
Abstract
Trichomes on plants, similar to fine hairs on animal and human bodies, play important roles in plant survival and development. They also represent a useful model for the study of cell differentiation. Although the regulatory gene network of unicellular trichome development in Arabidopsis thaliana has been well studied, the genes that regulate multicellular trichome development remain unclear. We confirmed that Cucumis sativus (cucumber) trichomes are multicellular and unbranched, but identified a spontaneous mutant, trichome-less (tril), which presented a completely glabrous phenotype. We compared the transcriptome profilings of the tril mutant and wild type using the Illumina HiSeq 2000 sequencing technology. A total of 991 genes exhibited differential expression: 518 were up-regulated and 473 were down-regulated. We further identified 62 differentially expressed genes that encoded crucial transcription factors and were subdivided into seven categories: homeodomain, MADS, MYB, and WRKY domains, ethylene-responsive, zinc finger, and other transcription factor genes. We further analyzed the tissue-expression profiles of two candidate genes, GLABRA2-like and ATHB51-like, using qRT-PCR and found that these two genes were specifically expressed in the epidermis and trichomes, respectively. These results and the tril mutant provide useful tools to study the molecular networks associated with multicellular trichome development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plant trichomes are specialized organs originating from the epidermal cells. They may be uni- or multicellular, glandular or glandless, and branched or unbranched (Werker 2000). Trichomes provide an excellent model system for studying cell differentiation and cell morphogenesis at the single-cell level (Hülskamp 2004; Szymanski et al. 2000). Trichomes are also involved in many developmental processes including deterrence of insects, herbivores, and microbes, maintenance of leaf temperature, reflectance of visible and UV light, and transpiration regulation (Bennett and Wallsgrove 1994; Wagner et al. 2004). They may also be involved in water absorption, secretion of heavy metals, and pollen collection (Choi et al. 2001; Küpper et al. 2000; Wagner et al. 2004; Werker 2000).
Unicellular trichome differentiation in Arabidopsis thaliana is regulated by a series of competing transcription factors that either up- or down-regulate relevant activities. The down-regulating activity inhibits trichome differentiation and is passed through neighboring cells (Larkin et al. 1997; Ohashi et al. 2002; Schnittger et al. 1999). The four crucial positive transcription factors are GL1 (GLABRA1), an R2R3 MYB protein (Kirik et al. 2005), GL3 (GLABRA3) and EGL3 (ENHANCER OF GLABRA3), which are both basic helix–loop–helix proteins (Payne et al. 2000), TTG1 (TRANSPARENT TESTA GLABRA1), a WD40-repeat protein (Walker et al. 1999), and a MYB–bHLH–WD40 complex that activates GL2 (GLABRA2), a homeodomain protein that initiates trichome differentiation (Pesch and Hülskamp 2009). Proteins in the small R3 single-repeat MYB family involved in the down-regulation of unicellular trichome differentiation include TRY (TRIPTYCHON), CPC (CAPRICE) and ETC (ENHANCER OF TRY AND CPC). These proteins compete with the R2R3 MYB protein GL1 and bind to bHLH proteins including GL3 or EGL3, inhibiting their activity (Kirik et al. 2004; Wester et al. 2009).
Cucumis sativus (cucumber) is an annual species that is commercially important worldwide. C. sativus has diverse sex types and is a useful model plant for sex determination studies (Tanurdzic and Banks 2004). Trichomes are commonly found on most organs of wild-type C. sativus plants, and fruit spines (trichomes on the fruits) are valued and commercially important (Fig. 1a, b). We report here a spontaneous mutant, CGN19839, which appeared completely glabrous (Fig. 1c, d); we named this mutant “trichome-less (tril)”. The regulatory mechanisms of multicellular trichome development have not been extensively studied. Here, we confirmed that C. sativus trichomes are multicellular and unbranched. We compared the transcriptome changes between the tril mutant and wild type with the Illumina HiSeq 2000 sequencing technology. From our results, we identified a series of candidate genes encoding crucial transcription factors that can be associated with multicellular trichome development.

Phenotypes of leaves, branches, flowers, fruits, and stems of various C. sativus lines. a, b Wild type. c, d The tril mutant. The tril mutant is characterized by the absence of leaf, branch, flower, fruit, and stem trichomes. e, f F1 progeny from a cross between tril and wild type. Scale bars represent 5 mm
Materials and methods
Plant materials
C. sativus North China inbred line 06-1 was used as the wild-type background. The spontaneous mutant CGN19839 was used as the tril mutant. The F1 descendants and F2 segregating population were generated from a cross between 06-1 and CGN19839. All C. sativus plants were grown in a greenhouse under a natural photoperiod.
Scanning electron microscopy analysis
Three centimeter-long juvenile leaf samples were fixed in aqueous formaldehyde–acetic acid–ethanol (FAA) containing 50 % (v/v) ethanol, 5 % (v/v) acetic acid, and 3.7 % (v/v) formaldehyde at 4 °C for 24 h, dehydrated through a seven-step ethanol series (50–100 %, v/v) and critical point dried in a Leica EM CPD030 desiccator. The dried specimens were sputter-coated with gold–palladium and observed under JSM-6360LV and JEM-2010HT scanning electron microscopes.
Illumina HiSeq 2000 transcriptome sequencing
Poly-A RNA was isolated with a TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA) and fragmented into 100-bp inserts to create cDNA libraries. Quality control used Pico green fluorescence spectrophotometry and an Agilent 2100 bioanalyzer.
Gene annotation, expression, classification, and enrichment analyses
The spliced reads were mapped using TopHat (http://ccb.jhu.edu/software/tophat/), and transcripts were assembled with Cufflinks (http://cufflinks.cbcb.umd.edu/). Gene assemblies were annotated with BLAST searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi/) against the C. sativus database. The reference C. sativus genome was ftp://ftp.ncbi.nlm.nih.gov/genomes/Cucumis_sativus/.
For each gene, the expression level was calculated from the baseMean value, which was the sequencing depth for each transcript normalized to the library size. The HTSeq (http://www-huber.embl.de/users/anders/HTSeq/) and DESeq (http://www-huber.embl.de/users/anders/DESeq/) programs were used to measure differential gene expression. The functional categories of genes were established with the eggNOG (evolutionary genealogy of genes: Non-supervised orthologous groups) (http://eggnog.embl.de/). GO (Gene ontology) and KEGG (Kyoto encyclopedia of genes and genomes) enrichment analyses were performed via http://www.geneontology.org/ and http://www.genome.jp/kegg/. Analyses were conducted as described by Powell et al. (2012), Ashburner et al. (2000), and Kanehisa et al. (2004), respectively.
Extraction of nucleic acids and qRT-PCR
Total RNA was extracted using RNeasy Plant Mini Kit (Qiagen, Hilden, Germany), and first-strand cDNA was prepared according to PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, Kyoto, Japan) protocol. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was conducted using a SYBR Premix Ex Taq II Kit (TaKaRa). CsActin3 was used as the reference gene to correct gene expression values. The primers used in this study are listed in Supplementary Dataset S1.
Accession numbers
Transcriptome raw data can be found in the NCBI BioProject database under accession numbers: SAMN03276490 (WT) and SAMN03382548 (tril).
Results
Tril acts as a single dominant nuclear gene
A cross between the tril mutant and wild type generated F1 descendants that all had a wild-type trichome phenotype (Fig. 1e, f). The F2 segregating population contained 79 of a total of 296 F2 plants that exhibited the mutant phenotype. This closely fit an expected 3:1 segregation ratio (217 wild type, 79 mutant type, χ 2 = 0.450 < \(\chi_{0.05,1}^{2}\) = 3.84), indicating that the mutation was recessive and Trichome-less (Tril) acts as a single dominant nuclear gene.
Multicellular trichome structure controlled by Tril
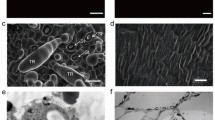
Scanning electron microscopy imaging showed that three distinct types of cells constitute a C. sativus trichome: (a) a non-glandular pyramid-shaped apical cell, (b) a two- to four-celled cylindrical-shaped stalk, and (c) a flat pie-shaped base cell, in contact with the epidermis (Fig. 2a, b). The tril mutant, however, exhibited a completely glabrous morphology, and only epidermal cells including stomata and encircling guard cells were visible (Fig. 2c, d) indicating that Tril functions in trichome cell fate determination.

Scanning electron microscopy images of wild-type and tril leaves. a, b Trichomes on the wild-type leaf. Arrows indicate the apical cell (middle), stalk cell (upper), and base cell (lower). c, d The tril leaf
Read mapping and gene annotation
The transcriptomes of 21-day-old leaves (trichomes attached with leaf epidermis) from both the tril mutant and wild type (each sample was mixed with three individuals) were sequenced on the Illumina HiSeq 2000 platform. The sequences of the cDNA libraries generated 40.41 and 105.40 million high-quality reads (reads obtained after data quality filtering from raw reads), respectively, with an average read length of 100 bp. A total of 36.74 (90.90 %) and 98.57 (93.52 %) million reads were mapped to the C. sativus genome, including 34.17 (93.01 %) and 92.04 (93.38 %) million unique reads and 2.57 (6.99 %) and 6.53 (6.62 %) million multiple reads (reads can be mapped to various genes), among which 35.54 (81.77 %) and 95.44 (82.91 %) million were mapped to genes, 34.99 (98.43 %) and 92.87 (97.31 %) million were mapped to exons, and 7.92 (18.23 %) and 19.68 (17.09 %) million were mapped to intergenic regions, respectively (Supplementary Dataset S2). All high-quality reads were assembled by the Cufflinks program and annotated via BLAST searches against the C. sativus database. As a result, 17,148 genes were predicted with annotations for each sample (Supplementary Dataset S3 and S4).
Functional category analysis by eggNOG
The functions of orthologous genes were classified with eggNOG. A total of 12,885 (75.14 %) genes were categorized into 25 groups (Fig. 3). “Function unknown” and “General function prediction only” represented the largest clusters in C. sativus species, containing 3528 (27.38 %) and 2041 (15.84 %) genes, respectively. These were followed by “Signal transduction mechanisms” (934 genes, 7.25 %), “Posttranslational modification, protein turnover, chaperones” (906 genes, 7.03 %), and “Transcription” (690 genes, 5.36 %) clusters that contained slightly fewer genes. “Extracellular structures” (10 genes, 0.08 %) and “Cell motility” (6 genes, 0.05 %) clusters had the least orthologous genes (Supplementary Dataset S5).

eggNOG functional category analysis. A total of 12,885 (75.14 %) genes were categorized into 25 eggNOG. The details of eggNOG functional category analysis can be found in Supplementary Dataset S5
Differential expression and enrichment analyses
Gene expression levels were calculated by baseMean values. Differential gene expression was defined with the following statistical parameters: P < 0.05 with a fold change >2 or <−2. Following this analysis, a total of 991 genes were identified that exhibited differential expression, including 518 up-regulated genes and 473 down-regulated genes (Fig. 4, Supplementary Dataset S6).

Differential gene expression analysis. a Volcano plot of log2(fold change) versus −log10(P-value); the horizontal line represents P = 0.05. b MA plot of log2(fold change) versus baseMean fold change. Genes in blue are differentially expressed, and genes in orange show no significant difference in expression. The details of differential expression analysis can be found in Supplementary Dataset S6 (colour figure online)
The biological functions of these genes were determined by GO enrichment analysis. Among all 53 GO terms, “Thylakoid” (P = 2.04E−30; 71 up-regulated genes, 1 down-regulated), “Plastid” (P = 5.32E−15; 120 up-regulated genes, 4 down-regulated), and “Extracellular region” (P = 8.09E−08; 25 up-regulated genes, 10 down-regulated) were the top three significantly enriched clusters. “Sequence-specific DNA binding transcription factor activity” (P = 2.25E−05; 24 up-regulated genes, 34 down-regulated), “Response to biotic stimulus” (P = 2.12E−04; 24 up-regulated genes, 12 down-regulated), and “Response to external stimulus” (P = 5.99E−04; 27 up-regulated genes, 17 down-regulated) clusters were also significantly enriched (Fig. 5, Supplementary Dataset S7).

GO enrichment analysis of differentially expressed genes. The red line represents P = 0.05. The details of GO enrichment analysis can be found in Supplementary Dataset S7 (colour figure online)
KEGG enrichment analysis was carried out to determine if trichome-related genes were involved in specific pathways. A total of 257 differentially expressed genes were classified into the 24 KEGG categories. The most enriched category was “Energy metabolism” (P = 3.22E−12; 46 differentially expressed genes), followed by “Biosynthesis of other secondary metabolites” (P = 3.11E−03; 14 differentially expressed genes) category (Fig. 6, Supplementary Dataset S8–S10).

KEGG enrichment analysis of differentially expressed genes. The red line represents P = 0.05. The details of KEGG enrichment analysis can be found in Supplementary Dataset S8–S10 (colour figure online)
Candidate genes associated with multicellular trichome development
A total of 62 differentially expressed genes were screened out. These genes encoded crucial transcription factors and were identified as candidates associated with multicellular trichome development. Twenty-two genes were not expressed at all (baseMean = 0) in the tril mutant, but were highly expressed in the wild-type background. The 62 genes were subdivided into seven categories according to the protein structure: homeodomain, MADS, MYB, and WRKY domains, ethylene-responsive, zinc finger, and other transcription factor genes (Table 1). For example, we identified a “Homeodomain-leucine zipper protein ATHB51-like” gene. The ATHB51 protein combines with LEAFY and together act as meristem regulators that induce CAULIFLOWER expression; the ATHB51 gene also controls leaf morphogenesis, floral meristem determinacy, and bract formation in A. thaliana (Saddic et al. 2006). We also found a “Homeodomain-leucine zipper protein GLABRA2-like” gene. The GLABRA2 gene regulates unicellular trichome and root hair development in A. thaliana (Pesch and Hülskamp 2009). Additionally, the 62 differentially expressed genes included a “Floral homeotic protein APETALA1-like” gene. The APETALA1 transcription factor acts as a MADS-domain protein and floral meristem regulator, and also interacts with LEAFY to regulate the flowering time genes SVP (SHORT VEGETATIVE PHASE) and AGL24 (AGAMOUS-LIKE24) (Gregis et al. 2006, 2008; Pastore et al. 2011). Finally, a “Transcription factor RAX2-like” gene was identified. The RAX2 (REGULATOR OF AXILLARY MERISTEMS2) gene belongs to the class R2R3 MYB family and regulates axillary meristem formation (Müller et al. 2006; Stracke et al. 2001).
Tissue-specific qRT-PCR validation of GLABRA2-like and ATHB51-like identified from the transcriptomic sequencing
Homeodomain-leucine zipper genes are unique to plants and participate in a wide variety of biological roles including trichome development (Ariel et al. 2007). Previous studies have shown that the class IV homeodomain-leucine zipper subfamily members, such as the A. thaliana GLABRA2 gene and its homologs, e.g., the Gossypium hirsutum (cotton) GaHOX1 gene and the Solanum lycopersicum (tomato) Wo (Woolly) gene, all regulate trichome development (Guan et al. 2008; Pesch and Hülskamp 2009; Yang et al. 2011); and the class I homeodomain-leucine zipper subfamily members have widely diverse roles that are specific to different tissues and organs in different species (Ariel et al. 2007), such as the A. thaliana ATHB51 gene regulates floral meristem determinacy, bract formation, and leaf morphology (Saddic et al. 2006), and its homolog, e.g., the Pisum sativum (pea) Tl (Tendril-less) gene, regulates tendril formation (Hofer et al. 2009). These results implied that homologs in the class I homeodomain-leucine zipper subfamily have acquired distinct functions over evolution. In the present study, both the GLABRA2 homolog, GLABRA2-like, and the ATHB51 homolog, ATHB51-like, were not expressed at all in the tril mutant, but were highly expressed in the wild-type background, indicating that GLABRA2-like and ATHB51-like might be linked to multicellular trichome development in C. sativus.
We conducted qRT-PCR for GLABRA2-like and ATHB51-like using leaf samples (with trichomes attached to leaves; leaf trichomes could not be stripped perfectly from leaves) at the same developmental stage as those in the transcriptomic sequencing. There was good agreement between the two methods for the expression of these genes; neither GLABRA2-like nor ATHB51-like was expressed in the tril mutant, but both were highly expressed in the wild-type background (Fig. 7a, b; Table 2).

Tissue-specific qRT-PCR validation of GLABRA2-like and ATHB51-like identified from the transcriptomic sequencing. The tissues examined included WT (wild-type) leaves (trichomes plus epidermis), tril leaves, WT branch trichomes, WT branch epidermis, tril branches, WT fruit trichomes, WT fruit epidermis, tril fruits, WT stem trichomes, WT stem epidermis, tril stems, WT cotyledons, tril cotyledons, WT roots, and tril roots. CsActin3 was used as the reference gene to correct gene expression values. Error bars represent the standard deviation of three biological replicates
Second, tissue-specific qRT-PCR with total RNA extracted from detached trichomes and trichome-stripped epidermis of various tissues including branches, fruits, and stems was conducted to detect the spatial expression patterns of GLABRA2-like and ATHB51-like. The results showed that the expression level of GLABRA2-like in the epidermis was significantly higher than in trichomes in these tissues (Fig. 7a). Conversely, ATHB51-like had a significantly higher expression level in trichomes compared with the epidermis (Fig. 7b). Neither of them was expressed in these tissues in the tril mutant (Fig. 7a, b). Given that trichomes develop from epidermal cells, these results indicated that both GLABRA2-like and ATHB51-like function in a tissue-specific manner in epidermal and trichome tissues, respectively.
Third, tissue-specific qRT-PCR with trichome-less tissues including cotyledons and roots was conducted. The results showed that neither GLABRA2-like nor ATHB51-like was expressed in the tril mutant, but both exhibited low-level expression in the wild-type background (Fig. 7a, b). In particular, ATHB51-like was barely expressed in the root tissues, which was in accordance with the previous results showing that ATHB51-like was expressed specifically in trichomes (Fig. 7b).
Thus, the wild-type and tril mutant transcriptomes represent a useful reference for further studies on multicellular trichome development.
Discussion
To date, there have been few reports of regulatory genes that control multicellular trichome development in plants. The regulatory mechanisms of multicellular trichome development differ from that in species such as A. thaliana and G. hirsutum. It appears that both A. thaliana and G. hirsutum use similar genes to control unicellular trichome development. For example, the G. hirsutum GaMYB2 gene controls cotton fiber development and is homologous to the A. thaliana GL1 gene. The GaMYB2 gene successfully rescued the trichome phenotype of the A. thaliana gl1 mutant (Wang et al. 2004). Similarly, the G. hirsutum GaHOX1 gene, which is homologous to the A. thaliana GL2 gene, rescued the trichome phenotype of the A. thaliana gl2 mutant (Guan et al. 2008).
However, plants with multicellular trichomes function with a different set of genes. For example, the Antirrhinum majus MIXTA gene, a MYB-like gene, regulates floral papillae development. This gene could not rescue the trichome phenotype of the A. thaliana gl1 mutant. However, this gene regulates trichome differentiation in Nicotiana tabacum (tobacco) (Payne et al. 1999). The S. lycopersicum Wo gene is a class IV homeodomain-leucine zipper gene. This gene is homologous to the A. thaliana GL2 and PDF2 (PROTODERMAL FACTOR2) genes, but Wo has an additional role in embryo development and the homozygous mutant is embryo lethal (Yang et al. 2011). Thus, the research literature suggests that the developmental processes of unicellular and multicellular trichomes are controlled by different regulatory genes. We concentrated on a new set of 62 transcription factor genes identified in our transcriptome analysis.
The genes identified as being associated with trichome development all appear to encode transcription factors. In this study, a total of 62 genes identified as candidates encoding crucial transcription factors associated with multicellular trichome development could be subdivided into seven categories: homeodomain, MADS, MYB, and WRKY domains, ethylene-responsive, zinc finger, and other transcription factor genes. In A. thaliana, responses to environmental conditions and developmental regulation of floral meristems, vascular systems, and lateral organs all involve homeodomain-leucine zipper transcription factor genes (Ariel et al. 2007; Baima et al. 2001; Henriksson et al. 2005; Otsuga et al. 2001; Williams et al. 2005). These transcription factor genes are unique to flowering plants and are involved in a range of activities; e.g., the GLABRA2 gene regulates unicellular trichome and root hair development in A. thaliana (Pesch and Hülskamp 2009). We identified a “Homeodomain-leucine zipper protein GLABRA2-like” gene which was not expressed in the tril mutant, but expressed strongly in the wild-type epidermis tissues. The A. thaliana ATHB51 gene regulates several different processes including floral meristem determinacy, bract formation, and leaf morphology (Saddic et al. 2006). The P. sativum Tl gene is homologous to the A. thaliana ATHB51 gene and regulates leaf tendril formation (Hofer et al. 2009). However, we found that a “Homeodomain-leucine zipper protein ATHB51-like” gene was expressed specifically in the wild-type trichome tissues, but was not expressed in the tril mutant. It is likely that the C. sativus ATHB51-like gene is associated with multicellular trichome development or another species-specific developmental process.
The MADS-domain transcription factors, initially identified as floral meristem regulators, play important roles especially in flower and fruit development (Smaczniak et al. 2012). It has been reported that this family of proteins or genes can interact with or be regulated by homeodomain or MYB-domain transcription factors. For example, the A. thaliana APETALA1 protein, a MADS-domain transcription factor, interacts with LEAFY to regulate floral meristem and sepal development (Gregis et al. 2006; William et al. 2004); in addition, APETALA1 is also regulated by LMI2 (LATE MERISTEM IDENTITY2), which belongs to the R2R3 MYB family (Pastore et al. 2011). In our analysis, an APETALA1-like gene and an R2R3 MYB RAX2-like gene exhibited significantly differential expression between the tril mutant and wild type.
In addition to transcriptional regulators, trichome development may also be regulated directly by phytohormones; e.g., salicylic and jasmonic acid decrease and increase the number of trichomes on leaves, respectively, in A. thaliana (Traw and Bergelson 2003). Ethylene gas can stimulate epidermal cell division in C. sativus, resulting in aberrant guard cell and trichome formation (Kazama et al. 2004). Cytokinin and gibberellin signals, which regulate inflorescence trichome initiation in A. thaliana, are integrated by ZFP6 (Zinc Finger Protein 6), a new zinc finger transcription factor gene (Zhou et al. 2013). A series of ethylene-responsive and zinc finger genes were also identified in our transcriptome analysis. These genes may be associated with phytohormone-related regulation of multicellular trichome development in C. sativus.
In conclusion, the loss of Tril function led to the identification of a group of 62 candidate genes that appear to be associated with multicellular trichome development in C. sativus. Our approach and the transcriptome profiling of the tril mutant provide useful tools to study the relevant molecular networks of multicellular trichome development in plants.
References
Ariel FD, Manavella PA, Dezar CA, Chan RL (2007) The true story of the HD-Zip family. Trends Plant Sci 12:419–426
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29
Baima S, Possenti M, Matteucci A, Wisman E, Altamura MM, Ruberti I, Morelli G (2001) The Arabidopsis ATHB-8 HD-Zip protein acts as a differentiation-promoting transcription factor of the vascular meristems. Plant Physiol 126:643–655
Bennett RN, Wallsgrove RM (1994) Secondary metabolites in plant defence mechanisms. New Phytol 127:617–633
Choi YE, Harada E, Wada M, Tsuboi H, Morita Y, Kusano T, Sano H (2001) Detoxification of cadmium in tobacco plants: formation and active excretion of crystals containing cadmium and calcium through trichomes. Planta 213:45–50
Gregis V, Sessa A, Colombo L, Kater MM (2006) AGL24, SHORT VEGETATIVE PHASE, and APETALA1 redundantly control AGAMOUS during early stages of flower development in Arabidopsis. Plant Cell 18:1373–1382
Gregis V, Sessa A, Colombo L, Kater MM (2008) AGAMOUS-LIKE24 and SHORT VEGETATIVE PHASE determine floral meristem identity in Arabidopsis. Plant J 56:891–902
Guan XY, Li QJ, Shan CM, Wang S, Mao YB, Wang LJ, Chen XY (2008) The HD-Zip IV gene GaHOX1 from cotton is a functional homologue of the Arabidopsis GLABRA2. Physiol Plant 134:174–182
Henriksson E, Olsson AS, Johannesson H, Johansson H, Hanson J, Engström P, Söderman E (2005) Homeodomain leucine zipper class I genes in Arabidopsis. Expression patterns and phylogenetic relationships. Plant Physiol 139:509–518
Hofer J, Turner L, Moreau C, Ambrose M, Isaac P, Butcher S, Weller J, Dupin A, Dalmais M, Le Signor C, Bendahmane A, Ellis N (2009) Tendril-less regulates tendril formation in pea leaves. Plant Cell 21:420–428
Hülskamp M (2004) Plant trichomes: a model for cell differentiation. Nat Rev Mol Cell Biol 5:471–480
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32:D277–D280
Kazama H, Dan H, Imaseki H, Wasteneys GO (2004) Transient exposure to ethylene stimulates cell division and alters the fate and polarity of hypocotyl epidermal cells. Plant Physiol 134:1614–1623
Kirik V, Simon M, Wester K, Schiefelbein J, Hulskamp M (2004) ENHANCER of TRY and CPC2 (ETC2) reveals redundancy in the region-specific control of trichome development of Arabidopsis. Plant Mol Biol 55:389–398
Kirik V, Lee MM, Wester K, Herrmann U, Zheng Z, Oppenheimer D, Schiefelbein J, Hulskamp M (2005) Functional diversification of MYB23 and GL1 genes in trichome morphogenesis and initiation. Development 132:1477–1485
Küpper H, Lombi E, Zhao FJ, McGrath SP (2000) Cellular compartmentation of cadmium and zinc in relation to other elements in the hyperaccumulator Arabidopsis halleri. Planta 212:75–84
Larkin JC, Marks MD, Nadeau J, Sack F (1997) Epidermal cell fate and patterning in leaves. Plant Cell 9:1109–1120
Müller D, Schmitz G, Theres K (2006) Blind homologous R2R3 Myb genes control the pattern of lateral meristem initiation in Arabidopsis. Plant Cell 18:586–597
Ohashi Y, Oka A, Ruberti I, Morelli G, Aoyama T (2002) Entopically additive expression of GLABRA2 alters the frequency and spacing of trichome initiation. Plant J 29:359–369
Otsuga D, DeGuzman B, Prigge MJ, Drews GN, Clark SE (2001) REVOLUTA regulates meristem initiation at lateral positions. Plant J 25:223–236
Pastore JJ, Limpuangthip A, Yamaguchi N, Wu MF, Sang Y, Han SK, Malaspina L, Chavdaroff N, Yamaguchi A, Wagner D (2011) LATE MERISTEM IDENTITY2 acts together with LEAFY to activate APETALA1. Development 138:3189–3198
Payne T, Clement J, Arnold D, Lloyd A (1999) Heterologous myb genes distinct from GL1 enhance trichome production when overexpressed in Nicotiana tabacum. Development 126:671–682
Payne CT, Zhang F, Lloyd AM (2000) GL3 encodes a bHLH protein that regulates trichome development in Arabidopsis through interaction with GL1 and TTG1. Genetics 156:1349–1362
Pesch M, Hülskamp M (2009) One, two, three…models for trichome patterning in Arabidopsis? Curr Opin Plant Biol 12:587–592
Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, Jensen LJ, von Mering C, Bork P (2012) eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res 40:D284–D289
Saddic LA, Huvermann B, Bezhani S, Su Y, Winter CM, Kwon CS, Collum RP, Wagner D (2006) The LEAFY target LMI1 is a meristem identity regulator and acts together with LEAFY to regulate expression of CAULIFLOWER. Development 133:1673–1682
Schnittger A, Folkers U, Schwab B, Jürgens G, Hülskamp M (1999) Generation of a spacing pattern: the role of TRIPTYCHON in trichome patterning in Arabidopsis. Plant Cell 11:1105–1116
Smaczniak C, Immink RG, Angenent GC, Kaufmann K (2012) Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139:3081–3098
Stracke R, Werber M, Weisshaar B (2001) The R2R3-MYB gene family in Arabidopsis thaliana. Curr Opin Plant Biol 4:447–456
Szymanski DB, Lloyd AM, Marks MD (2000) Progress in the molecular genetic analysis of trichome initiation and morphogenesis in Arabidopsis. Trends Plant Sci 5:214–219
Tanurdzic M, Banks JA (2004) Sex-determining mechanisms in land plants. Plant Cell 16:S61–S71
Traw MB, Bergelson J (2003) Interactive effects of jasmonic acid, salicylic acid, and gibberellin on induction of trichomes in Arabidopsis. Plant Physiol 133:1367–1375
Wagner GJ, Wang E, Shepherd RW (2004) New approaches for studying and exploiting an old protuberance, the plant trichome. Ann Bot 93:3–11
Walker AR, Davison PA, Bolognesi-Winfield AC, James CM, Srinivasan N, Blundell TL, Esch JJ, Marks MD, Gray JC (1999) The TRANSPARENT TESTA GLABRA1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in Arabidopsis, encodes a WD40 repeat protein. Plant Cell 11:1337–1349
Wang S, Wang JW, Yu N, Li CH, Luo B, Gou JY, Wang LJ, Chen XY (2004) Control of plant trichome development by a cotton fiber MYB gene. Plant Cell 16:2323–2334
Werker E (2000) Trichome diversity and development. Adv Bot Res 31:1–35
Wester K, Digiuni S, Geier F, Timmer J, Fleck C, Hülskamp M (2009) Functional diversity of R3 single-repeat genes in trichome development. Development 136:1487–1496
William DA, Su Y, Smith MR, Lu M, Baldwin DA, Wagner D (2004) Genomic identification of direct target genes of LEAFY. Proc Natl Acad Sci USA 101:1775–1780
Williams L, Grigg SP, Xie M, Christensen S, Fletcher JC (2005) Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development 132:3657–3668
Yang C, Li H, Zhang J, Luo Z, Gong P, Zhang C, Li J, Wang T, Zhang Y, Lu Y, Ye Z (2011) A regulatory gene induces trichome formation and embryo lethality in tomato. Proc Natl Acad Sci USA 108:11836–11841
Zhou Z, Sun L, Zhao Y, An L, Yan A, Meng X, Gan Y (2013) Zinc Finger Protein 6 (ZFP6) regulates trichome initiation by integrating gibberellin and cytokinin signaling in Arabidopsis thaliana. New Phytol 198:699–708
Acknowledgments
We thank San-Wen Huang for providing us with the tril mutant. We thank Li-Da Zhang for his help with bioinformatics analysis. This study was funded by the China 973 Program (No. 2012CB113900), National Natural Science Foundation of China (No. 31271291, 31471156), Shanghai Municipal Committee of Science and Technology (No. 13JC1403600), Specialized Research Fund for the Doctoral Program of Higher Education (No. 20120073110051), China Innovative Research Team, Ministry of Education, and Shanghai Graduate Education and Innovation Program (Horticulture).
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by C. Gebhardt.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhao, JL., Wang, YL., Yao, DQ. et al. Transcriptome profiling of trichome-less reveals genes associated with multicellular trichome development in Cucumis sativus . Mol Genet Genomics 290, 2007–2018 (2015). https://doi.org/10.1007/s00438-015-1057-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-015-1057-z