
Abstract
A quantitative trait locus (QTL) mapping was conducted to better understand the genetic control of plant architecture (PA), yield components (YC), and fiber properties (FP) in the two cultivated tetraploid species of cotton (Gossypium hirsutum L. and G. barbadense L.). One hundred and fifty-nine genomic regions were identified on a saturated genetic map of more than 2,500 SSR and SNP markers, constructed with an interspecific recombinant inbred line (RIL) population derived from the genetic standards of the respective cotton species (G. hirsutum acc. TM-1 × G. barbadense acc. 3-79). Using the single nonparametric and MQM QTL model mapping procedures, we detected 428 putative loci in the 159 genomic regions that confer 24 cotton traits in three diverse production environments [College Station F&B Road (FB), TX; Brazos Bottom (BB), TX; and Shafter (SH), CA]. These putative QTL loci included 25 loci for PA, 60 for YC, and 343 for FP, of which 3, 12, and 60, respectively, were strongly associated with the traits (LOD score ≥ 3.0). Approximately 17.7 % of the PA putative QTL, 32.9 % of the YC QTL, and 48.3 % of the FP QTL had trait associations under multiple environments. The At subgenome (chromosomes 1–13) contributed 72.7 % of loci for PA, 46.2 % for YC, and 50.4 % for FP while the Dt subgenome (chromosomes 14–26) contributed 27.3 % of loci for PA, 53.8 % for YC, and 49.6 % for FP. The data obtained from this study augment prior evidence of QTL clusters or gene islands for specific traits or biological functions existing in several non-homoeologous cotton chromosomes. DNA markers identified in the 159 genomic regions will facilitate further dissection of genetic factors underlying these important traits and marker-assisted selection in cotton.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus of Gossypium (cotton) consists of at least 45 diploid and 5 allotetraploid species grouped into nine genome types with the designations A, B, C, D, E, F, G, K, and AD (Campbell et al. 2010; Fryxell 1979; Percival et al. 1999; Wendel et al. 2009). The AD-genome tetraploid cottons originated from the hybridization between A-genome and D-genome diploid cottons approximately 1–2 million years ago (MYA) (Beasley 1940; Cronn et al. 1999; Wendel and Cronn 2003). Within the Gossypium genus, only two diploid species (2n = 2X = 26) from the Old World, G. herbaceum L. (A1) and G. arboreum L. (A2), and two tetraploid species (2n = 4X = 52) from the New World, G. hirsutum L. (AD1) and G. barbadense L. (AD2), are cultivated to produce the world’s leading natural fiber and the second most important oilseed crop (Campbell et al. 2010; Kantartzi et al. 2009; Lee 1984; Ulloa et al. 2013; Wendel et al. 1992; Percy et al. 2014). The complexity of Gossypium genomes has afforded research opportunities on the evolution and diversity among diploid (Ma et al. 2010) and allotetraploid species (Brubaker et al. 1999; Jiang et al. 1998; Paterson et al. 2012). Compared to other crops (Gao et al. 2013; Huang et al. 2012; Paran and Zamir 2003), the cotton research community has less information and inadequate knowledge of the intraspecific genetic variation within Gossypium species, including the two cultivated tetraploid species (G. hirsutum and G. barbadense), that would allow precise manipulation of agronomic traits (Abdurakhmonov et al. 2012; Campbell et al. 2010; Kantartzi et al. 2009; Percy et al. 2014). Understanding the underlying genetic control of biological processes for favorable traits is relevant to a range of research objectives. Such understanding has potential applications for cotton crop improvement (An et al. 2010; Cao et al. 2014; Lacape et al. 2013; Ning et al. 2014; Rong et al. 2007; Ulloa et al. 2005, 2013).
Genetic mapping with polymerase chain reaction (PCR)-based markers, such as simple sequence repeats (SSR) and single-nucleotide polymorphisms (SNP), has facilitated portable applications across different mapping populations and research facilities (Guo et al. 2007; Lacape et al. 2009; Nguyen et al. 2004; Reddy et al. 2001; Van Deynze et al. 2009; Xiao et al. 2009; Yu et al. 2012a). To date, several thousands of SSR and SNP markers have been developed in cotton (Blenda et al. 2012; Fang and Yu 2012; Guo et al. 2007; Lacape et al. 2009; Yu et al. 2012a) and they have been surveyed for polymorphism against a 12-genotype germplasm standard (diversity panel) of six Gossypium species (Yu et al. 2012b). The localization of SSR and SNP markers to all 26 chromosomes and the creation of high-density genetic maps for the tetraploid cotton genome provide opportunities to identify genetic factors controlling and regulating the expression of traits of interest (Lacape et al. 2013; Li et al. 2013; Marathi et al. 2012; Zhu et al. 2011). However, due to the allopolyploid nature of the cotton genome, certain chromosomal structures remain to be resolved. These include deletions, duplications, or rearrangements that may be specific to species, or to particular crosses within or between species (Brubaker et al. 1999; Guo et al. 2007; Lacape et al. 2009; Rong et al. 2004; Ulloa et al. 2013; Wang et al. 2013; Yu et al. 2012a).
Most traits of interest in crop plants are likely the product of complex biological processes in which multiple genes and environments interact to quantitatively determine phenotypic expression (An et al. 2010; Collard and Mackill 2008; Huang et al. 2012; Lacape et al. 2010, 2013; Paran and Zamir 2003; Paterson et al. 1988, 1991; Rong et al. 2007). A quantitative trait locus (QTL) mapping approach can be informative for studying quantitative inheritance, for detecting genomic regions associated with morphological, yield and fiber quality traits, and for identifying molecular markers linked to the genes conferring the phenotypic expression of the important traits (Draye et al. 2005; Kohel et al. 2001; Lacape et al. 2005; Li et al. 2013; Luan et al. 2009; Park et al. 2005; Shen et al. 2005; Song and Zhang 2009; Ulloa et al. 2007; Zhu et al. 2011). Molecular markers located in the cotton genome are used to address fundamental questions regarding the genetic factors for these traits (Lacape et al. 2010, 2013; Mohan et al. 1997). Markers can be used to improve elite cotton cultivar productivity, fiber and seed quality properties, and stress tolerance through marker-assisted selection (MAS) (Cao et al. 2014; Said et al. 2013). Using MAS, the breeding process can be accelerated, labor costs and time reduced, and selection effectiveness increased in identification of improved genotypes (Buckler et al. 2009; Ulloa et al. 2010, 2011).
Many cotton QTL mapping studies were conducted using either F2 or BC1 populations (An et al. 2010; Draye et al. 2005; Jiang et al. 1998; Kohel et al. 2001; Lacape et al. 2005; Li et al. 2013; Shen et al. 2005). These QTL studies also employed either single marker regression without a genetic map or interval mapping with an established linkage group (An et al. 2010; Li et al. 2013). In some cases, multiple environments were used but QTL analyses were conducted on the basis of individual locations (Lacape et al. 2010). Recently, an interspecific backcross inbred line population was used to identify 67 QTLs for fiber properties and yield components (Yu et al. 2013). Another study used a random-mated recombinant inbred population to identify 131 QTLs for fiber quality with insignificant interaction between genotype and environment (Fang et al. 2014). In our study, we used a recombinant inbred line (RIL) population derived from an interspecific cross between Upland G. hirsutum acc. TM-1 and Pima G. barbadense acc. 3-79 to provide better estimates of QTLs. Since its initial development and at different generations, these RILs have facilitated QTL mapping of important traits with immortal and genetically stable plant materials that were phenotyped simultaneously across multiple growth environments or geographical locations (Frelichowski et al. 2006; Lacape et al. 2010, 2013; Marathi et al. 2012; Park et al. 2005; Wang et al. 2012a). In addition, these two mapping parents (TM-1 and 3-79) are currently being sequenced to produce reference genomes for eventual analysis of all potential genes of interest in cotton (unpublished data). Our single seed decent (SSD) RIL progeny of an interspecific hybrid also provided us with an opportunity of having larger numbers of DNA markers that were polymorphic between the two parents (two species standards). The utilization of diverse production environments increases our capacity to elucidate the phenotypic differences present in morphological traits of agronomic importance, yield components and differences in fiber quality under a wide range of cultivation conditions.
Our objectives in this study were to map genomic loci of plant architecture (PA, 4), yield components (YC, 6), and fiber properties or quality (FP, 14) traits obtained from three environments using the QTL mapping approach, and to utilize a recently developed saturated genetic map of the tetraploid cotton genome and molecular markers (Yu et al. 2012a; Fang and Yu 2012), and the RIL population derived from a cross between TM-1 and 3-79. The 24 cotton traits from the RIL population were obtained in three diverse production environments [College Station F&B Road (FB) TX, Brazos Bottom (BB) TX, and Shafter (SH) CA]. This study was part of our efforts to integrate the cotton genetic and genomic information that include many genetic and cytogenetic stocks and isogenic mutant lines already developed with the two cotton genetic standards (TM-1 and 3-79).
Materials and methods
Plant materials
As an immortal mapping population maintained by USDA-ARS in College Station, TX, USA, 186 RILs were developed from selfing by SSD original individual plants of an interspecific F2 population derived from a cross of the cultivated cotton species G. hirsutum (acc. TM-1) and G. barbadense (acc. 3-79) (Kohel et al. 2001; Yu et al. 2012a). TM-1 is a long-term inbred (>40 years) derived from an Upland cotton cultivar Deltapine 14 (Kohel et al. 1970), and it is the basis for a series of near-isogenic lines (NILs) for various traits. 3-79 originated as doubled haploid line of the extra long staple (ELS) cultivar 3-79. The cultivar 3-79 predates any known human-directed introgression with G. hirsutum and thus is a “pure” representative of the G. barbadense species (Niles and Feaster 1984). TM-1 and 3-79, of which genomes are currently being sequenced (unpublished data), are recognized as the global genetic standards of two Gossypium species that have many unique contrasting phenotypes (high productivity and wide adaptability for TM-1, and superior fiber properties for 3-79), allowing us to identify those genomic regions that underlie genetic control of PA, YC, and FP traits. Difficulties with stand establishment resulted in different numbers of RIL progeny at the different environment locations, and different numbers of progeny that yielded data for all traits (Tables 1, 2, 3). These difficulties have been reported in similar studies with other interspecific populations (Lacape et al. 2010, 2013; Marathi et al. 2012; Yu et al. 2013; Zhu et al. 2011).
Field phenotypic evaluations
The RIL progeny (F7 in average at the time of this study), together with the parents TM-1 and 3-79, were germinated in a greenhouse pellet at each study location in 2005. After the development of the true leaves, plants from each greenhouse were transplanted into the cotton production field in College Station F&B Road (FB) TX, Brazos Bottom (BB) TX, and Shafter (SH) CA, respectively. Based on soil type and other conditions, FB location is described as an upland area not adapted for commercial cotton production, and it requires supplemental irrigation but has low yield potential. Although only about 10 miles away, BB location is an alluvial river bottom area with supplemental irrigation and high yield potential. SH location is a desert environment that is a sandy-loam soil field site, uses furrow irrigation for production, and has high yield potential. Our genetic research standard procedure was followed to use transplants for stand establishment in order to maximize the use of limited seed availability. At each location, the parents and RIL progeny were grown in 1-row plots 5 m long with 1-m row spacing between rows, averaging 2–5 plants in an incomplete randomized block design with two replications per environment (Fehr 1991; Li et al. 2012).
Data from four morphological or PA traits (Table 1) were collected from these three different environments: plant height (cm), number of nodes, average internode length (cm), and main stem diameter (mm) at cotyledonous nodes. In addition, data from six YCs (Table 2) were collected: seed weight per plant (g), lint weight per plant (g), lint percent, gin turnout, seed index, and lint index. Lint percent was calculated by lint weight/(lint weight + seed weight) × 100. Gin turnout was total lint weight recovered and it was portion of lint ginned to the whole harvested seed-cotton sample. Seed Index was based on the weight of 100 seeds. Lint index was calculated based on the lint obtained from 100 seeds and the average yield obtained per seed for each RIL. Moreover, lint obtained from hand-harvested open bolls from the two replicate plots per RIL at each location was used to analyze data from 14 FPs (Table 3) by the advanced fiber information system (AFIS®, USTER, Charlotte, NC, USA) instrument in the fiber laboratory at the Cotton Incorporated, Cary, NC, USA. Open bolls were multiply harvested to prevent any field deterioration because diverse maturity of the interspecific segregating material did not allow boll sampling as in a uniform breeding nursery with uniform genotypes. AFIS, a more accurate fiber measuring technology, was used in this study because the smaller sample weight needed for AFIS allowed more individuals to be measured than with HVI technology that would otherwise require a larger sample (at least 20 g of fiber) in this genetic mapping population. The following 14 traits were collected: nep size g. (no. g), number of neps (g−1), number of seed coats, upper quartile of fiber length by weight (mm), short fiber content by weight g (kg−1), average length of all fibers by weight (mm), 5.0 % fiber span length (mm), 2.5 % fiber span length (mm), VFM percent, fiber fineness (mTx), immature fiber content by weight g (kg−1), maturity ratio (unit), mean tenacity (unit), and mean elongation (unit).
This RIL population was previously used for developing a saturated genetic map (Yu et al. 2012a). The number of RIL progeny evaluated depended on seed viability and germination at each location. It is well known the existing problems of fertility and seed production on interspecific progeny. The numbers of RILs used in each location for different traits were as follows: for PA traits, 175 (FB), 146 (BB) and 170 (SH); for YC, 145 (FB), 95 (BB) and 152 (H); and for FP, 110 (FB), 40 (BB) and 98 (SH) (Tables 1, 2, 3).
Differences among observed traits of the RILs within each location and between locations were evaluated for each experiment using PROC GLM (SAS, ver. 9.2, SAS Institute, Cary, NC, USA). Mean separations in the various examinations of main effects were conducted using the Waller–Duncan k-ratio procedure (Ott 1988). Correlation analyses were performed to examine the similarity of responses of the RIL entries at the different environments. All correlations were performed using PROC CORR (SAS ver. 9.2, SAS Institute, Cary, NC, USA).
Marker data and linkage map
Our previously published linkage maps that saturate the tetraploid cotton genome with more than 2,500 SSR and SNP markers were used in this study (Yu et al. 2012a; Fang and Yu 2012). The individual marker scores of the RILs that were evaluated phenotypically in the field served as a foundation matrix to generate linkage groups and trait association. The present study was the first to include the SNP markers that are mapped to the tetraploid cotton genome. Many markers were developed from the Unigenes of cotton expressed sequence tags (ESTs) and they would offer new opportunities for further functional analysis of cotton traits.
Quantitative trait locus analyses
Single marker analysis was conducted using a nonparametric mapping test [Kruskal–Wallis analysis (K*)] equivalent to a one-way analysis of variance (Van Ooijen 2004). In the nonparametric analysis, no assumptions were made for the probability distribution(s) of each quantitative trait, and if the data were distributed normally, the nonparametric test was as powerful as parametric methods. Also, all markers genotyped on the RIL population regardless of their linkages were used in the nonparametric test. Markers were tested at each locus separately without the use of the linkage map. QTL analyses were also conducted using MapQTL 5.0 with interval mapping and the MQM QTL model mapping procedures (Van Ooijen 2004; Arends et al. 2010). Threshold values for LOD were determined empirically after 1,000 permutation tests for all traits (Churchill and Doerge 1994). The threshold for a QTL was determined at P < 0.05 using the nonparametric and at LOD ≥ 3.0 using the MQM analyses.
PA and YC traits are known to be affected by the environments, which tend to produce genotype (G) × environment (E) interactions. In addition, although FP traits are more stable with high heritability, fiber from open bolls were obtained from multiple harvests to prevent any field deterioration because of the diverse maturity of the interspecific segregating material, which is typical of these types of interspecific genome mapping populations. This boll harvesting process did not allow uniformity as in most breeding nurseries. Means were obtained across locations (FB, BB, and SH) from common RILs and were used to examine trait effects and GxE interactions (data not shown). However, QTL analyses were performed on the RIL population from the individual datasets of the three locations (BB, FB, and SH). QTL comparisons were made among all locations after the individual analyses.
A strong QTL is named as follows: the first letter of the QTL if it derives from FP is capitalized ‘F’, and if the trait is YC, the first part of the trait name is capitalized. The second letter is also capitalized of the second name of the trait, and the third letter of the name is a small letter from the rest of the trait name. The first three letters of the trait name are followed by ‘Qtl’ and followed by the c = chromosome and the number. Finally, an underline was scribed before the number of QTLs (1h or 1b) denoted by a small letter that represents the positive allele or high value of the QTL phenotypic effect from the parents of this interspecific RIL population [G. hirsutum (h) and G. barbadense (b)].
Results
Phenotypic variation
The mapping parents, TM-1 and 3-79, and their RIL progeny were examined for the 24 traits under three diverse production environments: FB, BB, and SH. Contrasting phenotypic differences were observed between TM-1 and 3-79, on respective average, 85.57 and 96.77 cm for plant height (Ph), 28.50 and 26.75 for number of nodes (NA), 3.23 and 3.89 cm for internode length (InL), 28.65 and 24.80 mm for main stem diameter (MsD), 177.65 and 118.17 g for seed weight per plant (Swp), 100.22 and 64.18 g for lint weight per plant (Lwp), 44.00 and 43.00 for lint percent (LP), 0.36 and 0.33 for gin turnout (GT), 14.85 and 11.28 for seed index (SI), 8.37 and 6.20 for lint index (LI), 824.33 and 669.33 g (no. g) for nep size (Ns), 129.83 and 144.33 g−1 for number of neps (Nn), 33.00 and 5.50 for number of seed coats (SCN), 32.76 and 36.32 mm for upper quartile of fiber length by weight (UQL), 62.8 and 59.0 g (kg−1) for short fiber content by weight g (SFC), 21.34 and 22.61 mm for average length of all fibers by weight (ALFw), 36.83 and 40.64 mm for 5.0 % fiber span length (5.0L), 39.62 and 43.94 mm for 2.5 % fiber span length (2.5L), 1.62 and 1.51 for VFM percent (VFM), 174 and 149 mtx for fiber fineness (FTX), 5.73 and 7.32 kg−1 for immature fiber content by weight g (IFC), 0.95 and 0.91 for maturity ratio (MR), 21.2 and 29.86 for mean tenacity (MT), and 6.5 and 7.79 for mean elongation (ME).
While differences among observed traits of the RILs within each location and between locations were observed with mean separations in the various examinations of main effects, there was a similarity of overall responses of the RIL entries at the different environments (detailed data not shown). A phenotypic data summary of means, standard deviations, and ranges for PA, YC, and FP traits observed in the RIL population is presented, respectively, in Tables 1, 2 and 3 for each trait group. Although the data of the above 24 traits were collected at different time intervals among the different environments, significant phenotypic differences (P < 0.05) were observed among the RILs for all traits. While the RIL plants showed a normal distribution for the PA traits, they grew much taller, with longer but fewer internodes in the California location (SH) than in the two Texas locations (FB and BB) (Table 1). For example, the plant height of the RIL population at the SH environment had the high mean value with 100.20 cm while FB environment had the low value with 54.78 cm. Average internode length ranged from 0.14 to 7.28 cm among the three different locations FB, BB, and SH. For the YC traits, the lint weight per plant ranged from 0.10 to 103.21 g, and seed index ranged from 6.2 to 19.2 (Table 2). For the FP traits, the 2.5 % fiber span length ranged from 27.5 to 47.5 mm, and immature fiber content by weight ranged from 4.3 to 12.2 (g/kg) (Table 3). The phenotypic variation also included poor or no germination from some of the RILs under the field conditions, and lack of fiber production, causing challenges for complete data analyses.
Identification of QTLs and QTL clusters
The profound phenotypic variability of the RIL population was reflected in the identification of large numbers of putative QTLs for the PA, YC, and FP traits (Fig. 1; Supplementary Tables S1–S3). More than 600 putative QTLs, many expressed in multiple environments, were detected by single marker analysis (P < 0.05) using a nonparametric mapping test (Fig. 1; complete data not shown). For the PA traits, we detected more than 23 putative QTLs at FB, 17 QTLs at BB, and 22 QTLs at SH, explaining from 5 to 15 % of the phenotypic variation and involving 23 of the 26 cotton chromosomes. For the YC traits, we detected more than 32 putative QTLs at FB, 12 QTLs at BB, and 35 QTLs at SH, explaining from 5 to 31 % of the phenotypic variation and involving 25 of the 26 cotton chromosomes. For the FP traits, we detected more than 125 putative QTLs at FB, 110 QTLs at BB, and 135 at SH, explaining from 7 to 62 % of the phenotypic variation and involving all the 26 cotton chromosomes. More than 500 putative QTLs by trait group from the three environments were detected by interval mapping: around 62 for PA, 79 for YC, and 360 for FP (data not shown). The MQM QTL model mapping analysis identified and confirmed 72 strong QTLs from the above putative QTLs with a LOD > 3.0 threshold value at each environment (Table 4; Fig. 1). For comparison between environments, however, putative QTLs with P < 0.05 and LOD > 1.7 were included in this study (Supplementary Tables S1–S3).







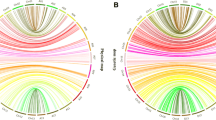
Genomic locations of putative QTL loci among 26 allotetraploid cotton chromosomes that are presented in 13 At and Dt subgenome homoeologous pairs. All SSR and SNP markers shown on the right were previously developed and genetically mapped using the same TM-1 × 3-79 RIL mapping population (Yu et al. 2012a; Fang and Yu 2012). The position of the markers shown in Kosambi (1944) centiMorgan (cM) on the left reflects the calculation of marker distance and order, and chromosome orientation. A line bar connects a marker that is putatively associated with a single trait or multiple traits, and only one trait is listed if more than one environment location show a response to QTL effect. The number (in parenthesis) following the trait abbreviation indicates the environment location (1-FB, 2-BB, and 3-SH). A description of individual trait abbreviation is presented in Table 1. An asterisk denotes a strong QTL locus with the LOD score greater than 3.0 (Table 4; Supplementary Tables S1–S3)
A total of 428 putative QTLs were significant (P < 0.05 or LOD ≥ 2.0) at one location and they were distributed in 159 genomic regions across the tetraploid cotton genome with several clusters located in a few chromosomes (Fig. 1; Supplementary Tables S1–S3). Such chromosomes included QTL clusters for fiber length on chromosome 10 (A10) and fiber maturation on chromosomes 5 (A05) and 15 (D01). The QTLs for most traits were largely located on non-homoeologous chromosomes of the A and D subgenomes. However, the pair of homoeologous chromosomes 5 (A05) and 19 (D05) each harbored 12 and 6 putative fiber QTLs, respectively (Fig. 1; Supplementary Table S3). For the 72 strong QTLs with a LOD score greater than 3.0, the pattern of being located largely on non-homoeologous chromosomes is similar to that of the putative QTLs while four and five strong QTL clusters were located on homoeologous chromosomes 5 (A05) and 19 (D05), respectively (Fig. 1; Table 4).
Comparison of putative QTLs among the environments and between the subgenomes
When putative QTLs identified using the MQM model procedure were compared among FB, BB and SH environments, 25 of the 62 PA QTLs were identified in more than one environment. The 25 putative QTLs were associated with the same trait or with another PA trait with at least P < 0.05 or LOD ≥ 2.0 at one location. Nineteen putative QTLs on six chromosomes belonged to the At subgenome (chromosomes 1–13), representing a 72.7 % contribution for the At subgenome, while six putative QTLs on three chromosomes belonged to the Dt subgenome (chromosomes 14–26), representing a 27.3 % contribution for the Dt subgenome (Fig. 1; Supplementary Table S1). When putatively identified QTLs of YC traits were compared among the three environments, 60 of the 79 YC putative QTLs were identified in more than one environment. The 60 putative QTLs were associated with the same trait or with another YC trait with at least P < 0.05 or LOD ≥ 2.0 at one location. Twenty-six putative QTLs on 10 chromosomes belonged to the At subgenome, representing a 46.2 % contribution for the At subgenome, while 44 QTLs on 9 chromosomes belonged to the Dt subgenome, representing a 53.8 % contribution for the Dt subgenome (Fig. 1; Supplementary Table S2). When putatively identified QTLs of FP traits were compared among the three environments, 343 of the 360 FP putative QTLs were identified in more than one environment. The 343 putative QTLs were associated with the same trait or with another FP trait with at least P < 0.05 or LOD ≥ 2.0 at one location. One hundred seventy-two putative QTLs on the 13 chromosomes belonged to the At subgenome, representing 50.4 % of the At subgenome contribution, and 171 putative QTLs on the 13 chromosomes belonged to the Dt subgenome, representing 49.6 % of the Dt subgenome contribution (Fig. 1; Supplementary Table S3). Overall, we observed almost even contribution to YC and FP traits from At and Dt subgenomes but significantly more contribution to PA traits from At than Dt subgenome for the detected QTLs (Fig. 1). Furthermore, the putative QTLs for the various traits tend to reside at or near the same locus for certain chromosomes from at least two different environments on this RIL population, confirming a cluster network with alleles showing heterogeneous phenotypic effects.
Phenotypic effect of QTLs under multiple environments
Although heterogeneous QTL results were observed, suggesting a complex network of genes for the variation of PA, YC, and FP traits, the nonparametric mapping (Van Ooijen 2004) compiled with the MQM test identified several DNA markers that are associated with strong QTLs (LOD > 3.0) for different traits or for same traits under multiple environments with P < 0.05 (Fig. 1; Table 4; Supplementary Tables S1–S3).
SSR marker TMB 1496 located on chromosome 5 (A05) explained up to 12.4, 15.5, 14.5, and 40.3 % of four fiber properties: average length of all fibers by weight, upper quartile of fiber length by weight, 5.0 % fiber span length, and mean tenacity, respectively. Alleles with the large effect on average length of all fibers by weight (FAlQtlc05_1b), upper quartile of fiber length by weight (FUqQtlc05_1b), and 5.0 % fiber span length (F5lQtlc05_1b) were contributed by 3-79, while that on mean tenacity (FMtQtlc05_1h) was contributed by TM-1 (Table 4). SSR marker TMB0694b located on chromosome 15 (D01) explained up to 18.1 and 25.6 % of the lint weight per plant (LWpQtlc15_1b) variation of the YC traits in FB and SH environments, respectively. SSR marker TMB0400 located on chromosome 21 (D11) explained 18.9 and 16.0 % of seed weight per plant (SWpQtlc21_1b) variation in FB and BB environments, respectively. Alleles with the large effect on both lint weight per plant (LWpQtlc21_1b) and seed weight per plant (SWpQtlc21_1b) were contributed by 3-79 (Table 4; Supplementary Table S2). SNP marker UCcgs10033_191 on chromosome 11 (A11) were associated with putative QTLs that contributed about 7–8 % of the phenotypic variation of lint percentage, gin turnout, and lint index at the SH environment (Supplementary Table S2). SSR marker MUCS145b on chromosome 13 (A13) associated with a strong QTL for fiber fineness of the FP traits explained up to 12.5 and 27.5 % of the fiber fineness (FFiQtlc13_1b) variation at the FB and SH environments, respectively (Table 4; Supplementary Table S3). In addition, a major contribution for fiber fineness variation also was made by NAU2140, located on chromosome 5 (A05), explaining from 17 to 24 % of the phenotypic variation in BB and SH environments (Supplementary Table S3).
Based on this study, QTLs for specific traits, discerned in at least two different environments, tended to reside at or near the same locus of the same chromosomes. Progeny homozygous for alleles identified by the markers on the chromosomes from the parents (TM-1 and 3-79) generally agreed with their contribution to YC and FP traits in their RIL progeny. The observation of improved progeny carrying alleles from both parents is an indication of additive effect of the contributing genes that could be subject to MAS.
Discussion
The identification of DNA markers that are associated with traits of agronomic significance provides a valuable tool for understanding the genetic control in the plant genome and for facilitating marker-assisted breeding of the crop plant (Collard and Mackill 2008; Lacape et al. 2013; Said et al. 2013). In this study, QTL mapping was conducted under three diverse production environments (FB, BB, and SH) to identify the genetic factors or genomic loci for PA, YC, and FP with an interspecific cotton (G. hirsutum acc. TM-1 × G. barbadense acc. 3-79) RIL population. New QTLs unique to this TM-1 × 3-79 RIL mapping population and/or a particular environmental condition were detected while others appeared to express across multiple environments and/or different mapping populations (Lacape et al. 2010, 2013; Rong et al. 2007; Song and Zhang 2009). Identification of 159 genomic regions or QTL-bearing markers from more than 2,500 mapped SSR and SNP loci of the tetraploid cotton genome would facilitate further dissection of genetic factors underlying these important traits and MAS for the cotton crop.
Although much used in linkage mapping efforts, the present investigation is one of the first studies that use the interspecific RIL population to identify and locate the genomic loci for all three sets of morphological, yield, and fiber traits in the tetraploid cotton genome. This TM-1 × 3-79 RIL population is a genetic standard resource that has been distributed by USDA-ARS among the cotton research community (Park et al. 2005; Wang et al. 2012a). Another major interspecific cotton RIL population used to investigate such traits is derived from a cross between Guazuncho 2 (G. hirsutum) and VH8-4602 (G. barbadense) (Lacape et al. 2010). In that study, subsets of 140 RILs, originated from some of approximately 600 F2 plants, were used for trait observation among different environments (Lacape et al. 2013). In addition, an interspecific backcross inbred population of 146 lines was used to identify QTLs for fiber properties and yield components (Yu et al. 2013). In our present study, the parental genotypes (TM-1 and 3-79) used in making this population represent highly divergent global genetic standards for G. hirsutum and G. barbadense species, respectively, to maximize the chance of QTL identification for all these traits. Characterizing the phenotypic variation of morphological traits, yield components, and fiber quality properties facilitates the utilization of this population by other researchers who wish to further examine the co-segregation of any type of molecular markers and phenotypes (Wang et al. 2012a; Said et al. 2013). Obtaining the information that quantifies the phenotypic variation within the TM-1 × 3-79 RIL population allows us to determine what morphological, yield and fiber quality traits may be improved through introgression using the RIL population.
While our interspecific RIL population exhibited profound variation in many traits including PA, YC, and FP, some individual RILs of the population have reduced fertility and productivity, as commonly observed in RIL and other populations of interspecific origin (Lacape et al. 2010, 2013). In this study, the allotetraploidy of cultivated parental cottons (G hirsutum and G. barbadense) provides an opportunity to investigate putative QTLs or genetic factors between the homoeologous chromosomes (Buyyarapu et al. 2013; Xu et al. 2008a). The two subgenomes of the allotetraploid cottons may contribute differently but complementarily to functional networks of cotton genes (Xu et al. 2010).
A problem arising from the variable fertility of RILs has been seed shortages, compounded with the poor germination of some RILs. Regardless of these difficulties, such segregating populations were used to map cotton yield and quality QTLs, and comparable data were obtained for traits across the multiple environment locations in other studies (Lacape et al. 2013; Yu et al. 2013). RIL populations are usually extended beyond F6-F9 generations which have provided better genetic uniformity of trait response (homozygous alleles), reducing the external or environmental effects as compared to early generations on such traits as fiber yield that may not be highly precise due to the limited information in this area. However, the general information is important for breeders who are using interspecific introgression breeding via molecular markers. The two parental species used in our study represent unique contrasting yield and quality traits that are desired in the other species. G hirsutum cotton is known for its high yield and wide adaptation while G. barbadense cotton is known for its superior fiber quality (length, fineness and strength). Even though the data of the 24 traits were collected at different time intervals among the different environments, significant phenotypic differences (P < 0.05) were observed among the RILs for all traits within each location. RIL plants showed a normal distribution for traits with large environmental effect such as PA. As expected, plants grew much taller, with longer but fewer internodes in the California location than in the two Texas locations (Table 1), and the lint weight per plant ranged from 0.10 to 103.21 g and seed index from 6.2 to 19.2 (Table 2). Although data were collected from 2–5 RIL plants per row, phenotypic variation also included poor or no germination from some of the RILs under the field conditions and lack of fiber production, causing challenges for complete data analyses. The identification of QTLs for these traits would advance our understanding and increase our knowledge in future introgression breeding efforts that have not been very successful in cotton. For more effective and precise use of the information in cotton molecular breeding, however, further research at a higher resolution of the genome sequence is desired to validate and exploit putative QTLs identified in the present study. This is especially important for complex YC traits that can largely be affected by the environment but can advance our understanding and knowledge for future breeding efforts.
The data from our study indicate that most of the 428 putative QTLs for the traits measured are located in non-homoeologous chromosomes of the tetraploid cotton genome. A study, using different populations and linkage maps, analyzed 432 cotton fiber QTLs that were largely found in non-homoeologous regions of the tetraploid subgenomes (Rong et al. 2007). In another study with an interspecific RIL population, Lacape et al. (2010) analyzed 651 cotton fiber QTLs that basically agree to the findings of Rong et al. (2007). Both studies reported the exceptional pair of chromosomes 5 (A05) and 19 (D05), in which we identified 12 and 6 putative QTLs (of which 4 and 5 QTLs were strongly expressed), respectively. Said et al. (2013) recently compiled over 1,000 reported QTLs from dozens of cotton studies, which showed an uneven distribution of QTLs in the cotton genome. Of particular interest were specific 20-cM genomic regions of these two chromosomes for Micronaire hotspots. Our result supports and augments the prior evidence of this particular pair of gene-enriched chromosomes (Lacape et al. 2010; Said et al. 2013). In chromosome 5, several gene islands for fiber initiation and fiber fineness were previously reported (Rong et al. 2007; Xu et al. 2008b). DNA markers associated with such gene islands or QTL clusters would be valuable for further investigations including MAS.
Many putative QTLs associated with different, correlated traits or with the same trait measured in different ways, co-resided in the same genomic regions, and these QTLs tended to be expressed in a particular environmental condition (Fig. 1). For example, QTLs for fiber fineness (FFiQtlc19_1b), immature fiber content (FIfQtlc19_1b), and maturity ratio (FMrQtlc19_1h) co-resided in a genomic region of chromosome 19 (D05), associated with SSR marker BNL3875 at the SH location (Table 4). Another SSR marker CIR176 on the same chromosome 19 (D05) is associated with the putative QTLs conferring six FP traits: mean elongation, 2.5 % fiber span length, 5.0 % fiber span length, upper quartile of fiber length, average length of all fibers, and nep size (Supplementary Table S3). These putative QTLs are primarily expressed for fiber traits at the BB location. In this study, DNA markers used to identify cotton QTLs include those that were developed from the bacteria artificial chromosome (BAC) clones (Yu et al. 2012a). For example, a TM-1 BAC-derived marker, TMB0799, on chromosome 12 (A12), is strongly associated with the QTLs conferring five FP traits primarily at the SH location: fiber fineness (FFiQtlc12_1h), short fiber content (FSfQtlc12_1b), average length of all fibers (FAlQtlc12_1b), immature fiber content (FIfQtlc12_1b), and maturity ratio (FMrQtlc12_1h) (Fig. 1; Table 4). With the development of integrated physical map and genome sequence map of the tetraploid cotton chromosome 12 (A12), future research with this BAC-derived marker would shed more light on the QTL cluster or gene island to better understand the genetic mechanisms underlying these fiber traits (Buyyarapu et al. 2013; Xu et al. 2008a, b).
As shown in this study and others, both superior and inferior parents may possess genetic factors that control the performance of contrasting traits otherwise undetectable by phenotype alone (Paterson et al. 1991; Tanksley and McCouch 1997; Wang et al. 2012a). TM-1 alleles at the TMB1496 locus contributed to fiber tenacity (FMtQtlc05_1h) and 3-79 alleles at the TMB0400 locus contributed to lint weight (LWpQtlc21_1b) and seed weight (SWpQtlc21_1b), which is counter intuitive for these parents by phenotypic observation. To further dissect the 159 genomic regions that harbor the 428 putative QTL loci identified in this study, for eventual use in cotton molecular breeding programs, a graphical display of each QTL locus or QTL cluster for cotton PA, YC, and FP traits needs to be developed, and a subset of the selected TM-1 × 3-79 RILs that best represent genomic regions of interest needs to be identified for detail characterization.
The functional network of QTL clusters in cultivated cottons is complex, and distinguishing orthologs from paralogs is challenging in the absence of complete genome sequences. This study is the first to include SNP markers to identify the QTLs in cotton. For example, an SNP marker UCcg11114_430 mapped on chromosome 8 (A08) is associated with the putative QTLs conferring number of neps, immature fiber content, maturity ratio, and nep size (Fig. 1; Supplementary Table S3). Another SNP marker UCcg10645_71, mapped approximately 20 cM apart from TMB0799 (discussed above) on chromosome 12 (A12), is associated with the putative QTLs conferring average length of all fibers, upper quartile of fiber length, 5.0 % fiber span length, and short fiber content (Fig. 1; Supplementary Table S3). With increasing numbers of SNP markers being developed in cotton, profound diversity can be exploited between TM-1 and 3-79 that contrast in the traits of agronomic importance (Elshire et al. 2011).
The recent development of a complete genome sequence in cotton provides an unprecedented opportunity to scan or browse the QTL regions with the finest details (Li et al. 2014; Paterson et al. 2012; Wang et al. 2012b). The markers associated (putatively or strongly) with any QTLs can be aligned to the genome sequence (Gao et al. 2013). Because the parental lines of our TM-1 × 3-79 RIL mapping population represent the global genetic standards of G. hirsutum and G. barbadense, respectively, the international cotton genome research community is collaborating closely to sequence both Gossypium tetraploid reference genomes (i.e., TM-1 and 3-79). Assembly and annotation of the two parental genome sequences, along with re-sequencing of selected progeny RILs would facilitate detailed structural, functional, and evolutionary analyses of the cotton QTLs (Gao et al. 2013; Lacape et al. 2010, 2013; Rong et al. 2007; Said et al. 2013; Ulloa et al. 2013; Wang et al. 2012a). More precise studies of the putative QTLs identified in this study and in many others would follow in the future when the tetraploid cotton genome sequence becomes available.
References
Abdurakhmonov IY, Buriev ZT, Shermatov SE, Abdullaev AA, Urmonov K, Kushanov F, Egamberdiev SS, Shapulatov U, Abdukarimov A, Saha S, Jenkins JN, Kohel RJ, Yu JZ, Pepper AE, Kumpatala S, Ulloa M (2012) Genetic diversity in Gossypium genus. In: Çalişkan M (ed) Genetic diversity in plants. InTech, New York, pp 313–338
An C, Jenkins JN, Wu J, Guo Y, McCarty JC (2010) Use of fiber and fuzz mutants to detect QTL for yield components, seed, and fiber traits of upland cotton. Euphytica 172(1):21–34
Arends D, Prins P, Jansen RC, Broman KW (2010) R/qtl: high-throughput multiple QTL mapping. Bioinformatics 26(23):2990–2992. doi:10.1093/bioinformatics/btq565
Beasley JO (1940) The origin of American tetraploid Gossypium species. Am Nat 74:285–286
Blenda A, Fang DD, Rami J-F, Garsmeur O, Luo F, Lacape J-M (2012) A high-density consensus genetic map of tetraploid cotton that integrates multiple component maps through molecular marker redundancy check. PLoS ONE 7(9):e45739
Brubaker CL, Paterson AH, Wendel JF (1999) Comparative genetic mapping of allotetraploid cotton and its diploid progenitors. Genome 42(2):184–203
Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC et al (2009) The genetic architecture of maize flowering time. Science 325(5941):714–718
Buyyarapu R, Kantety RV, Yu JZ, Xu Z, Kohel RJ, Percy RG, Macmil S, Wiley GB, Roe BA, Sharma GC (2013) BAC-pool sequencing and analysis of large segments of A12 and D12 homoeologous chromosomes in Upland cotton. PLoS ONE 8(10):e76757. doi:10.1371/journal.pone.0076757
Campbell B, Saha S, Percy R, Frelichowski J, Jenkins J, Park W, Mayee C, Gotmare V, Dessauw D, Giband M, Du X, Jia Y, Constable G, Dillion S, Abdurakhmonov I, Abdukarimov A, Rizaeva S, Abdullaev A, Barroso P, Padua J, Hoffmann L, Podolnaya L (2010) Status of the global cotton germplasm resources. Crop Sci 50:1161–1179
Cao Z, Wang P, Zhu X, Chen H, Zhang T (2014) SSR marker-assisted improvement of fiber qualities in Gossypium hirsutum using G. barbadense introgression lines. Theor Appl Genet 127(3):587–594
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Collard BCY, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc B Biol Sci 363(1491):557–572
Cronn RC, Small RL, Wendel JF (1999) Duplicated genes evolve independently after polyploid formation in cotton. Proc Natl Acad Sci USA 96(25):14406–14411
Draye X, Chee P, Jiang C-X, Decanini L, Delmonte TA, Bredhauer R, Smith CW, Paterson AH (2005) Molecular dissection of interspecific variation between Gossypium hirsutum and G. barbadense (cotton) by a backcross-self approach: II. Fiber fineness. Theor Appl Genet 111(4):764
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6(5):e19379
Fang DD, Yu JZ (2012) Addition of four-hundred fifty-five microsatellite marker loci to the high-density Gossypium hirsutum TM-1 × G. barbadense 3-79 genetic map. J Cotton Sci 16:229–248
Fang DD, Jenkins JN, Deng DD, McCarty JC, Li P, Wu J (2014) Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in Upland cotton (Gossypium hirsutum L.). BMC Genom 15(1):397
Fehr WR (1991) Principles of cultivar development. Theory and technique, vol 1. Macmillan Publishing Company, Ames, p 672
Frelichowski JE Jr, Palmer M, Main D, Tomkins JP, Cantrell R, Stelly DM, Yu JZ, Kohel RJ, Ulloa M (2006) Cotton genome mapping with new microsatellites from Acala ‘Maxxa’ BAC-ends. Mol Genet Genom 275:479–491
Fryxell PA (1979) Natural history of the cotton tribe (Malvaceae, tribe Gossypieae). Texas A&M University Press, College Station, p 245
Gao Z-Y, Zhao S-C, He W-M, Guo L-B, Peng Y-L, Wang J-J, Guo X-S, Zhang X-M, Rao Y-C, Zhang C, Dong G-J, Zheng F-Y, Lu C-X, Hu J, Zhou Q, Liu H-J, Wu H-Y, Xu J, Ni P-X, Zeng D-L, Liu D-H, Tian P, Gong L-H, Ye C, Zhang G-H, Wang J, Tian F-K, Xue D-W, Liao Y, Zhu L, Chen M-S, Li J-Y, Cheng S-H, Zhang G-Y, Wang J, Qian Q (2013) Dissecting yield-associated loci in super hybrid rice by resequencing recombinant inbred lines and improving parental genome sequences. Proc Natl Acad Sci USA 110(35):14492–14497
Guo W, Cai C, Wang C, Han Z, Song X, Wang K, Niu X, Wang C, Lu K, Shi B, Zhang T (2007) A Microsatellite-based, gene-rich linkage map reveals genome structure, function and evolution in Gossypium. Genetics 176(1):527–541
Huang X, Kurata N, Wei X, Wang Z, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W, Guo Y, Lu Y, Zhou C, Fan D, Weng Q, Zhu C, Huang T, Zhang L, Wang Y, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan X, Xu Q, Dong G, Zhan Q, Li C, Fujiyama A, Toyoda A, Lu T, Feng Q, Qian Q, Li J, Han B (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature 490:497–501
Jiang C, Wright RJ, El-Zik KM, Paterson AH (1998) Polyploid formation created unique avenues for response to selection in Gossypium. Proc Natl Acad Sci USA 95:4419–4424
Kantartzi SK, Ulloa M, Sacks E, Stewart JM (2009) Assessing genetic diversity in Gossypium arboreum L. cultivars using genomic and EST-derived microsatellites. Genetica 136(1):141–147
Kohel R, Richmond T, Lewis C (1970) Texas Marker-1. A description of a genetic standard for Gossypium hirsutum L. Crop Sci 10:670–671
Kohel RJ, Yu JZ, Park YH, Lazo GR (2001) Molecular mapping and characterization of traits controlling fiber quality in cotton. Euphytica 121:163–172
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Lacape J-M, Nguyen T-B, Courtois B, Belot J-L, Giband M, Gourlot J-P, Gawryziak G, Roques S, Hau B (2005) QTL analysis of cotton fiber quality using multiple Gossypium hirsutum × Gossypium barbadense backcross generations. Crop Sci 45(1):123
Lacape J-M, Jacobs J, Arioli T, Derijcker R, Forestier-Chiron N, Llewellyn D, Jean J, Thomas E, Viot C (2009) A new interspecific, Gossypium hirsutum × G. barbadense, RIL population: towards a unified consensus linkage map of tetraploid cotton. Theor Appl Genet 119(2):281–292
Lacape J-M, Llewellyn D, Jacobs J, Arioli T, Becker D, Al-Ghazi Y, Liu S, Palaï O, Georges S, Giband M, Assunção JH, Barroso PAV, Claverie M, Gawryziak G, Jean J, Vialle M, Viot C (2010) Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum × G. barbadense RIL population. BMC Plant Biol 10:132. doi:10.1186/1471-2229-10-132
Lacape J-M, Gawryziak G, Cao T-V, Viot C, Llewellyn D, Liu S, Jacobs J, Becker D, Barroso PAV, Assunção JH, Palaï O, Georges S, Jean J, Giband M (2013) Mapping QTLs for traits related to phenology, morphology and yield components in an inter-specific Gossypium hirsutum × G. barbadense cotton RIL population. Field Crop Res 144:256–267
Lee JA (1984) Cotton as a world crop. In: Kohel RJ, Lewis CF (eds) Cotton, agronomy monograph, vol 24. American Society of Agronomy, Madison, pp 1–25
Li C, Wang C, Dong N, Wang X, Zhao H, Converse R, Xia Z, Wang R, Wang Q (2012) QTL detection for node of first fruiting branch and its height in upland cotton (Gossypium hirsutum L.). Euphytica 188(3):441–451
Li F, Fan G, Wang K, Sun F, Yuan Y, Song G, Li Q, Ma Z, Lu C, Zou C, Chen, Liang X, Shang H, Liu W, Shi C, Xiao G, Gou C, Ye W, Xu X, Zhang X, Wei H, Li Z, Zhang G, Wang J, Liu K, Kohel RJ, Percy RG, Yu JZ, Zhu Y-X, Wang J, Yu S (2014) Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet 46:567–572. doi:10.1038/ng.2987
Li X, Yuan D, Zhang J, Lin Z, Zhang X (2013) Genetic mapping and characteristics of genes specifically or preferentially expressed during fiber development in cotton. PloS ONE 8(1):e54444
Luan M, Guo X, Zhang Y, Yao J, Chen W (2009) QTL mapping for agronomic and fibre traits using two interspecific chromosome substitution lines of Upland cotton. Plant Breed 128(6):671–679
Ma XX, Zhou BL, Lu YH, Guo WZ, Zhang TZ (2010) Simple sequence repeat genetic linkage maps of A-genome diploid cotton (Gossypium arboreum). J Integr Plant Biol 50(4):491–502
Marathi B, Guleria S, Mohapatra T, Parsad R, Mariappan N, Kurungara VK, Atwal SS, Prabhu KV, Singh NK, Singh AK (2012) QTL analysis of novel genomic regions associated with yield and yield related traits in new plant type based recombinant inbred lines of rice (Oryza sativa L.). BMC Plant Biol 12:137. doi:10.1186/1471-2229-12-137
Mohan M, Nair S, Bhagwat A, Krishna TG, Yano M, Bhatia CR, Sasaki T (1997) Genome mapping, molecular markers and marker-assisted selection in crop plants. Mol Breed 3(2):87–103
Nguyen T-T, Giband M, Brottier P, Risterucci A-M, Lacape J-M (2004) Wide coverage of the tetraploid cotton genome using newly developed microsatellite markers. Theor Appl Genet 109:167–175
Niles GA, Feaster CV (1984) Breeding. In: Kohel RJ, Lewis CF (eds) Cotton. Monograph series agronomy, vol 24. American Society of Agronomy, Madison, pp 201–231
Ning Z, Chen H, Mei H, Zhang T (2014) Molecular tagging of QTLs for fiber quality and yield in the upland cotton cultivar Acala-Prema. Euphytica 195(1):143–156
Ott L (1988) An introduction to statistical methods and data analysis. PWS-Kent, Boston, MA, p 1296
Paran I, Zamir D (2003) Quantitative traits in plants: beyond the QTL. Trends Genet 19(6):303–306
Park Y-H, Alabandy MS, Ulloa M, Wilkins TA, Yu JZ, Stelly DM, Kohel RJ, Elshihy OM, Cantrell RG (2005) Genetic and QTL mapping of new fiber EST-derived microsatellites on an interspecific recombinant inbred line (RIL) cotton population. Mol Genet Genom 274:428–441
Paterson A, Lander E, Hewitt J, Peterson S, Lincoln S, Tanksley S (1988) Resolution of quantitative traits into Mendelian factors by using a complete map of restriction fragment length polymorphisms. Nature 335:721–726
Paterson AH, Tanksley SD, Sorrells ME (1991) DNA markers in plant improvement. In: Donald LS (ed) Advances in agronomy, vol 46. Academic press, London, pp 39–90
Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, Jin D, Llewellyn D, Showmaker KC, Shu S, Udall J, Yoo M, Byers R, Chen W, Faigenboim AD, Duke MV, Gong L, Grimwood J, Grover C, Grupp K, Hu G, Lee T, Li J, Lin L, Liu T, Marler BS, Page JT, Roberts AW, Romanel E, Sanders WS, Szadkowski E, Tan X, Tang H, Xu C, Wang J, Wang Z, Zhang D, Zhang L, Ashrafi H, Bedon F, Bowers JE, Brubaker CL, Chee PW, Das S, Gingle AR, Haigler CH, Harker D, Hoffmann LV, Hovav R, Jones DC, Lemke C, Mansoor S, Rahman M, Rainville LN, Rambani A, Reddy UK, Rong J, Saranga Y, Scheffler BE, Scheffler JA, Stelly DM, Triplett BA, Van Deynze A, Vaslin MFS, Waghmare VN, Walford SA, Wright RJ, Zaki EA, Zhang T, Dennis ES, Mayer KFX, Peterson DG, Rokhsar DS, Wang X, Schmutz J (2012) Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492:423–427
Percival AE, Wendel JF, Stewart JM (1999) Taxonomy and germplasm resources. In: Smith CW, Cothren JT (eds) Cotton: origin, history, technology, and production. Wiley, New York, pp 33–63
Percy RG, Frelichowski JE, Arnold M, Campbell BT, Dever J, Fang DD, Hinze LL, Main D, Scheffler JA, Sheehan M, Ulloa M, Yu J, Yu JZ (2014) The U.S. national cotton germplasm collection—its contents, preservation, characterization, and evaluation. In: Abdurakhmonov I (ed) World cotton germplasm resources. InTech, Rijeka, pp 167–201
Reddy OUK, Pepper AE, Abdurakhmonov I, Saha S, Jenkins J, Brooks T, El-Zik KM (2001) New dinucleotide and trinucleotide microsatellite marker resources for cotton genome research. J Cotton Sci 5:103–113
Rong J, Abbey C, Bowers JE, Brubaker CL, Chang C, Chee PW, Delmonte TA, Ding X, Garza JJ, Marler BS, Park C, Pierce GJ, Rainey KM, Rastogi VK, Schulze SR, Trolinder NL, Wendel JF, Wilkins TA, Williams-Coplin TD, Wing RA, Wright RJ, Zhao X, Zhu L, Paterson AH (2004) A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166(1):389–417
Rong J, Feltus FA, Waghmare VN, Pierce GJ, Chee PW, Draye X, Saranga Y, Wright RJ, Wilkins TA, May OL, Smith CW, Gannaway JR, Wendel JF, Paterson AH (2007) Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176:2577–2588
Said JI, Lin Z, Zhang X, Song M, Zhang J (2013) A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom 14(1):776
Shen X, Guo W, Zhu X, Yuan Y, Yu JZ, Kohel RJ, Zhang T (2005) Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol Breed 15(2):169
Song X, Zhang T (2009) Quantitative trait loci controlling plant architectural traits in cotton. Plant Sci 177(4):317–323
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: Unlocking genetic potential from the wild. Science 277(5329):1063–1066
Ulloa M, Brubaker C, Chee P (2007) Cotton. In: Kole C (ed) Genome mapping and molecular breeding. Technical crops, vol 6. Springer, New York, pp 1–50
Ulloa M, Wang C, Roberts PA (2010) Gene action analysis by inheritance and QTL mapping of resistance to root-knot nematodes in cotton. Plant Breed 129(5):541–550
Ulloa M, Wang C, Hutmacher RB, Wright SD, Davis RM, Saski CA, Roberts PA (2011) Mapping Fusarium wilt race 1 resistance genes in cotton by inheritance, QTL and sequencing composition. Mol Genet Genom 286:21–36
Ulloa M, Hutmacher RB, Roberts PA, Wright SD, Nichols RL, Davis RM (2013) Inheritance and QTL mapping of Fusarium wilt race 4 resistance in cotton. Theor Appl Genet 126:1405–1418
Ulloa M, Saha S, Jenkins JN, Meredith WR Jr, McCarty JC, Stelly MD (2005) Chromosomal assignment of RFLP linkage groups harboring important QTLs on an intraspecific cotton (Gossypium hirsutum L.) joinmap. J Hered 96:132–144
Van Deynze A, Stoffel K, Lee M, Kozik A, Wilkins TA, Cantrell RG, Yu JZ, Kohel RJ, Stelly DM (2009) Sampling nucleotide diversity in cotton. BMC Plant Biol 9:125
Van Ooijen JW (2004) MapQTL® 5. Software for the mapping of quantitative trait loci in experimental populations. Kyazma B.V., Wageninggen, Netherlands
Wang C, Ulloa M, Mullens TR, Yu JZ, Roberts PA (2012a) QTL analysis for transgressive resistance to root-knot nematode in interspecific cotton (Gossypium spp.) progeny derived from susceptible parents. PLoS ONE 7(4):e34874. doi:10.1371/journal.pone.0034874
Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S, Zou C, Li Q, Yuan Y, Lu C, Wei H, Gou C, Zheng Z, Yin Y, Zhang X, Liu K, Wang B, Song C, Shi N, Kohel RJ, Percy RG, Yu JZ, Zhu Y-X, Wang J, Yu S (2012b) The draft genome of a diploid cotton Gossypium raimondii. Nat Genet 44(10):1098–1104
Wang Z, Zhang D, Wang X, Tan X, Guo H, Paterson AH (2013) A whole-genome DNA marker map for cotton based on the D-genome sequence of Gossypium raimondii L. G3 Genes Genom Genet 3(10):1759–1767
Wendel JF, Brubaker C, Alvarez I, Cronn R, Stewart JM (2009) Evolution and natural history of the cotton genus. In: Paterson AH (eds), Genetics and genomics of cotton. Springer, New York, pp 3–22
Wendel JF, Brubaker CL, Percival AE (1992) Genetic diversity in Gossypium hirsutum and the origin of Upland cotton. Am J Bot 79(11):1291–1310
Wendel JF, Cronn RC (2003) Polyploidy and the evolutionary history of cotton. In: Advances in agronomy, vol 78. Academic Press, London, pp 139–186
Xiao J, Wu K, Fang DD, Stelly DM, Yu JZ, Cantrell RG (2009) New DNA markers for the use in cotton (Gossypium spp.) improvement. J Cotton Sci 3(2):75–157
Xu Z, Kohel RJ, Song G, Cho J, Yu J, Yu S, Tomkins J, Yu JZ (2008a) An integrated genetic and physical map of homoeologous chromosomes 12 and 26 in Upland cotton (G. hirsutum L.). BMC Genom 9:108. doi:10.1188/1471-2164-9-108
Xu Z, Kohel RJ, Song G, Cho J, Alabady M, Yu J, Koo P, Chu J, Yu S, Wilkins TA, Zhu Y, Yu JZ (2008b) Gene-rich islands for fiber development in the cotton genome. Genomics 92:173–183
Xu Z, Yu JZ, Cho J, Yu J, Kohel RJ, Percy RG (2010) Polyploidization altered gene functions in cotton (Gossypium spp.). PLoS ONE 5(12):e14351. doi:10.1371/journal.pone.0014351
Yu J, Zhang K, Li S, Yu S, Zhai H, Wu M, Li X, Fan S, Song M, Yang D, Li Y, Zhang J (2013) Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. TAG Theor Appl Genet 126(1):275–287
Yu JZ, Kohel RJ, Fang DD, Cho J, Van Deynze A, Ulloa M, Hoffman SM, Pepper AE, Stelly DM, Jenkins JN, Saha S, Kumpatla SP, Shah, Hugie WV, Percy RG (2012a) A high-density simple sequence repeat and single nucleotide polymorphism genetic map of the tetraploid cotton genome. G3 Genes Genom Genet 2:43–58
Yu JZ, Fang DD, Kohel RJ, Ulloa M, Hinze LL, Percy RG, Zhang J, Chee P, Scheffler BE, Jones DC (2012b) Development of a core set of SSR markers for the characterization of Gossypium germplasm. Euphytica 187:203–213
Zhu H, Han X, Lu J, Zhao L, Xu X, Zhang T, Guo W (2011) Structure, expression differentiation and evolution of duplicated fiber developmental genes in Gossypium barbadense and G. hirsutum. BMC Plant Biol 11:40
Acknowledgments
J. Z. Yu, M. Ulloa, and S. M. Hoffman contributed equally to the work. This research was supported by U. S. Department of Agriculture, Agricultural Research Service projects 6202-21000-030-00D, 5303-21220-003-00D, and Cotton Incorporated fellowship to S. M. Hoffman (project 02-260). The authors thank J. Harris for his assistance in maintaining the TM-1 × 3-79 RIL mapping population, S. Young for his check on output analyses of comparative data sets, and J. Frelichowski for his assistance in field study and his review of the manuscript. Mention of trade names or commercial products in this manuscript is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture that is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, J.Z., Ulloa, M., Hoffman, S.M. et al. Mapping genomic loci for cotton plant architecture, yield components, and fiber properties in an interspecific (Gossypium hirsutum L. × G. barbadense L.) RIL population. Mol Genet Genomics 289, 1347–1367 (2014). https://doi.org/10.1007/s00438-014-0930-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-014-0930-5