clear, reproducible amplicons. These 85 markers were then applied to 48 spinach accessions from worldwide origins, resulting in 389 alleles with 89% polymorphism. The average gene diversity (GD) value of the markers (based on a GD calculation that ranges from 0 to 0.5) was 0.25. Our results demonstrated that the newly developed SSR markers are suitable for assessing genetic diversity and population structure of spinach germplasm. The markers also revealed clustering of the accessions based on geographical origin with clear separation of Far Eastern accessions which had the overall highest genetic diversity when compared with accessions from Persia, Turkey, Europe, and the USA. Thus, the SSR markers have good potential to provide valuable information for spinach breeding and germplasm management. Also they will be helpful for genome mapping and core collection establishment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinach (Spinacia oleracea L.) is an edible member of the Amaranthaceae family and is believed to have originated in Persia (modern Iran and neighboring countries) (Boswell 1949). Spinach arrived in China via Nepal during the seventh century but did not reach Europe until the twelfth century (Kuwahara et al. 2014). From there, spinach spread to the United States during the nineteenth century (Kuwahara et al. 2014). It is currently produced in more than 50 countries (Kuwahara et al. 2014), however, 91% of spinach is cultivated in China (21 million tons FAO, 2013). The USA and Japan rank second and third in production with approximately 300,000 tons each per year.
Spinach is a popular leafy green vegetable due to its nutritional composition (Ito 2000). It contains high concentrations of vitamins A, E, C, and K; folic acid and oxalic acid. A 100 g serving of fresh spinach provides 20% of the recommended daily intake of ascorbic acid (vitamin C), β-carotene (provitamin A), lutein, folate (vitamin B9), phylloquinone (vitamin K1), and α-tocopherol (vitamin E) (Lester et al. 2013).
Although spinach is not a model organism, its diploid, average-sized genome makes it easy to study. Spinach has 12 chromosomes (2n) with an estimated genome size of 980 Mb (Arumuganathan and Earle 1991). Unlike many model vegetable crops, spinach is cross-pollinated. Despite its popularity and nutritional importance, very little research has examined the genetic diversity of spinach germplasm. Indeed, a literature search up to and including 2016 reveals only six major studies in this area. Hu et al. (2007) studied 38 spinach accessions and 10 commercial hybrids and demonstrated that target region amplification polymorphism (TRAP) markers were useful in determining their genetic diversity. However, they were unable to correlate spinach genetic diversity with geographical origin. In the same year, Khattak et al. (2007) analyzed 33 spinach hybrids using 13 polymorphic simple sequence repeat (SSR) markers. These individuals grouped into three clusters. While two clusters contained European spinach cultivars, the third cluster was a mixture of European and Asian spinach types. Kuwahara et al. (2014) screened 50 accessions from geographically diverse regions with six previously described markers (Khattak et al. 2007). In this study, West Asian accessions (from Afghanistan, Iran, Iraq, and Syria) had the highest level of diversity. Feng et al. (2015) designed pairs of primers for 100 SSRs and 24 retrotransposons from BAC end sequences, but did not use the markers to study genetic diversity. Chitwood et al. (2016) analyzed population structure and genetic diversity of 288 accessions with 1733 single nucleotide polymorphisms (SNPs) using a genotyping by sequencing (GBS) approach. Similarly, Ma et al. (2016) and Shi et al. (2016) determined population structure and/or genetic diversity of more than 300 spinach accessions using GBS-generated SNP data. All three of these most recent studies also did association mapping in spinach for important traits including bolting, height, leaf morphology, and oxalate concentration (Chitwood et al. 2016; Ma et al. 2016; Shi et al. 2016).
The first objective of the present study was to develop new SSR markers by sequencing the spinach nuclear genome using MiSeq Next Generation Sequencing Technology. A draft genome sequence was assembled and used to identify SSRs and appropriate primers for marker development. SSRs are 1–6 nucleotide long genomic repeats and are one of the most informative DNA repeats in plant genetic research (Zalapa et al. 2012). These sequences are found throughout eukaryotic genomes and are highly polymorphic (Park et al. 2009). Unlike SNPs which are diallelic, SSRs are often multiallelic. Moreover, SSRs do not require sequencing or the design of specific assays such as kompetitive allele specific PCR (KASP) for genotyping. The second aim of this study was to verify the effectiveness of the SSRs for studying genetic diversity. Thus, 85 selected primer pairs were applied to 48 spinach (Far East, Persia, Turkey, Europe, and USA) accessions.
Materials and methods
Plant material and DNA isolation
A total of 48 S. oleracea accessions from 20 countries were used as plant material (Table 1). All accessions were provided by the United States Department of Agriculture (USDA, Plant Introduction Research Unit, Ames, IA, USA). Five seeds from each accession were planted and grown in soil containing perlite and natural fertilizer in the greenhouse at Izmir Institute of Technology. Genomic DNA extraction was performed from the youngest leaves. Total DNA was extracted using CTAB extraction buffer according to Doyle and Doyle (1990). DNA quality was checked by agarose gel electrophoresis with quantification done by spectrophotometer (Thermo Scientific, Multiskan GO).
DNA sequencing
The spinach cultivar S. oleracea Universal was sequenced by next generation sequencing (MiSeq) technology by the Biotechnology Center at the University of Wisconsin–Madison, USA (https://www.biotech.wisc.edu). This technology produced 300 nucleotide long, paired-end reads. Further information can be found at the SRA database of NCBI (SRX2266012).
Data pre-processing
Adapter and linker sequences were removed from reads with cutadapt version 1.8.3 software (Martin 2011). Reads that were shorter than 50 nucleotides were removed from the dataset using default settings with minimum-length switch (−m = 50). Cleaned reads were mapped to the human genome using Bowtie version 2.1.0 with default settings to remove possible human contaminant sequences that may come from sequencing or library preparation steps (Langmead and Salzberg 2012).
Sequence assembly
AbySS version 1.3.6, a de novo, parallel, paired-end sequence assembler, was used for sequence assembly (Simpson et al. 2009). To find the best assembly, more than 100 runs were performed with different parameters such as changing kmer (all possible substrings of length k contained in reads). Various measurements were considered for selecting the best assembly: N50 (weighted median of contig lengths), assembly nucleotide length (closeness to the estimated size of the S. oleracea genome), length of largest contig and contig number. The settings that were finally chosen to create contigs were the default settings with kmer k:175.
SSR detection, annotation, and primer design
Any contig smaller than 1000 nucleotides was removed from the dataset. The dataset was then used for SSR detection with our in-house tool SiSeer (http://bioinformatics.iyte.edu.tr/index.php?n=Softwares.SiSeeR). The minimum number of repeats required to identify perfect SSRs were: ten for mononucleotides, four for dinucleotides, and three for motifs that comprised three or more nucleotides. Identified SSR sequences were padded with 100 nucleotides and extracted as FASTA sequences. These sequences were used as query sequences against the Uniprot non-redundant plant protein database (Taxonomy = Viridiplantae) with BLASTX version 2.2.30 (Altschul et al. 1990). For all SSRs, primer pairs were designed using Primer3 (primer_core) version 2.3.6 using the following settings: primer task = generic, primer optimum size = 20, primer minimum size = 18, primer maximum size = 24, primer product size = 100–300, primer minimum TM = 50, primer maximum TM = 60, and primer optimum TM = 55.
Validation of genomic SSR markers
Genomic DNA of PI 175312 (accession 14) was used for SSR validation using the dye-terminator sequencing method. Polymerase chain reaction (PCR) products were used as template in the dye-terminator sequencing reaction and were purified with the DNA Clean & Concentrator–5 Kit (Zymo Research). Sequencing reactions were prepared using GenomeLab DTCS Quick Start Kit (Beckman Coulter) according to the manufacturer’s instructions. Thermal cycling conditions were 30 cycles of 96 °C for 20 s, 50 °C for 20 s, 60 °C for 4 min. ZR DNA Sequencing Clean-up Kit (Zymo Research) was used to purify the reaction mixture for each SSR amplicon. Products were then suspended in 30 μL sample loading solution (Beckman Coulter) and run on a Beckman CEQ8800 capillary electrophoresis device using the LFR-c method (injection voltage 2.0 kV for 10–15 s, separation temperature 60 °C, separation voltage 7.4 kV, separation time 45 min).
SSR amplification
SSR amplification for each primer pair was carried out in a final volume of 20 µl and contained 2 μl 10X PCR buffer (50 mM KCl, 10 mM Tris–HCl, 1.5 mM MgCl2, pH: 8.3), 1 μl MgCl2 (1.5 mM), 0.75 μl dNTP (0.2 mM), 0.75 μl each forward and reverse primers (10 pmol), 0.75 μl Taq polymerase (0.25 U), 13 μl sterile double-distilled water, and 1 μl DNA (10 ng/μl). PCR conditions were 95 °C for 5 min for one cycle, followed by 35 cycles of 30 s at 94 °C for denaturation, 30 s at 55 °C for annealing and 30 s at 72 °C for extension, and the final extension cycle was at 72 °C for 10 min. PCR reactions were performed in a Veriti 96-Well Thermal Cycler (Applied Biosystems). PCR products were separated using a capillary electrophoresis instrument (Fragment Analyzer Automated CE System, Advanced Analytical) with the DNF-900 dsDNA Reagent Kit (Advanced Analytical); SSR alleles were visualized and scored using PROSize 2.0 software version 1.2.1.1 (Advanced Analytical).
SSR data analysis
SSR alleles were scored for presence (1) or absence (0). Average gene diversity (Nei 1973) was calculated depending on the frequency of the allele for each SSR marker using the GDdom online computer program (Abuzayed et al. 2016). This program uses the formula of Roldan-Ruiz et al. (2000): GDi = 2fi (1—fi). Where GDi is the gene diversity of marker ‘i,’ fi is the frequency of the amplified allele (band presence), and 1—fi is the frequency of the null allele.
Marker data were used to infer population structure of the 48 spinach accessions with the Structure computer program (Structure 2.3.4 version) (Pritchard et al. 2000). Models with 1–20 subpopulations (K) were tested for ten iterations. Burn-in period was 10,000 and the number of Monte Carlo Markov Chain repeats was 300,000. Structure Harvester computer program (Earl and Von Holdt 2012) was used to calculate ΔK values for each model based on posterior probabilities. The model with the highest ΔK was selected as the best. Inferred ancestry threshold was set as ≥0.60 to assign the accessions to subpopulations. Accessions with lower probabilities were assigned to the admixed group. To study genetic diversity, the binary presence/absence data were used to generate a dissimilarity matrix using the Dice coefficient in the Darwin5 computer program (Perrier and Jacquemoud-Collet 2006). The distance data were used to construct a dendrogram of the 48 spinach accessions using the unweighted neighbor-joining method. Principal coordinate analyses (PCoA) based on genetic distances between accessions was also carried out with Darwin5 software.
Discriminant analyses were performed with the SPSS 22.0 software program for Windows (SPSS Science, Chicago, IL, USA). Differences in the amount of diversity between different locations (Europe, Far East, Persia, Turkey, and USA) were determined by analysis of variance (ANOVA) and Tukey’s HSD (honest significant difference) test at P < 0.05.
Results
Sequence assembly and simple sequence repeat identification
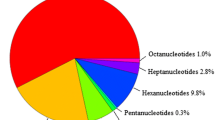
The sequencing raw data output consisted of 15,925,882 reads comprising about 5000 Mb. After cleaning, the sequence reads resulted in 4,736 Mb with an average sequence length of 297.5 nucleotides. Sequencing quality was established using FastQC. These sequences were assembled using Abyss (Simpson et al. 2009). Assembly fragments longer than 1000 nucleotides were treated as contigs and used for further procedures. The total number of contigs was 18,545 which encompassed 24.18 Mb, representing 2.5% of the spinach genome (Table S1). These contigs were mined for SSRs, resulting in the identification of 3,852 SSRs. SSR lengths varied from 6 to 124 nucleotides with an average of 15.4 nucleotides. Among all identified SSRs, the most frequent type was mononucleotide repeats, representing 54.3% of all SSRs. The second most common repeat type was dinucleotide repeats, representing 15.7% of the total. Hexanucleotide repeats were the third most common, representing 11.8% of all SSRs (Fig. 1). A/T repeats were the most frequent among mononucleotide repeats (98.9%). Among dinucleotide repeats, TA was most frequently observed (21.8%) followed by AG/CT and GA/TC repeats (14.4 and 13.7%, respectively) (Table 2).

Simple sequence repeat types in S. oleracea genome
Primer design and validation
A total of 3275 primer pairs were successfully developed for the 3,852 newly identified SSRs. The remaining 577 SSRs did not have enough flanking sequence to design suitable primers. These SSRs were located at the ends or very close to the ends of the contigs.
To validate the presence of the expected SSR motifs, six amplicons from PI 175,312 (accession 14) were sequenced using the dye-terminator method. All six sequences contained the expected SSR motifs, proving the identity of our primers as SSR markers (Table S2).
Gene diversity, population structure, and genetic diversity analysis
We tested 100 of the designed primers for their amplification efficiency using accessions 13, 14, 15, and 16. A total of 85 primers (85%) successfully amplified PCR products from these four accessions. These 85 SSR primers were then applied to 48 spinach accessions from throughout the world (Table 1). Turkey, Persia, Europe, the USA, and the Far East (China, Japan, South Korea, and Thailand) were represented by 8, 12, 10, 9, and 9 accessions, respectively. In all, 77 of the primers (91%) produced polymorphic bands and generated 389 alleles, 346 of which were polymorphic (89%; Table S3). The average number of amplified fragments per genomic SSR marker was 4.6. The average gene diversity value of the markers (based on a GD calculation that ranges from 0 to 0.5) was 0.25, with the highest value calculated for soSSR2173 (0.49). The lowest value was zero for the monomorphic markers: soSSR64, soSSR976, soSSR1003, soSSR1016, soSSR1667, soSSR1088, soSSR2141, and soSSR2389.
The SSR data were used to study the population structure and genetic diversity of the 48 spinach accessions. According to population structure analysis, the spinach accessions fell into three subpopulations (Table 1) with a relatively high ΔK value at K = 3 (Figure S1). The standard deviation (SD) for each value of K is also important when deciding the correct number of subpopulations and these values supported the choice of K = 3 (Figure S1). Subpopulation 1 had eight individuals, subpopulation 2 had 13 individuals, subpopulation 3 had six individuals, and the admixed group had 21 individuals (Table 1).
Principal coordinate analysis (PCoA) of the SSR data plotted in the first two dimensions (Fig. 2) showed clear separation between the three subpopulations with the admixed individuals occupying the space between the three groups. The analysis also suggested that subpopulation 1 was more diverse than the others as it had a wider distribution in the plot.

PCoA for the three subpopulations of spinach. Subpopulation 1, subpopulation 2, subpopulation 3, and admixed are shown in purple, orange, red, and green, respectively
A dendrogram was drawn using the Dice coefficient and the unweighted neighbor-joining algorithm (Figure S2). A strong correlation between the neighbor-joining dendrogram and the distance matrix was evident from the Mantel test result of r = 0.94. Average pairwise dissimilarity among the 48 spinach accessions was 0.24, with the highest value (0.37) calculated between accessions PI 179508 (Iraq) and PI 165504 (India). The lowest dissimilarity was 0.13, calculated between accessions NSL 184379 (USA) and NSL 42771 (USA). The spinach accessions were grouped into three clusters (A, B, and C) in the dendrogram (Figure S2). Cluster A of the neighbor-joining dendrogram included 25 accessions. Genetic diversity in Cluster A ranged between 0.16 and 0.37 with an average pairwise dissimilarity of 0.26 (data not shown). All of the Far East accessions were in Cluster (A). Accessions from Persia were generally located in Cluster A (67%) with only four found outside this cluster. In addition to the eight accessions from Persia, Cluster A also contained five from Turkey, two from USA, and one from Europe. Cluster B contained 21 accessions, generally those from the USA and Europe. Genetic diversity of Cluster B ranged between 0.13 and 0.27 with an average pairwise dissimilarity of 0.20 (data not shown). All but two accessions from the USA were in Cluster B (78%). Similarly, only one accession from Europe fell outside of Cluster (B). Cluster B also contained four accessions from Persia and three from Turkey. Cluster C had only two accessions: one from Persia and one from Turkey.
When the population structure and genetic diversity results were compared, there was a 38% overlap of accessions belonging to subpopulation 1 and Cluster A of the dendrogram, a 52% overlap in subpopulation 2 and Cluster B, and a 50% overlap between subpopulation 3 and Cluster C.
Genetic diversity and geographical origin
The spinach accessions were grouped based on origin in order to compare the average diversity (based on the Dice coefficient) of material from different geographical regions. According to Tukey’s HSD test, there was no significant difference between diversity of the European and USA accessions which had average values of 0.19 and 0.20, respectively (Table 3). The Persian, Turkish, and Far Eastern accessions had significantly higher diversity than those from Europe and the USA with average diversities ranging from 0.24 to 0.26. Different amounts of genetic diversity in the material from different origins were also evident in PCoA analysis (Fig. 3). Accessions from Europe and USA were more tightly grouped than those from Persia, Turkey, and the Far East. The US and European accessions overlapped with each other and with accessions from Persia and Turkey. Those from the Far East were widely spread and only overlapped with accessions from Persia. Persian and Turkish accessions were fairly well-distributed and occupied very similar locations in the plot.

PCoA for the five groups of spinach. Accessions from Persia, Far East, Europe, USA, and Turkey are shown in green, purple, blue, yellow, and red, respectively
Discussion
NGS technology is widely used to develop SSR markers for non-model organisms. It is more cost-effective and faster than Sanger sequencing and also does not require SSR enrichment (Zalapa et al. 2012). In previous work, NGS was used to obtain approximately 50% coverage of the spinach cv. Viroflay genome (Dohm et al. 2014). These sequencing data were used for gene prediction in the species (Minoche et al. 2015; Yang et al. 2016) as well as for SNP and SSR identification (Yang et al. 2016). However, none of the 57,519 SSRs for which primers were designed were tested (Yang et al. 2016). Indeed, before NGS sequencing, only 113 spinach-specific SSR markers had been published with only 13 SSR markers used for genetic diversity analysis (Khattak et al. 2006, 2007; Feng et al. 2015). Thus, the goal of the present work was to develop and test additional SSR markers for use in diversity studies and breeding of spinach and closely related species. We obtained a draft genome assembly covering 2.5% of the estimated 980 Mb spinach genome through NGS and identified 3,852 non-redundant SSR markers. Thus, one SSR marker was detected every 6.3 Kb (on average) in the 24 Mb of contigs. Although Yang et al. (2016) identified many more SSRs in their analysis of the spinach genome sequence (Dohm et al. 2014), they did not provide frequency statistics. However, our results are consistent with those of Cardle et al. (2000) who identified one SSR every 6.8 Kb in genomic DNA for many plant species.
Among the 3,852 SSRs, the most frequent were mononucleotide repeats, representing 54.3% of all SSRs. Dinucleotides ranked second and represented 15.7% of all SSRs. Our results are in agreement with Yang et al. (2016) who studied spinach and Cardle et al. (2000) who reported that mononucleotide repeats are the most abundant repeats in several plant genomes. Among mononucleotides, A/T repeats were the most frequent (98.9%) with TA repeats (21.9%) the most common for dinucleotides. Among trinucleotides, ATA/TAT repeats were the most frequent (13.2%). In this study, AT-rich repeats were the most common repeat type especially for mononucleotides, dinucleotides, and trinucleotides. These findings are in agreement with Yang et al. (2016) and Cardle et al. (2000) who showed that A/T repeats and TA repeats are the most common repeat type for mononucleotides and dinucleotides, respectively.
In our study, the newly developed SSR markers revealed geographical clustering among spinach accessions from different origins. In a previous study that used 48 accessions, spinach germplasm did not show tight clustering based on geographically proximity (Hu et al. 2007). However, Khattak et al. (2007) found clustering of European spinach germplasm according to breeding station source. Kuwahara et al. (2014) were also successful in correlating geographical origin and genetic diversity of spinach samples collected from diverse locations.
It is believed that spinach originated in Persia (Boswell 1949). As expected, Persian accessions had relatively high genetic diversity. In the dendrogram, accessions from Persia mostly grouped together in Cluster A near to those from Turkey. Similar results were observed in the PCoA in which the Persian and Turkish accessions overlapped. This is not surprising because, compared to the other sampled regions, Persia and Turkey are geographically close. In addition, Anatolia and Persia have had active commercial trade throughout history. Therefore, it is reasonable to expect that the two regions have similar spinach germplasm and levels of diversity. Accessions from the Far East also fell into Cluster A of the dendrogram with most of the other Asian accessions. Interestingly, these accessions were tightly clustered in the dendrogram and yet were highly diverse and showed very little overlap with other material in the PCoA. Indeed, the only significant overlap was with accessions from Persia, the most likely source of spinach in the Far East. The Far Eastern accessions probably had tight grouping and little coincidence with other genetic material because access to this region is restricted by the Gobi Desert in Mongolia and the Himalayas in India. The relatively high genetic diversity in the material may be explained by adoption of a wider variety of spinach genotypes than in the USA or Europe, where spinach is a very minor crop. Most accessions from the USA grouped closely together in the dendrogram and were found in the same cluster as most of the European and a few Persian accessions in the two-dimensional PCoA. This is expected because accession in the USA probably originated in Europe. European accessions were intermixed with some Persian and Turkish accessions. This result is not a surprise because spinach reached Europe from Persia via Anatolia. Turkish spinach germplasm did not demonstrate tight grouping. They were spread throughout the dendrogram, and often associated with Persian accessions. Overall, the results showed that Turkish spinach has high genetic diversity compared to material from Europe and the USA.
In conclusion, our results demonstrated that the newly developed SSR markers are suitable for assessing genetic diversity and population structure of spinach germplasm. The study supports the proposed distribution route for spinach from Persia to Asia and from Persia to Europe via Turkey and then to America. Moreover, the SSR markers revealed that Far Eastern accessions may be an interesting source of new variability for spinach. Thus, the newly developed and tested SSR markers will provide valuable information for spinach breeding and germplasm management and will be helpful for genome mapping and core collection establishment.
References
Abuzayed M, El-Dabba N, Frary A, Doganla r S (2016) GDdom: an online tool for calculation of dominant marker gene diversity. Biochem Genet 43:1–3
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Rep 9(3):208–218
Boswell VR (1949) Garden peas and spinach from the Middle East. Reprint of “Our Vegetable Travelers” Natl Geogr 96:2
Cardle L, Milbourne D, Macaulay M, Marshall D, Waugh R (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156:847–854
Chitwood J, Shi A, Mou B, Evans M, Clark J, Motes D, Chen P, Hensley D (2016) Population structure and association analysis of bolting, plant height, and leaf erectness in spinach. HortScience 51(5):481–486
Dohm JC, Minoche AE, Holtgräwe D, Capella-Gutiérrez S, Zakrzewski F, Tafer H, Rupp O, Sörensen TR, Stracke R, Reinhardt R, Goesmann A, Kraft T, Schulz, Stadler PF, Schmidt T, Gabaldón T, Lehrach H, Weisshaar B, Himmelbauer H (2014) The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature 505(7484):546–549
Doyle JJ, Doyle JE (1990) Isolation of plant DNA from fresh tissue. Focus (12):13–15
Earl DA, Von Holdt BM (2012) Structure Harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Feng C, Bluhm BH, Correll JC (2015) Construction of a spinach bacterial artificial chromosome (BAC) library as a resource for gene identification and marker development. Plant Mol Biol Rep 33(6):1996–2005
Food and Agriculture Organization of the United Nati1ons, FAOSTAT (2013) http://www.fao.org/corp/statistics/en. Accessed 04 July 2016
Hu J, Mou B, Vick BA (2007) Genetic diversity of 38 spinach (Spinacia oleracea L.) germplasm accessions and 10 commercial hybrids assessed by TRAP markers. Genet Resour Crop Evol 54(8):1667–1674
Ito M, Ohmido N, Akiyama Y, Fukui K, Koba T (2000) Characterization of spinach chromosomes by condensation patterns and physical mapping of 5 S and 45 S rDNAs by FISH. J Am Soc Hortic Sci 125(1):59–62
Khattak JZK, Torp AM, Andersen SB (2006) A genetic linkage map of Spinacia oleracea and localization of a sex determination locus. Euphytica 148:311–318
Khattak JZK, Christiansen JL, Torp AM, Andersen SB (2007) Genic microsatellite markers for discrimination of spinach cultivars. Plant Breeding 126(4):454–456
Kuwahara K, Suzuki R, Ito Y, Mikami T, Onodera Y (2014) An analysis of genetic differentiation and geographical variation of spinach germplasm using SSR markers. Plant Genet Resour 12(02):185–190
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359
Lester GE, Makus DJ, Hodges DM, Jifon JL (2013) Summer (Subarctic) versus winter (Subtropic) production affects spinach (Spinacia oleracea L.) leaf bionutrients: Vitamins (C, E, Folate, K1, provitamin A), lutein, phenolics, and antioxidants. J Agric Food Chem 61(29):7019–7027
Ma J, Shi A, Mou B, Evans M, Clark JR, Motes D, Correll JC, Xiong H, Qin J, Chitwood J, Weng Y (2016) Association mapping of leaf traits in spinach (Spinacia oleracea L.). Plant Breeding 404:1–6
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17(1):10–12
Minoche AE, Dohm JC, Schneider J, Holtgräwe D, Viehöver P, Montfort M, Sörensen TR, Weisshaar B, Himmelbauer H (2015) Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome Biol 16(1):184
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70(12):3321–3323
Park YJ, Lee JK, Kim NS (2009) Simple sequence repeat polymorphisms (SSRPs) for evaluation of molecular diversity and germplasm classification of minor crops. Molecules 14(11):4546–4569
Perrier X, Jacquemoud-Collet JP (2006) DARwin software. http://darwin.cirad.fr
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Roldan-Ruiz I, Dendauw J, Bockstaele EV, Depicker A, Loose MD (2000) AFLP markers reveal high polymorphic rates in ryegrasses (Lolium spp.). Mol Breed 6:125–134
Shi A, Beiquan M, James CC (2016) Association analysis for oxalate concentration in spinach. Euphytica 212(1):17–28
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ BL (2009) AbySS: a paralel assembler for short read sequence data. Genome Res 19(6):1117–1123
Yang XD, Tan HW, Zhu WM (2016) SpinachDB: a well-characterized genomic database for gene family classification and SNP information of spinach. PloS One 11(5):e0152706
Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, McCown B, Harbut R, Simon P (2012) Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot 99(2):193–208
Acknowledgements
This research was supported by funding from an Izmir Institute of Technology Scientific Research Project, IYTE-BAP2012-2014.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Data availability
Sequence data are available at the SRA database of NCBI (SRX2266012).
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Göl, Ş., Göktay, M., Allmer, J. et al. Newly developed SSR markers reveal genetic diversity and geographical clustering in spinach (Spinacia oleracea). Mol Genet Genomics 292, 847–855 (2017). https://doi.org/10.1007/s00438-017-1314-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-017-1314-4