
Abstract
The association between common variations (rs10937405, rs4488809) on 3q28 and lung cancer has been widely evaluated in various ethnic groups, since it was first identified through genome-wide association approach. However, the results have been inconclusive. To derive a more precise estimation of the relationship and the effect of factors that might modify the risk, we performed this meta-analysis. The random-effects model was applied, addressing heterogeneity and publication bias. A total of 10 articles involving 36,221 cases and 58,108 controls were included. Overall, the summary per-allele OR of 1.19 (95 % CI 1.14–1.25, P < 10−5) and 1.17 (95 % CI 1.10–1.23, P < 10−5) was found for the rs10937405 and rs4488809 polymorphisms, respectively. Significant results were also observed in heterozygous and homozygous when compared with wild genotype for these polymorphisms. Significant results were found in East Asians when stratified by ethnicity, whereas no significant associations were found among Caucasians. After stratifying by sample size, study design, control source and sex, significant associations were also obtained. In addition, our data indicate that these polymorphisms are involved in lung cancer susceptibility and confer its effect primarily in lung adenocarcinoma when stratified by histological subtype. Furthermore, significant associations were also detected both never-smokers and smokers for these polymorphisms. In conclusion, this meta-analysis demonstrated that rs10937405 and rs4488809 are a risk factor associated with increased non-small cell lung cancer susceptibility, particularly for East Asian populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer (LC) is a major cause of cancer death worldwide with >1 million deaths each year (Jemal et al. 2011). Although >80 % of the population attributable risk of lung cancer can be ascribed to tobacco smoking, several lines of evidence indicate that inherited genetic factors influence the development and progression of lung cancer; in particular, epidemiologic studies have consistently shown an elevated risk of lung cancer in relatives of lung cancer cases after adjustment for smoking (Tokuhata and Lilienfeld 1963). Lung cancer is classified into two main histologic groups: small cell lung cancer (SC) and non-small cell lung cancer; the latter includes adenocarcinoma (AD) and squamous cell carcinoma (SQ), along with rarer subtypes (Sun et al. 2007). Each type has different pathophysiological and clinical features, suggesting that their mechanisms of carcinogenesis differ (Daigo and Nakamura 2008). Despite much investigation, only a few genes identified through the candidate gene approach have been confirmed to be associated with LC (e.g., CYP1A1, XRCC1, GSTs, and TP53) (Ye et al. 2006; Chen et al. 2011; Li et al. 2013; Zhou et al. 2013). However, because the pathogenesis of LC is yet to be elucidated completely, the candidate gene approach is limited in power to detect novel disease-susceptibility genes.
Genome-wide association studies (GWAS) of lung cancer with a full range of histological types have been conducted in European populations, and associations at 15q25.1 (CHRNA5-CHRNA3-CHRNB3), 5p15.33 (TERT-CLPTM1L) and 6p21.33 (BAT3-MSH5) have been identified (Amos et al. 2008; Hung et al. 2008; McKay et al. 2008; Wang et al. 2008; Broderick et al. 2009). Recently, a GWAS conducted among subjects from Japan and South Korea replicated the association between lung adenocarcinoma and the CLPTM1L-TERT locus in East Asians and identified a novel association between lung adenocarcinoma and the TP63 locus on 3q28 (Miki et al. 2010). However, the association was less clear as it did not achieve genome-wide significance (P ≤ 10−7) when restricted to never-smoking males (P = 0.012) and never-smoking females (P = 3.5 × 10−6). Subsequently, considerable efforts have been devoted to further exploring the strength of this association with lung cancer risk in the absence of tobacco use among various ethnic populations. However, a proportion of them have produced inconsistent results. These disparate findings may be due partly to insufficient power, phenotypic heterogeneity, population stratification, small effect of the polymorphism on LC risk, and even publication biases. We therefore performed a meta-analysis of the published studies to clarify this inconsistency and to establish a comprehensive picture of the relationship between two common variants (rs10937405 and rs4488809) on chromosome 3q28 and lung cancer.
Materials and methods
Identification and eligibility of relevant studies
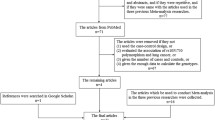
Electronic databases (Pubmed, EMBASE, ISI Web of Science, EBSCO, Cochrane Library databases) were searched up to Jan 2014 for all genetic association studies evaluating polymorphism at 3q28 and lung cancer in humans in all languages. The search strategy contained both medical subject heading terms and text words as follows: ‘‘3q28’’ or ‘‘tp63’’ or ‘‘rs10937405’’ or ‘‘rs4488809’’, in combination with ‘‘lung cancer’’ or ‘‘lung carcinoma’’ or ‘‘lung neoplasm’’ and combined with ‘‘genetic’’ or “polymorphism(s)’’ or ‘‘variations(s)’’ or ‘‘genotype’’ or ‘‘gene(s)’’. Articles were selected on the basis of the abstract, before examining the full text. In addition, the reference lists of selected articles were hand-searched to identify additional relevant reports. Case reports, case-only studies, editorials, and review articles were excluded. Articles in languages other than English were translated. The included studies have to meet the following criteria: (1) case–control or cohort studies to evaluate the association between polymorphism at 3q28 and LC risk, (2) identification of lung cancer patients was confirmed histologically or pathologically, (3) original papers containing independent data which have been published in peer-reviewed journal, (4) genotype distribution information in cases and controls or odds ratios (ORs) with its 95 % confidence intervals (CIs) and P value. If multiple published reports from the same study population were available, we included only the one with largest sample size and the most detailed information. Studies with different ethnic groups were considered as individual studies for our analyses.
Quality assessment and data extraction
Each article was read and assessed according to the score scale for randomized controlled association study proposed by Li and He (2008). In brief, papers were rated according to several items on the scale in relation to two areas: experiment design to minimize potential bias and data analysis. The quality score categorizes studies as of “high”, “median” or “low” quality.
Data extraction was performed independently by two reviewers. Review reports from the two were then compared to identify any inconsistency, and differences were resolved by further discussion among all authors. For each included study, the following information was extracted from each report according to a fixed protocol: first author, year of publication, ethnicity, identification of cancer cases, age, sex, smoking status (never-smoker and smoker), histological types (adenocarcinoma, squamous cell carcinoma, and small cell carcinoma), study design (GWAS or candidate gene study), source of control groups (population-based controls and hospital-based controls), Hardy–Weinberg equilibrium (HWE) status among controls, number of cases and controls, genotype frequency, and genotyping methods. Where essential information was not presented in articles, every effort was made to contact the authors.
Statistical analysis
We first assessed HWE in the controls for each study using goodness-of-fit test (Chi-square or Fisher’s exact test) and a P < 0.05 was considered as significant disequilibrium. The strength of the association between LC and common variations at 3q28 was estimated using ORs, with the corresponding 95 % CIs. The meta-analysis examined the association between the polymorphism and the risk of LC for the: (1) allele contrast, (2) heterozygous, and (3) homozygote (Pan et al. 2013). Cochran’s Chi square-based Q statistic test was performed to assess possible heterogeneity between the individual studies, and thus to insure that each group of studies was suitable for meta-analysis (Cochran et al. 1954). Both fixed-effects (Mantel–Haenszel method) (Mantel and Haenszel 1959) and random-effects (DerSimonian–Laird method) (DerSimonian and Laird 1986) models were performed to calculate the pooled ORs. Owing to a priori assumptions about the likelihood of heterogeneity between primary studies, the random-effects model, which usually is more conservative, was reported in the text. Subsidiary analyses included subgroup analyses or random-effects meta-regression with restricted maximum likelihood (Thompson and Sharp 1999). Ethnicity (East Asian vs. Caucasian), study design (GWAS vs. candidate gene studies), source of controls (population-based vs. hospital-based studies), and sample size (≥1,000 cases or, <1,000 cases) were pre-specified as characteristics for the assessment of heterogeneity. Ethnicity, sample size, study design, source of controls, histological types of LC, smoking behavior, and sex distribution in cases and controls were analyzed as covariates in meta-regression. The Z test was used to determine the significance of the pooled OR (Tang et al. 2010). Sensitivity analyses were performed to assess the stability of the results, namely, a single study in the meta-analysis was deleted each time to reflect the influence of the individual dataset to the overall OR. The potential publication bias was estimated using Egger’s linear regression test by visual inspection of the funnel plot (Egger et al. 1997). If publication bias existed, the Duval and Tweedie nonparametric ‘‘trim and fill’’ method was used to adjust for it (Taylor and Tweedie 1998). All P values are two-sided at the P = 0.05 level. All of the statistical tests used in this meta-analysis were performed by STATA version 10.0 (Stata Corporation, College Station, TX) and SAS (version 9.1; SAS Institute, Cary, NC, USA).
Results
Characteristics of included studies
The combined search yielded 91 references. In all, we included 10 studies in this meta-analysis (Table 1; Supplementary Fig. 1), with a total of 36,221 cases and 58,108 controls (Miki et al. 2010; Hu et al. 2011; Wang et al. 2011; Hosgood et al. 2012; Lan et al. 2012; Shiraishi et al. 2012; Timofeeva et al. 2012; Yin et al. 2013; Hu et al. 2014). The main study characteristics were summarized in Table 1. For the rs10937405 polymorphism, 24 data sets from 8 studies involved a total of 31,369 cases and 52,317 controls. For the rs4488809 polymorphism, 20 data sets from 9 studies involved a total of 21,511 cases and 28,014 controls. These two polymorphisms were found to occur in frequencies consistent with Hardy–Weinberg equilibrium in the control populations of all included studies. Of the cases, 70 % were East Asian, and 30 % were Caucasian. The studies finally included were of median-to-high quality and included no “poor quality” study.
rs10937405 polymorphism and lung cancer
There was a wide variation in the risk C allele frequency of the rs10937405 polymorphism among the controls across different ethnicities, ranging from 0.55 to 0.72 (Supplementary Fig. 2). For East Asian controls, the C allele frequency was 0.70 (95 % CI 0.67–0.72), which was lower than that in Caucasian controls (0.57; 95 % CI 0.55–0.59) indicating a significant difference among East Asians as compared with Caucasians (P < 10−5).
Significant associations were found in the pooled analysis between rs10937405 polymorphism and increased risk of LC (C allele OR = 1.19, 95 % CI = 1.14–1.25, P < 10−5; heterozygous OR = 1.28, 95 % CI = 1.18–1.40, P < 10−5; homozygote OR = 1.25, 95 % CI = 1.19–1.31, P < 10−5) (Fig. 1). Significant heterogeneity was present among the included studies of the 3q28-rs10937405 polymorphism (P < 0.05). In the stratified analysis by ethnicity, significant associations were detected among East Asians in all genetic models (C allele OR = 1.19, 95 % CI = 1.14–1.25; heterozygous OR = 1.32, 95 % CI = 1.19–1.46; homozygote OR = 1.27, 95 % CI = 1.21–1.34). However, we failed to detect any association to LC risk for Caucasians in all genetic models (Table 2). When stratifying for sample size, an OR of 1.27 (95 % CI 1.18–1.36, P < 10−5) resulted for the risk allele among small studies. Analysis restricted to the 11 studies with at least 1,000 cases, which should be less prone to selective publication than smaller studies, yielded an OR of 1.16 (95 % CI 1.10–1.22, P < 10−5). In the subgroup analyses by control source, the per-allele OR for population-based study of the C variant was 1.16 (95 % CI 1.08–1.25, P < 10−5) and for hospital-based study was 1.21 (95 % CI 1.16–1.27, P < 10−5). Further stratified according to study design, significant results were found for GWAS and candidate gene study in all genetic models (Table 2). After adjusting for multiple testing using Bonferroni correction, all significant associations for rs10937405 under different genetic models remained.

Per-allele ORs and 95 % CIs for the association between 3q28-rs10937405 and lung cancer risk stratified by ethnicity
We further performed analyses to test for differences in the associations of the polymorphism with lung cancer risk with respect to different clinical factors (Table 3). Given the biological differences between the histological forms of LC, we examined the association between the polymorphism and risk by histological subtype. A subgroup analysis by histology revealed strong heterogeneity (P < 10−5) with the strongest association for adenocarcinoma (OR = 1.26, 95 % CI = 1.21–1.30, P < 10−5). rs10937405 also showed an association with squamous cell carcinoma in the (OR = 1.14, 95 % CI = 1.06–1.22, P < 10−4). The association between the polymorphism and the risk of LC was further examined by stratifying the subjects according to smoking behavior. Never-smokers with the C allele of the polymorphism had similar increased LC risk compared to smoker cancer cases with an OR of 1.27 (95 % CI 1.21–1.33, P < 10−5) and 1.26 (95 % CI 1.18–1.34, P < 10−5), respectively. We next analyzed the effect of rs10937405 according to sex, this SNP tended to have similar OR for both females and males (Table 3).
As the formal test for heterogeneity may not be powerful enough, we conducted meta-regression as there were also grounds for considering the ethnicity, sample size, histological subtype, study design, and source of controls as potential sources of heterogeneity. The meta-regression showed that none of these covariates significantly contributed to the heterogeneity among the individual study results except for ethnicity (P = 0.001) and histological subtype (P = 0.02). Galbraith plot analyses of all included studies were used to assess the potential sources of heterogeneity. The study by Timofeeva et al. (2012), the study by Shiraishi et al. (2012) and one dataset from the study by Miki et al. (2010) were found to be the contributors of heterogeneity for rs10937405 polymorphism (Supplementary Fig. 3).
Sensitivity analysis was performed by excluding one study at a time (Supplementary Fig. 4). The results confirmed the significant association between the rs10937405 polymorphism and the risk of LC, with ORs and 95 % CIs ranging from 1.18 (95 % CI 1.13–1.23) to 1.20 (95 % CI 1.14–1.26). A funnel plot of these included studies suggested a possibility of the preferential publication of positive findings in smaller studies (Egger test, P = 0.04, Supplementary Fig. 5) for rs10937405. The Duval and Tweedie nonparametric “trim and fill” method was used to adjust for publication bias. Meta-analysis with “trim and fill” method did not draw different conclusion (OR = 1.14, 95 % CI = 1.09–1.19, P < 10−5; Supplementary Fig. 6), indicating that our results were statistically robust.
rs4488809 polymorphism and lung cancer
In the overall analysis, the risk C allele of rs4488809 was significantly associated with elevated LC (OR = 1.17, 95 % CI = 1.10–1.23, P < 10−5; Fig. 2). Significant associations were also found for heterozygous (OR = 1.21, 95 % CI = 1.09–1.35, P < 10−5) and homozygous (OR = 1.22, 95 % CI = 1.12–1.33, P < 10−5) when compared with wild genotype. In view of significant heterogeneity and to seek for its potential sources, we performed a panel of subgroup analyses on ethnicity, sample size, study design and source of controls (Table 4). Significant associations were found in East Asians (OR = 1.18, 95 % CI = 1.12–1.25, P < 10−5), while no significant associations were observed in Caucasians. When the analyses were performed by sample size, increased risk of LC was found only in larger studies in all genetic models (Table 4). Subgroup analysis showed a significant association in the studies using hospital-based control with pooled OR of 1.23 (95 % CI 1.19–1.27, P < 10−5), as compared with population-based control study (OR = 1.12, 95 % CI = 1.00–1.24, P = 0.045). Subsidiary analyses of study design yielded a per-allele OR for GWAS of 1.12 (95 % CI 0.94–1.35) and for candidate gene study of 1.17 (95 % CI 1.11–1.25). In addition, for rs4488809, associations were maintain statistically significant after Bonferroni correction for multiple genetic models. Meta-regression was used to explore the cause of heterogeneity, and it was found that ethnicity (P = 0.05), study design (P = 0.07) and source of controls (P = 0.10) did not significantly correlated with the magnitude of the genetic effect. However, a slight effect in heterogeneity for ethnicity (P = 0.04) and histological subtype (P = 0.007) was found. One dataset from the study by Hu et al. (2011), one dataset from the study by Hosgood et al. (2012) and the study by Timofeeva et al. (2012) were found to be contributors of heterogeneity for rs4488809 polymorphism (Supplementary Fig. 7).

Per-allele ORs and 95 % CIs for the association between 3q28-rs4488809 and lung cancer risk stratified by ethnicity
In considering histological types, the overall per-allele OR of the C variant for lung adenocarcinoma was 1.19 (95 % CI 1.10–1.29, P < 10−5), with corresponding results for squamous cell carcinoma and small cell carcinoma of 1.08 (95 % CI 0.87–1.34, P = 0.36) and 1.05 (95 % CI 0.95–1.17, P = 0.46), respectively. Tobacco smoking is the major risk factor for lung cancer, and we further performed analyses to test for differences in the associations of the polymorphism with lung cancer risk with respect to different smoking behavior. Among never-smokers, there was significant association between rs4488809 and the risk of lung cancer (OR = 1.14, 95 % CI = 1.04–1.25, P = 0.003). Among smokers, this SNP tended to have a higher OR of 1.21 (95 % CI 1.14–1.29, P < 10−5). In the subgroup analyses by sex, we observed a sex difference for rs4488809 and LC risk, with a stronger association in male than in female (Table 5).
For the rs4488809 polymorphism, sensitivity analysis indicated that no single study influenced the pooled OR qualitatively, suggesting that the results of this meta-analysis are stable (Supplementary Fig. 8). The publication bias was evaluated with asymmetry tests. The shape of the funnel plots was symmetrical for these polymorphisms (Supplementary Fig. 9). The Egger test provided evidence that there was no publication bias among the studies included for rs4488809 (Egger’s test, P = 0.21).
Discussion
Multiple lines of evidence support an important role for genetics in determining risk for LC, and association studies are appropriate for searching susceptibility genes involved in LC (Risch and Merikangas 1996). Nevertheless, small sample sized association studies lack statistical power and have resulted in apparently contradicting findings (Lohmueller et al. 2003). Via a comprehensive meta-analysis, we evaluated the association of two common polymorphisms on 3q28 with the risk of LC. Overall results demonstrated that rs10937405-C allele and rs4488809-C allele might be risk-conferring factors for the development of LC in East Asians, but not in Caucasians. Although potential sources of heterogeneity could not be easily eliminated, the present study, to our knowledge, is the first meta-analysis which involved a total of 36,221 cases and 58,108 controls from 10 studies to date dealing with the association of these two polymorphisms with LC susceptibility.
Genetic heterogeneity is inevitable in disease identification strategy (Hemminki et al. 2006). We identified ethnicity as a potential source of between-study heterogeneity by subgroup analysis and meta-regression. In the subgroup analysis by ethnicity, significant associations were found in East Asians but not for Caucasians. There are some points that should be concerned for such inconsistent results. First, ethnic differences may attribute to these different results, since the distributions of these polymorphisms were different between various ethnic populations. For instance, the frequency of risk-C allele of rs10937405 differs from 56 % in Whites (Wang et al. 2011), to 72 % in Chinese population (Hosgood et al. 2012; Yin et al. 2013). Second, this conflicting association could also be explained by study design or small sample size. This is particularly true for SNP rs4488809 because only one study conducted among Caucasians was included in the present meta-analysis which had insufficient statistical power to detect a slight effect or different linkage disequilibrium (LD) pattern of the polymorphism among Caucasians. Furthermore, it is possible that variation at this locus has modest effects on LC, while combinations of multiple genes and environmental factors finally lead to the disease, it would not be observed, because environmental factors may predominate in the development of LC, like air pollution (Zhao et al. 2006) and smoking that have been already well studied in recent years all around the world (Hecht 2002; Vineis et al. 2004). Moreover, clinical heterogeneity like age, sex ratio, dietary, years from onset and disease severity may also explain the discrepancy. Therefore, well-designed studies in different ethnic populations focused on other loci which are in LD with these variations are needed to further validate ethnic difference in their effects on LC.
Stratification of tumors by histological subtype indicated that rs10937405 and rs4488809 confer risk, preferentially for lung adenocarcinoma; thereby confirming the recent observation made by Miki and colleagues in an analysis of East Asian populations (Miki et al. 2010). While it will be challenging to identify the precise mechanism by which 3q28 variation affects lung adenocarcinoma development, accumulation of DNA damage and lack of response to genotoxic stress is recognized to contribute to lung carcinogenesis. rs10937405 and rs4488809 were located at the first intron of TP63 at chromosome 3q28. TP63 is a member of the tumor suppressor TP53 gene family, which transcriptionally regulates genes involved in DNA repair (Flores 2007; Lin et al. 2009), and it is important for normal development and differentiation of stratified epithelial tissues as well as for human carcinogenesis (Tomkova et al. 2008; Vousden and Prives 2009). Further, p63 has been found to play an important role in cancer development and progression through its interaction with mutant p53 (Melino 2011). Exposure of cells to DNA damage leads to induction of TP63 and both isoforms have the ability to transactivate TP53 target genes, hence impacting on cellular responsiveness to DNA damage (Katoh et al. 2000; Petitjean et al. 2008). TP63 is expressed mainly in two isoforms, the TA and N-terminal-truncated (ΔN) forms. The TAp63 isoforms are transcribed using a promoter-located upstream of exon 1 of the gene, whereas expression of the ΔNp63 isoforms are regulated by a promoter within intron 3 of TP63 (Moll and Slade 2004). Miki et al. show that rs10937405 and rs4488809 appear to define a single risk haplotype to which a functional variant maps (Miki et al. 2010). If the association annotated by this haplotype reflects a single risk variant, it does preclude the possibility that the haplotype may capture multiple functional risk alleles. Although elucidating a functional basis for the SNP associations will be contingent on fine mapping, it is entirely plausible that they may impact either directly or through LD on TP63 expression.
Cigarette smoking directly causes lung cancer development by several mechanisms (Hecht 1999; Zhou et al. 2006; Jorgensen et al. 2010), including inactivation of tumor suppressor genes (Liu et al. 2005; Lee et al. 2008), induction of oxidative stress and DNA damage, and activation of signaling pathways that underlie apoptosis and autophagy (Maiuri et al. 2009; Essick and Sam 2010). Furthermore, ΔNp63α and tobacco smoking have a synergetic effect on carcinogenesis (Ratovitski 2010). Therefore, it is difficult to determine whether these loci are associated with lung cancer risk, tobacco use, or perhaps both.
An important source of bias in every meta-analysis is publication bias because the likelihood of publishing a study could be related to the results of that study. However, among our meta-analysis, there have been many studies published with negative findings (Timofeeva et al. 2012; Yin et al. 2013). Although the funnel plot for East Asians is not symmetric, the overall results of both ethnic groups are concordant, indicating that this bias cannot affect the final result. On the other hand, funnel plot asymmetry is not always caused by publication bias. True heterogeneity may also lead to funnel plot asymmetry. For example, significant difference may be seen only in high-risk individuals, and these high-risk people are usually more likely to be included in small studies. This is particularly true in our meta-analysis because all the significant associations in East Asians have been observed among the studies from high-risk populations. Language bias or citation bias also could be an important source in this group of studies, meaning that the studies without significant findings are preferentially published in languages other than English and less likely to be cited in other articles. Finally, it is possible that an asymmetrical funnel plot arises simply by chance.
In summary, our meta-analysis showed that rs10937405 and rs4488809 at 3q28 might be risk-conferring factor for the development of non-small cell LC in East Asians, but not in Caucasians. As studies among Caucasian, African populations are currently limited, further studies including a wider spectrum of subjects to investigate the role of these variants in different populations will be needed.
References
Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, Sullivan K, Matakidou A, Wang Y, Mills G, Doheny K, Tsai YY, Chen WV, Shete S, Spitz MR, Houlston RS (2008) Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet 40:616–622
Broderick P, Wang Y, Vijayakrishnan J, Matakidou A, Spitz MR, Eisen T, Amos CI, Houlston RS (2009) Deciphering the impact of common genetic variation on lung cancer risk: a genome-wide association study. Cancer Res 69:6633–6641
Chen Z, Li Z, Niu X, Ye X, Yu Y, Lu S, Chen Z (2011) The effect of CYP1A1 polymorphisms on the risk of lung cancer: a global meta-analysis based on 71 case–control studies. Mutagenesis 26:437–446
Cochran WG (1954) The combination of estimates from different experiments. Biometrics 10:101–129
Daigo Y, Nakamura Y (2008) From cancer genomics to thoracic oncology: discovery of new biomarkers and therapeutic targets for lung and esophageal carcinoma. Gen Thorac Cardiovasc Surg 56:43–53
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7:177–188
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315:629–634
Essick EE, Sam F (2010) Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid Med Cell Longev 3:168–177
Flores ER (2007) The roles of p63 in cancer. Cell Cycle 6:300–304
Hecht SS (1999) Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 91:1194–1210
Hecht SS (2002) Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol 3:461–469
Hemminki K, Lorenzo Bermejo J, Försti A (2006) The balance between heritable and environmental aetiology of human disease. Nat Rev Genet 7:958–965
Hirschhorn JN, Lohmueller K, Byrne E (2002) A comprehensive review of genetic association studies. Genet Med 4:45–61
Hosgood HD 3rd, Wang WC, Hong YC, Wang JC, Chen K, Chang IS, Chen CJ, Lu D, Yin Z, Wu C, Zheng W, Qian B, Park JY, Kim YH, Chatterjee N, Chen Y, Chang GC, Hsiao CF, Yeager M, Tsai YH, Wei H, Kim YT, Wu W, Zhao Z, Chow WH, Zhu X, Lo YL, Sung SW, Chen KY, Yuenger J, Kim JH, Huang L, Chen YH, Gao YT, Kim JH, Huang MS, Jung TH, Caporaso N, Zhao X, Huan Z, Yu D, Kim CH, Su WC, Shu XO, Kim IS, Bassig B, Chen YM, Cha SI, Tan W, Chen H, Yang TY, Sung JS, Wang CL, Li X, Park KH, Yu CJ, Ryu JS, Xiang Y, Hutchinson A, Kim JS, Cai Q, Landi MT, Lee KM, Hung JY, Park JY, Tucker M, Lin CC, Ren Y, Perng RP, Chen CY, Jin L, Chen KC, Li YJ, Chiu YF, Tsai FY, Yang PC, Fraumeni JF Jr, Seow A, Lin D, Zhou B, Chanock S, Hsiung CA, Rothman N, Lan Q (2012) Genetic variant in TP63 on locus 3q28 is associated with risk of lung adenocarcinoma among never-smoking females in Asia. Hum Genet 131:1197–1203
Hu Z, Wu C, Shi Y, Guo H, Zhao X, Yin Z, Yang L, Dai J, Hu L, Tan W, Li Z, Deng Q, Wang J, Wu W, Jin G, Jiang Y, Yu D, Zhou G, Chen H, Guan P, Chen Y, Shu Y, Xu L, Liu X, Liu L, Xu P, Han B, Bai C, Zhao Y, Zhang H, Yan Y, Ma H, Chen J, Chu M, Lu F, Zhang Z, Chen F, Wang X, Jin L, Lu J, Zhou B, Lu D, Wu T, Lin D, Shen H (2011) A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat Genet 43:792–796
Hu QY, Jin TB, Wang L, Zhang L, Geng T, Liang G, Kang LL (2014) Genetic variation in the TP63 gene is associated with lung cancer risk in the Han population. Tumour Biol 35:1863–1866
Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Chen C, Goodman G, Field JK, Liloglou T, Xinarianos G, Cassidy A, McLaughlin J, Liu G, Narod S, Krokan HE, Skorpen F, Elvestad MB, Hveem K, Vatten L, Linseisen J, Clavel-Chapelon F, Vineis P, Bueno-de-Mesquita HB, Lund E, Martinez C, Bingham S, Rasmuson T, Hainaut P, Riboli E, Ahrens W, Benhamou S, Lagiou P, Trichopoulos D, Holcátová I, Merletti F, Kjaerheim K, Agudo A, Macfarlane G, Talamini R, Simonato L, Lowry R, Conway DI, Znaor A, Healy C, Zelenika D, Boland A, Delepine M, Foglio M, Lechner D, Matsuda F, Blanche H, Gut I, Heath S, Lathrop M, Brennan P (2008) A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452:633–637
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61:69–90
Jorgensen ED, Zhao H, Traganos F, Albino AP, Darzynkiewicz Z (2010) DNA damage response induced by exposure of human lung adenocarcinoma cells to smoke from tobacco- and nicotine-free cigarettes. Cell Cycle 9:2170–2176
Katoh I, Aisaki KI, Kurata SI, Ikawa S, Ikawa Y (2000) p51A (TAp63gamma), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene 19:3126–3130
Lan Q, Hsiung CA, Matsuo K, Hong YC, Seow A, Wang Z, Hosgood HD 3rd, Chen K, Wang JC, Chatterjee N, Hu W, Wong MP, Zheng W, Caporaso N, Park JY, Chen CJ, Kim YH, Kim YT, Landi MT, Shen H, Lawrence C, Burdett L, Yeager M, Yuenger J, Jacobs KB, Chang IS, Mitsudomi T, Kim HN, Chang GC, Bassig BA, Tucker M, Wei F, Yin Z, Wu C, An SJ, Qian B, Lee VH, Lu D, Liu J, Jeon HS, Hsiao CF, Sung JS, Kim JH, Gao YT, Tsai YH, Jung YJ, Guo H, Hu Z, Hutchinson A, Wang WC, Klein R, Chung CC, Oh IJ, Chen KY, Berndt SI, He X, Wu W, Chang J, Zhang XC, Huang MS, Zheng H, Wang J, Zhao X, Li Y, Choi JE, Su WC, Park KH, Sung SW, Shu XO, Chen YM, Liu L, Kang CH, Hu L, Chen CH, Pao W, Kim YC, Yang TY, Xu J, Guan P, Tan W, Su J, Wang CL, Li H, Sihoe AD, Zhao Z, Chen Y, Choi YY, Hung JY, Kim JS, Yoon HI, Cai Q, Lin CC, Park IK, Xu P, Dong J, Kim C, He Q, Perng RP, Kohno T, Kweon SS, Chen CY, Vermeulen R, Wu J, Lim WY, Chen KC, Chow WH, Ji BT, Chan JK, Chu M, Li YJ, Yokota J, Li J, Chen H, Xiang YB, Yu CJ, Kunitoh H, Wu G, Jin L, Lo YL, Shiraishi K, Chen YH, Lin HC, Wu T, Wu YL, Yang PC, Zhou B, Shin MH, Fraumeni JF Jr, Lin D, Chanock SJ, Rothman N (2012) Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat Genet 44:1330–1335
Lee JM, Yanagawa J, Peebles KA, Sharma S, Mao JT, Dubinett SM (2008) Inflammation in lung carcinogenesis: new targets for lung cancer chemoprevention and treatment. Crit Rev Oncol Hematol 66:208–217
Li D, He L (2008) Meta-study on association between the monoamine oxidase A gene (MAOA) and schizophrenia. Am J Med Genet B Neuropsychiatr Genet 147B:174–178
Li Y, Huang Y, Cao YS, Zeng J, Tong WN, Xu SL, Zhuo AS (2013) Assessment of the association between XRCC1 Arg399Gln polymorphism and lung cancer in Chinese. Tumour Biol 34:3681–3685
Lin YL, Sengupta S, Gurdziel K, Bell GW, Jacks T, Flores ER (2009) p63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS Genet 5:e1000680
Liu G, Zhou W, Christiani DC (2005) Molecular epidemiology of non-small cell lung cancer. Semin Respir Crit Care Med 26:265–272
Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN (2003) Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 33:177–182
Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, Kroemer G (2009) Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ 16:87–93
Mantel N, Haenszel W (1959) Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22:719–748
McKay JD, Hung RJ, Gaborieau V, Boffetta P, Chabrier A, Byrnes G, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, McLaughlin J, Shepherd F, Montpetit A, Narod S, Krokan HE, Skorpen F, Elvestad MB, Vatten L, Njølstad I, Axelsson T, Chen C, Goodman G, Barnett M, Loomis MM, Lubiñski J, Matyjasik J, Lener M, Oszutowska D, Field J, Liloglou T, Xinarianos G, Cassidy A; EPIC Study, Vineis P, Clavel-Chapelon F, Palli D, Tumino R, Krogh V, Panico S, González CA, Ramón Quirós J, Martínez C, Navarro C, Ardanaz E, Larrañaga N, Kham KT, Key T, Bueno-de-Mesquita HB, Peeters PH, Trichopoulou A, Linseisen J, Boeing H, Hallmans G, Overvad K, Tjønneland A, Kumle M, Riboli E, Zelenika D, Boland A, Delepine M, Foglio M, Lechner D, Matsuda F, Blanche H, Gut I, Heath S, Lathrop M, Brennan P (2008) Lung cancer susceptibility locus at 5p15.33. Nat Genet 40:1404–1406
Melino G (2011) p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ 18:1487–1499
Miki D, Kubo M, Takahashi A, Yoon KA, Kim J, Lee GK, Zo JI, Lee JS, Hosono N, Morizono T, Tsunoda T, Kamatani N, Chayama K, Takahashi T, Inazawa J, Nakamura Y, Daigo Y (2010) Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat Genet 42:893–896
Moll UM, Slade N (2004) p63 and p73: roles in development and tumor formation. Mol Cancer Res 2:371–386
Pan Y, Wang F, Qiu Q, Ding R, Zhao B, Zhou H (2013) Influence of the Angiotensin converting enzyme insertion or deletion genetic variant and coronary restenosis risk: evidence based on 11,193 subjects. PLoS One 8:e83415
Petitjean A, Ruptier C, Tribollet V, Hautefeuille A, Chardon F, Cavard C, Puisieux A, Hainaut P, Caron de Fromentel C (2008) Properties of the six isoforms of p63: p53-like regulation in response to genotoxic stress and cross talk with DeltaNp73. Carcinogenesis 29:273–281
Ratovitski EA (2010) LKB1/PEA3/ΔNp63 pathway regulates PTGS-2 (COX-2) transcription in lung cancer cells upon cigarette smoke exposure. Oxid Med Cell Longev 3:317–324
Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517
Shiraishi K, Kunitoh H, Daigo Y, Takahashi A, Goto K, Sakamoto H, Ohnami S, Shimada Y, Ashikawa K, Saito A, Watanabe S, Tsuta K, Kamatani N, Yoshida T, Nakamura Y, Yokota J, Kubo M, Kohno T (2012) A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat Genet 44:900–903
Sun S, Schiller JH, Gazdar AF (2007) Lung cancer in never smokers-a different disease. Nat Rev Cancer 7:778–790
Tang K, Li Y, Zhang Z, Gu Y, Xiong Y, Feng G, He L, Qin S (2010) The PstI/RsaI and DraI polymorphisms of CYP2E1 and head and neck cancer risk: a meta-analysis based on 21 case-control studies. BMC Cancer 10:575
Taylor SJ, Tweedie RI (1998) Practical estimates of the effect of publication bias in meta-analysis. Australas Epidemiol 5:14–17
Thompson SG, Sharp SJ (1999) Explaining heterogeneity in meta-analysis: a comparison of methods. Stat Med 18:2693–2708
Timofeeva MN, Hung RJ, Rafnar T, Christiani DC, Field JK, Bickeböller H, Risch A, McKay JD, Wang Y, Dai J, Gaborieau V, McLaughlin J, Brenner D, Narod SA, Caporaso NE, Albanes D, Thun M, Eisen T, Wichmann HE, Rosenberger A, Han Y, Chen W, Zhu D, Spitz M, Wu X, Pande M, Zhao Y, Zaridze D, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Krokan HE, Gabrielsen ME, Skorpen F, Vatten L, Njølstad I, Chen C, Goodman G, Lathrop M, Benhamou S, Vooder T, Välk K, Nelis M, Metspalu A, Raji O, Chen Y, Gosney J, Liloglou T, Muley T, Dienemann H, Thorleifsson G, Shen H, Stefansson K, Brennan P, Amos CI, Houlston R, Landi MT; Transdisciplinary Research in Cancer of the Lung (TRICL) Research Team (2012) Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Hum Mol Genet 21:4980–4995
Tokuhata GK, Lilienfeld AM (1963) Familial aggregation of lung cancer in humans. J Natl Cancer Inst 30:289–312
Tomkova K, Tomka M, Zajac V (2008) Contribution of p53, p63, and p73 to the developmental diseases and cancer. Neoplasma 55:177–181
Vineis P, Alavanja M, Buffler P, Fontham E, Franceschi S, Gao YT, Gupta PC, Hackshaw A, Matos E, Samet J, Sitas F, Smith J, Stayner L, Straif K, Thun MJ, Wichmann HE, Wu AH, Zaridze D, Peto R, Doll R (2004) Tobacco and cancer: recent epidemiological evidence. J Natl Cancer Inst 96:99–106
Vousden KH, Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137:413–431
Wang Y, Broderick P, Webb E, Wu X, Vijayakrishnan J, Matakidou A, Qureshi M, Dong Q, Gu X, Chen WV, Spitz MR, Eisen T, Amos CI, Houlston RS (2008) Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet 40:1407–1409
Wang Y, Broderick P, Matakidou A, Vijayakrishnan J, Eisen T, Houlston RS (2011) Variation in TP63 is associated with lung adenocarcinoma in the UK population. Cancer Epidemiol Biomarkers Prev 20:1453–1462
Ye Z, Song H, Higgins JP, Pharoah P, Danesh J (2006) Five glutathione s-transferase gene variants in 23,452 cases of lung cancer and 30,397 controls: meta-analysis of 130 studies. PLoS Med 3:e91
Yin ZH, Cui ZG, Ren YW, Su M, Ma R, He QC, Zhou BS (2013) TP63 gene polymorphisms, cooking oil fume exposure and risk of lung adenocarcinoma in chinese non-smoking females. Asian Pac J Cancer Prev 14:6519–6522
Zhang Y, Gu C, Shi H, Zhang A, Kong X, Bao W, Deng D, Ren L, Gu D (2012) Association between C3orf21, TP63 polymorphisms and environment and NSCLC in never-smoking Chinese population. Gene 497:93–97
Zhao Y, Wang S, Aunan K, Seip HM, Hao J (2006) Air pollution and lung cancer risks in China–a meta-analysis. Sci Total Environ 366:500–513
Zhou W, Heist RS, Liu G, Asomaning K, Miller DP, Neuberg DS, Wain JC, Lynch TJ, Christiani DC (2006) Second hand smoke exposure and survival in early-stage non-small-cell lung cancer patients. Clin Cancer Res 12:7187–7193
Zhou C, Chen H, Wang A (2013) P53 codon 72 polymorphism and lung cancer risk: evidence from 27,958 subjects. Tumour Biol 34:2961–2969
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jin, Yx., Jiang, Gn., Zheng, H. et al. Common genetic variants on 3q28 contribute to non-small cell lung cancer susceptibility: evidence from 10 case-control studies. Mol Genet Genomics 290, 573–584 (2015). https://doi.org/10.1007/s00438-014-0934-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-014-0934-1