
Abstract
MicroRNAs (miRNAs) are a class of 21–24 nucleotide non-coding RNAs that down-regulate gene expression by cleaving or inhibiting the translation of target gene transcripts. miRNAs have been extensively analyzed in a few model plant species such as Arabidopsis, rice and Populus, and partially investigated in other non-model plant species. However, only a few conserved miRNAs have been identified in Chinese cabbage, a common and economically important crop in Asia. To identify novel and conserved miRNAs in Chinese cabbage (Brassica rapa L. ssp. pekinensis) we constructed a small RNA library. Using high-throughput Solexa sequencing to identify microRNAs we found 11,210 unique sequences belonging to 321 conserved miRNA families and 228 novel miRNAs. We ran a Blast search with these sequences against the Chinese cabbage mRNA database and found 2,308 and 736 potential target genes for 221 conserved and 125 novel miRNAs, respectively. The BlastX search against the Arabidopsis genome and GO analysis suggested most of the targets were involved in plant growth, metabolism, development and stress response. This study provides the first large scale-cloning and characterization of Chinese cabbage miRNAs and their potential targets. These miRNAs add to the growing database of new miRNAs, prompt further study on Chinese cabbage miRNA regulation mechanisms, and help toward a greater understanding of the important roles of miRNAs in Chinese cabbage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNA) are 21–24 nucleotide non-coding RNA molecules that have been shown to play a key role in the regulation of gene expression (Mallory and Vaucheret 2004; Jones-Rhoades et al. 2006). miRNAs regulate a number of biological processes, such as development, signal transduction, metabolism and responses to environmental stress (Aukerman and Sakai 2003; Palatnik et al. 2003; Park et al. 2002). The majority of miRNA genes exist as independent transcriptional units, and are transcribed by RNA polymerase II into long primary transcripts (pri-miRNA) that contain perfect foldback structures with 5′ caps and 3′ poly (A) tails (Chen 2005; Xie et al. 2005). An miRNA:miRNA* complex is generated from a longer hairpin precursor by the ribonuclease III-like enzyme Dicer (DCL1) and exported to the cytoplasm. In the cytoplasm, the miRNA:miRNA* complexes are separated into miRNA and miRNA* units (Bartel 2004). The miRNAs serve as a guide for the RNA-induced silencing complex (RISC), which cleaves the RNA of target genes at the paired region or interferes with translation, while the miRNA*s are degraded (Carrington and Ambros 2003; Llave et al. 2002). Compared to other mechanisms that regulate gene expression, identifying a gene targeted by an miRNA is a straightforward process in plants (Barakat et al. 2007). Since the mature miRNA and its target sequence have almost perfect complementarity, identifying an miRNA usually leads to the prediction and/or identification of its target (Barakat et al. 2007).
In recent years, miRNAs have been identified through both experimental and computational approaches (Bartel 2004). The genetic screen for aberrant development, an experimental approach, was first employed to identify lin-4 and let-7, the two founding members of miRNAs in Caenorhabditis elegans (Lee et al. 1993; Reinhart et al. 2000). This method is inefficient for miRNA gene discovery because it is expensive, time consuming and produces only a few miRNA genes (Abbott et al. 2005; Berezikov et al. 2006). The computational approach is used to predict miRNAs based on highly conserved sequences in mature miRNAs and long hairpin structures in pre-miRNA by Blastn searches against a genome sequence or other database like expressed sequence tags (EST) (Zhang et al. 2005, 2006), but the predicted miRNAs need to be validated by cloning or Northern blotting experiments (Zhang et al. 2006). To improve discovery of novel miRNAs, direct cloning and sequencing of small RNA libraries, another experimental approach, has been recently used by many groups (Sunkar et al. 2005; Lu et al. 2005; Berezikov et al. 2006). The development of high-throughput sequencing methods, including the Roche 454 Life Sciences system, the Illumina Genome Analyzer and Applied Biosystems’ SOLiD, has greatly improved this approach, which can identify low-abundance or tissue-specific miRNAs. The 454 system is usually used for genome and transcriptome sequencing, since it has a longer read length compared with the other two platforms (Collins et al. 2008; Parchman et al. 2010; Sun et al. 2010). Because the Illumina Genome Analyzer can yield a higher number of reads (Moxon et al. 2008) with a relatively low cost (Costa et al. 2010; Morozova and Marra 2008), it is preferred for miRNA discovery (Song et al. 2010), since the sequences are only 21–24 nt in length.
Chinese cabbage (Brassica rapa L. ssp. pekinensis) is a subspecies of B. rapa (AA, 2n = 2x = 20) in the Cruciferae family, and is a widely cultivated and economically important vegetable crop in Asia. To date, 19 miRNAs and 4 miRNA*s have been deposited in the miRBase database (Release 18.0, November 2011), and recently, from a comparative analysis strategy based on the evolutionary conservation between species, 168 potential miRNAs, derived from 22 ESTs and 119 genomic survey sequences (GSS) in Chinese cabbage, were identified using publicly available GSS and EST datasets such as NCBI (Wang et al. 2011a). Compared with the number of miRNAs that have been identified (miRNA Registry, Release 18.0, November 2011) in Arabidopsis thaliana (328), Oryza sativa (661), Zea mays (321), Populus trichocarpa (237), Glycine max (395), and Medicago truncatula (674), more miRNAs can be mined out of Chinese cabbage.
In this study, using high-throughput sequencing of a Chinese cabbage small RNA library we identified 11,210 unique sequences belonging to 321 conserved miRNAs families and 228 novel miRNAs based on either sequence similarity or the secondary structure of their precursors. We validated eight of these miRNAs using quantitative real-time PCR (RT-qPCR). Furthermore, we made a deep analysis of Chinese cabbage miRNA target functions based on the information provided by a Blastx search against the Arabidopsis genome and GO analysis.
Materials and methods
Plant materials
Chinese cabbage line BrapGC1 was grown in a winter greenhouse at a relative humidity of 75 % and 20/11 °C day/night temperatures. Leaves and roots from 1-month-old seedlings, and leaves, stems, flowers, and siliques from booting stage plants were collected and stored in liquid nitrogen for RNA extraction.
Small RNA library preparation and sequencing
Mixed Chinese cabbage tissues (leaf, root, stem, flower, silique and seed) were used for RNA extraction. To identify as many tissue- or developmental stage-specific miRNAs as possible, we pooled the total RNAs from leaf, root, stem, flower, silique and seed samples in an equal fraction ratio. Total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The samples were subjected to 15 % denaturing polyacrylamide gel electrophoresis, after which the small RNA fragments of 18–26 nt were isolated from the gel and purified. Small RNAs were ligated sequentially to 5′ and 3′ RNA/DNA chimeric oligonucleotide adapters, and converted to DNA by RT-PCR. Finally, approximately 20 μg of RT-PCR products were sequenced directly using an Illumina Genome Analyzer according to the manufacturer’s protocols (Beijing Genomics Institute, China).
Small RNA analysis
Small 35-nt RNA reads were produced using an Illumina 1G Genome Analyzer at the Beijing Genomics Institute (BGI; Shenzhen, China). We then went through the processes of data cleaning and length distribution analysis, including (1) elimination of low-quality reads (more than one base with SQ value ≤10 or more than two bases with SQ value ≤13); (2) elimination of reads with 5′ primer contaminants; (3) elimination of reads without 3′ primer; (4) elimination of reads without the insert tag; (5) elimination of reads with poly A; (6) elimination of reads shorter than 18 nt; (7) summarize the length distribution of the clean reads. The remaining reads ≥18 nt were mapped to the Chiifu Chinese cabbage genome (http://brassicadb.org/brad/) using SOAP v1.11 (Li et al. 2008) to analyze their expression and distribution on the genome. To annotate the small RNAs, the following processes were performed: (1) align small RNA tags to Genbank and Rfam to annotate the small RNA tags with rRNA, scRNA, snoRNA, snRNA and tRNA, and eliminate matched tags; (2) align small RNA tags to the miRNA precursor/mature miRNA of corresponding species or all plants deposited in miRBase database (Release 18.0, November 2011) to identify conserved miRNAs in Chinese cabbage, in which only perfectly matched or closely related (allowing up to two mismatches) sequences were considered conserved miRNAs, additionally, the sequences that could form duplex-like miRNA:miRNA* pairs and mapped to the conserved miRNA precursors were considered as founder miRNA*s; (3) align small RNA tags to repeat associated RNA to find matched tags in the sample; (4) align small RNA tags to exons and introns of mRNA to find the degraded fragments of mRNA in the small RNA tags. To make a unique small RNA mapped to only one annotation, we follow the following priority rule: rRNAetc (in which Genbank > Rfam) >known miRNA > repeat > exon > intron. The small RNAs cannot align to any databases were defined as unannotated tags.
Prediction of novel miRNA
The tags which unannotated and matched to intron were aligned to the Chiifu Chinese cabbage genome (http://brassicadb.org/brad/) for precursor sequences to identify non-conserved miRNAs. For each mappable sequence, hairpin folding was evaluated by the Mireap program developed by the Beijing Genome Institute (BGI; Shenzhen, China), and the folding energy was calculated by Mfold (Zuker 2003). If a sequence satisfied the criteria described by Allen et al. (2005), it was considered a candidate for a predicted miRNA precursor.
Prediction of miRNA targets
Putative target sites for miRNA candidates were identified by aligning the miRNA sequences with the annotated genes Chiifu Chinese cabbage database (http://brassicadb.org/brad/) using a custom Perl script. The rules for target prediction were based on those of Allen et al. (2005) and Schwab et al. (2005). The criteria were: (1) no more than four mismatches between sRNA and target (G-U bases count as 0.5 mismatches); (2) no more than two adjacent mismatches in the miRNA/target duplex; (3) no adjacent mismatches in positions 2–12 of the miRNA/target duplex (5′ of miRNA); (4) no mismatches in positions 10–11 of miRNA/target duplex; (5) no more than 2.5 mismatches in positions 1–12 of the of the miRNA/target duplex (5′ of miRNA); (6) minimum free energy (MFE) of the miRNA/target duplex should be ≥75 % of the MFE of the miRNA bound to its perfect complement. To understand their biological function, these target genes were subjected to an online (http://brassicadb.org/brad/searchAll.php) BlastX search against the Arabidopsis genome, since they both are the members of Brassicaceae. Additionally, to better understand miRNA target functions, the Blast2GO program (Conesa et al. 2005) was used to obtain GO annotations from the UniGene database based on Blastx hits against the NCBI Nr database with an E-value threshold of less than 10−5.
RT-qPCR validation of miRNA expression
Validation of mature miRNA expression by RT-qPCR was carried out as described in Fu et al. (2006). Briefly, total RNA was isolated from different tissues as described above, using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, and treated with RNase-free DNase I (Promega, Madison, WI, USA). First-strand cDNA synthesis was then performed using a miRcute miRNA first-strand cDNA synthesis kit (Tiangen, Beijing, China) according to the manufacturer’s instructions. The product was series diluted to 101×, 102×, 103×, 104× and 105×, with distilled water (DW) as a negative control. RT-qPCR was carried out using the IQ5 real-time PCR system (BIO-RAD) and the SYBR Green PCR master mix (miRcute miRNA qPCR detection kit, Tiangen, Beijing, China), which contains antisense adaptor primers, and using corresponding miRNA sequences (Table 1) as sense primers. The PCR-cycling conditions comprised an initial polymerase activation step at 95 °C for 1 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 20 s and 72 °C for 30 s. After each PCR run, a dissociation curve was generated to confirm the specificity of the product and to avoid the production of primer dimers.
Results
Sequence analysis of short RNAs
A cDNA library of small RNAs from Chinese cabbage was sequenced using the Solexa system, resulting in a total of 5,440,690 raw reads. After removing adaptor/acceptor sequences, filtering low-quality tags and cleaning up the contamination formed by adaptor–adaptor ligation, 5,179,988 (95.21 %) clean reads of 18–30 nt remained, comprising 2,259,611 unique sequences (Table 2). The unique sequences were mapped to the Chinese cabbage genome assembly using the SOAP program (Li et al. 2008), leading to 1,287,166 genome-matched reads (56.96 %). Removal of 133,665 (5.92 %) exon RNAs (sense and antisense), 96,875 (4.29 %) intron RNAs (sense and antisense), 63,381 (2.80 %) rRNAs, 1,554 (0.07 %) repeat regions, 1,578 (0.07 %) snRNAs, 697 (0.03 %) snoRNAs, 6,486 (0.29 %) tRNAs and 1,944,165 (86.04 %) unannotated RNAs, left a total of 11,210 (0.50 %) reads. These were screened as miRNA candidates used in subsequent analyses.
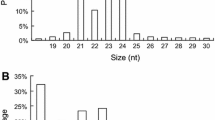
The size distribution of reads is shown in Fig. 1. The majority of small RNAs from the library were 24 nt long (Fig. 1) and accounted for 47.51 % of the total sequence length, followed by 23 nt (15.25 %), 21 nt (13.42 %) and 22 nt (7.76 %). This result was consistent with studies of A. thaliana, M. truncatula, O. sativa, Populus spp., Citrus trifoliate and Arachis hypogaea, where 24 nt sRNAs dominated the sRNA transcriptome (Moxon et al. 2008; Rajagopalan et al. 2006; Szittya et al. 2008; Song et al. 2010; Fahlgren et al. 2007; Morin et al. 2008; Chi et al. 2011). This could reflect the complexity of the Chinese cabbage genome, since 24-nt sequences dominated the small RNA library and 24-nt siRNAs are conserved because of their involvement in heterochromatin modification, especially for a genome with a high content of repetitive sequences (Herr 2005; Vazquez 2006). However, our sRNA size distribution differed from a previous study on B. rapa using a construct colony vector and custom sequencing method, where the majority of small RNAs were 21 nt long (He et al. 2008). The possible reason is that only 400 small RNAs were found in that study, a partial distribution of the small RNAs that exist in B. rapa.

Length distribution and abundance of the sequences
Identification of conserved miRNAs in Chinese cabbage
To identify conserved miRNAs in Chinese cabbage, sequence comparisons between Chinese cabbage miRNA candidates and the precursor/mature miRNA of all plants deposited in miRBase database (miRNA Registry, Release 18.0, November 2011) were carried out. Among the 5,179,988 sequences screened, 11,210 unique sequences were found to be orthologs of conserved miRNAs from plant species in the miRBase. Allowing one or two mismatches between sequences, these miRNAs represented 321 conserved miRNA families (Supplementary Table S1). Nineteen miRNAs and four miRNA*s belonging to 10 miRNA families in B. rapa have been previously deposited in the miRBase database (miRNA Registry, Release 18.0, November 2011). Here, except for bra-miR2111a*, these known miRNAs and miRNA*s were all successfully sequenced (Table 3). The reason bra-miR2111a* could not be sequenced may be because of low expression levels.
The number of miRNA reads in the library can be used as an index for estimating the relative abundance of miRNAs. Among the identified conserved miRNAs, the read number varied from one to 56,722 (Supplementary Table S1), indicating that expression varies significantly among different miRNAs. The most common read number of Chinese cabbage miRNAs was 1–100 (234, 72.9 %), followed by 101–500 (49, 15.26 %), 501–1,000 (11, 3.43 %), >10,000 (9, 2.80 %), 1,001–5,000 (5, 1.56 %) and 5,001–10,000 (3, 0.93 %). The most common (>20,000 reads) miRNAs in the library were: miR167 (56,722), miR157 (47,848), miR156 (38,692) and miR168 (28,381). These results indicate that different miRNAs have clearly different expression levels, probably because expression is tissue specific or developmental stage specific, similar to previous findings in A. hypogaea and Citrus trifoliate, in which miR167, miR157, miR156 and miR168 had relatively higher expression levels (Chi et al. 2011; Song et al. 2010).
To explore the evolutionary features of Chinese cabbage miRNAs, we compared the collection of conserved miRNAs with published miRNAs from other species. Nine were orthologs of conserved miRNAs from other species, with miR156 being the dominant family detected from 30 species, followed by miR159 (27 species), miR171-1 (27 species), miR160 (23 species), miR167-1 (23 species), miR172 (19 species), miR164 (18 species), miR2111 (8 species) and miR824 (5 species) (Supplementary Table S2). This indicated that these miRNAs could perform similar biological functions in diverse species. For example, miR156 was detected in A. thaliana, O. sativa and A. hypogaea, with similar expression patterns in each: strong expression during seedling development and weak expression in mature tissues (Axtell and Bartel 2005; Xie et al. 2006; Chi et al. 2011).
Identification of potential novel miRNAs in Chinese cabbage
In total, 228 small RNAs met our criteria as described in the “Materials and methods” section to be considered potential novel miRNAs in Chinese cabbage (Supplementary Table S3; Supplementary Fig. 1). These miRNAs were present on all of the Chinese cabbage chromosomes but unevenly distributed; chromosome A3 showed the most novel miRNAs (29), followed by chromosome A9 (27), chromosome A6 (25), and chromosome A7 (21), while chromosome A10 showed only seven novel miRNAs (Fig. 2a). One reason for this phenomenon could be that chromosome A3, A6 and A9 are longer in all B. rapa chromosomes (Wang et al. 2011a). Among these novel miRNAs, 28 candidates were supported by miRNA*s (Supplementary Table 3); detection of miRNA*s is a strong clue, albeit not infallible, for the formation of precursor hairpin structures (Fahlgren et al. 2007; Sunkar et al. 2008). The majority of the novel miRNAs were 23-nt long (118, 51.75 %), followed by 21 nt (83, 36.40 %), 22 nt (21, 9.21) and 20 nt (6, 2.63 %) (Fig. 2b). The predicted hairpin structures for the precursors of these miRNAs required 66–365 nt, with the majority of the identified miRNA precursors requiring 101–150 nt (97, 42.54 %), followed by 66–100 nt (43, 18.86 %), 151–200 nt (38, 15.35 %), 251–300 nt (17, 7.46 %), 301–350 nt (17, 7.46 %) and 351–365 nt (6, 2.63 %) (Fig. 2c). This is similar to what has been observed in A. thaliana, O. sativa and A. hypogaea (Zhang et al. 2006; Chi et al. 2011). A previous study on Chinese cabbage also showed the majority of pre-miRNAs were 60–150 nt in length (Wang et al. 2011a). The folding energy of the precursors of these novel miRNAs ranged from −181.92 to −18.10 kcal/mol, and mostly ranged from −80.00 to −40.01 kcal/mol (129, 56.58 %), followed by −40.00 to −18.10 kcal/mol (72, 31.58 %), −120.00 to −80.01 kcal/mol (18, 7.89 %), −160.00 to −120.01 kcal/mol (6, 2.63 %) and −181.92 to −160.01 kcal/mol (3, 1.32 %) (Fig. 2d). The average folding energy was about −52.27 kcal/mol, similar to the folding energy values of other plant miRNA precursors: −59.50, −71.00 and −50.01 kcal/mol in A. thaliana, O. sativa and A. hypogaea (Zhang et al. 2006; Chi et al. 2011), respectively. These values are much lower than the previously reported folding energies of tRNA (−27.5 kcal/mol) or rRNA (−33 kcal/mol).

Summary of identified novel potential miRNAs in Chinese cabbage. a Distribution of novel potential miRNAs in Chinese cabbage chromosomes. b Frequency of mature miRNAs length of novel potential miRNAs. c Frequency of precursor length of novel potential miRNAs. d Frequency of folding energy of novel potential miRNAs
In this study, except for nmiR172 (13,723) and nmiR153 (3,734) which had relatively high expression levels, the novel miRNAs (nmiRNAs) all had low expression levels (<1,000). Among these, 204 (89.47 %) nmiRNAs generated less than 100 reads, and 72 (31.58 %) nmiRNAs even generated less than 10 reads (Supplementary Table S3). This result was similar to the finding in A. hypogaea that non-conserved miRNAs are often expressed at a lower level than conserved miRNAs (Chi et al. 2011).
Validation of conserved and novel miRNAs using RT-qPCR
To validate the high-throughput sequencing results, RT-qPCR analysis was performed according to the method described in Fu et al. (2006). Although accurate validation of all the miRNAs detected in this experiment was beyond the scope of this investigation, we randomly selected two conserved and six novel miRNAs as examples. In all eight miRNA samples no amplification was detected when DW was used as the template (Supplementary Fig. 2), suggesting there was no primer dimer production. Additionally, the dissociation curve showed only one melt peak in all eight miRNAs (Supplementary Fig. 2), suggesting there was no non-specific amplification. miR158 and miR162 were both amplified when the template was diluted from 101× to 105×, while the other six miRNAs could be amplified when the template was diluted to a minimum of 103× or 104× (Supplementary Fig. 2). These results suggested that the data on identification of conserved and novel miRNAs was credible, and exhibited the same expression profiles as the original high-throughput sequencing results.
Prediction of conserved and novel miRNA targets genes in Chinese cabbage
To better understand the biological functions of the newly identified Chinese cabbage miRNAs, the putative target genes were identified according to the criteria described in the “Materials and methods” section. In total, we found 2,308 and 736 potential targets genes for 221 conserved and 125 novel miRNAs (Supplementary Table S4), with an average of 10.44 and 5.89 targets per miRNA molecule, respectively. This was higher than a previous finding on Chinese cabbage with a mean of 8.25 targets per conserved miRNA (Wang et al. 2011a). One reason for this higher ratio was that the prediction of target genes for the previous study was based on the EST (149,480) and GSS (179,678) databases which download from NCBI, and cannot cover the whole Chinese cabbage genome. Moreover, this result suggests that conserved miRNAs could perform a broader range of biological functions than novel miRNAs, since they contain more target genes than novel miRNAs.
For annotation of the targets genes, the targets were subjected to an online (http://brassicadb.org/brad/searchAll.php) BlastX search against the Arabidopsis genome (Supplementary Table S4). Among the targets of conserved miRNAs, many were transcription factors (16.3 %), such as NAC, WRKY, AP2, AGL, MYB and Zinc finger proteins (Supplementary Table S4). These results are similar to those of earlier studies (Wang et al. 2011a). The targets of novel miRNAs also contain some similar functional transcription factors, but the ratio (12.8 %) was lower than that in the targets of conserved miRNAs (Supplementary Table S4). In addition to transcription factors, there were other predicted targeted genes, including those encoding the disease resistance protein (TIR-NBS-LRR class), protein kinase family protein, nucleoporin family protein, ribosomal protein, ATPase, receptor-like protein, dehydration-responsive family protein, exostosin family protein, F-box family protein, glycosyl hydrolase family protein, leucine-rich repeat family protein, O-methyltransferase, ABC1 family protein, WD-40 repeat family protein and so on, which involved in a broad range of biological processes, such as stress response, metabolism and signal transduction. Additionally, we predicted a few genes with unknown function and hypothetical genes as conserved and novel miRNA targets (Supplementary Table S4) and that further investigation of these potential targets will contribute to better understanding of the role of miRNAs in Chinese cabbage.
All targets regulated by Chinese cabbage conserved and novel miRNAs identified in this study were subjected to GO analysis to investigate gene ontology. Under the category of biological processes, there were 22 and 31 GO categories (Supplementary Table S5) that were significantly enriched (corrected P value < 0.05) for conserved and novel miRNA targets, respectively. For conserved miRNAs, cellular processes (880), cellular metabolic processes (684), primary metabolic processes (663), nitrogen compound metabolic processes (323), nucleobase, nucleoside, nucleotide and nucleic acid metabolic processes (278) and multicellular organismal processes (204) were the main groups. This was consistent with a previous study on Chinese cabbage, in which cellular and metabolic processes were the dominant GO categories (Wang et al. 2011a). Unlike conserved miRNAs, the targets of the novel miRNAs were mainly associated with developmental processes (69), multicellular organismal processes (56), anatomical structure development (49), organ development (33), system development (33), root system development (13) and root development (13), suggests that the corresponding novel miRNAs possibly participate in different biological processes with conserved miRNAs in Chinese cabbage.
Discussion
miRNA-mediated RNA interference (RNAi) plays an important role in the silencing of selfish genetic elements and the formation of heterochromatin in plants, animals and fission yeasts (Lippman and Martienssen 2004). To date, 18,226 miRNAs (miRBase database, Release 18.0, September 2011) have been identified from various multicellular eukaryotes, including humans, flies, nematodes and plants. No systematic studies have yet been reported for Chinese cabbage, one of the most important vegetable crops cultivated in Asia. Recently, some miRNAs from Chinese cabbage have been identified by computational and direct cloning approaches (Wang et al. 2011a; He et al. 2008). However, the recently developed sequencing technology, Solexa (Illumina) could yield >5 million reads (up to 50 bp) per sample, (Sequenced at BGI, http://www.genomics.cn/en/index) and the recently sequenced Chiifu Chinese cabbage genome (Wang et al. 2011b) will help us to discover more miRNAs from this species.
Using high-throughput Solexa sequencing technology, 11,210 unique sequences belonging to 321 conserved miRNA families and 228 novel miRNAs were identified in Chinese cabbage. Sequence analysis revealed that the relative abundance of conserved miRNAs is higher than that of novel miRNAs. This result was similar to the finding in A. hypogaea that non-conserved miRNAs were often expressed at lower levels than conserved miRNAs (Chi et al. 2011). The low abundance of these novel miRNAs might suggest they play specific roles in specific tissues or developmental stages. Whether these low-abundance miRNAs are regulated by biotic or abiotic stresses remains to be investigated.
This study also discovered conserved miRNAs shared by other plants, including O. sativa, Triticum aestivum, A. thaliana, Glycine max, and M. truncatula, which will provide an opportunity to examine the evolution of these families during the divergence of plants. Furthermore, since these miRNAs are conserved among several species, it will help us to infer the functions of miRNAs in Chinese cabbage based on their conserved functions in other plants. Previous reports have shown that miR156 directs the cleavage of two SBP box genes (Arazi et al. 2005; Wu and Poethig 2006), miR166 directs the cleavage of members of the HD-ZIP transcription factor gene family (Floyd et al. 2006) and the miR159/319 family targets subsets of the MYB and TCP transcription factor gene families (Rhoades et al. 2002; Palatnik et al. 2003; Schommer et al. 2008; Nag et al. 2009). Here, the predicted target genes of miR156, miR166, miR159/319 and miR396 were consistent with earlier reports; some of these targets in Chinese cabbage are plant-specific transcription factors, such as SBP, HD-ZIP, MYB and TCP. Moreover, previous studies have illustrated that miR156 has similar expression patterns in A. thaliana, O. sativa and A. hypogaea, which is strongly expressed during seedling development and shows weak expression in mature tissues (Axtell and Bartel 2005; Xie et al. 2006; Chi et al. 2011). The results of the present study suggest that significant functional differentiation of miRNAs occurs at different developmental stages and some miRNA regulatory mechanisms maybe similar in Chinese cabbage and other higher plants.
In conclusion, high-throughput Solexa sequencing provides a good way for us to study small RNAs in Chinese cabbage, an important vegetable in Asia. This study led to the discovery of a large number of miRNAs, including 11,210 unique sequences belonging to 321 conserved miRNA families and 228 novel miRNAs from Chinese cabbage. The prediction of putative targets for these miRNAs was also performed. Going forward, it will be very important to experimentally characterize these miRNAs and their downstream targets to help us gain a better understanding of the functions, relationships and mechanisms of miRNAs in the regulation network.
References
Abbott AL, Alvarez-Saavedra E, Miska EA, Lau NC, Bartel DP, Horvitz HR, Ambros V (2005) The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev Cell 9:403–414
Allen E, Xie Z, Gustafson AM, Carrington JC (2005) microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121:207–221
Arazi T, Talmor-Neiman M, Stav R, Riese M, Huijser P, Baulcombe DC (2005) Cloning and characterization of micro-RNAs from moss. Plant J 43(6):837–848
Aukerman MJ, Sakai H (2003) Regulation of flowering time and floral organ identity by a microRNA 15(11):2730–2741
Axtell MJ, Bartel DP (2005) Antiquity of microRNAs and their targets in land plants. Plant Cell 17:1658–1673
Barakat A, Wall K, Leebens-Mack J, Wang YJ, Carlson JE, Depamphilis CW (2007) Large-scale identification of microRNAs from a basal eudicot (Eschscholzia californica) and conservation in flowering plants. Plant J 51:991–1003
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297
Berezikov E, van Tetering G, Verheul M, van de Belt J, van Laake L, Vos J, Verloop R, van de Wetering M, Guryev V, Takada S et al (2006) Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res 16(10):1289–1298
Carrington JC, Ambros V (2003) Role of microRNAs in plant and animal development. Science 301:336–338
Chen X (2005) MicroRNA biogenesis and function in plants. FEBS Lett 579(26):5923–5931
Chi X, Yang Q, Chen X, Wang J, Pan L, Chen M, Yang Z, He Y, Liang X, Yu S (2011) Identification and characterization of microRNAs from Peanut (Arachis hypogaea L.) by high-throughput sequencing. PLoS ONE 6:e27530
Collins LJ, Biggs PJ, Voelckel C, Joly S (2008) An approach to transcriptome analysis of non-model organisms using short-read sequences. Genome Inform 21:3–14
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21(18):3674–3676
Costa V, Angelini C, Feis ID, Ciccodicola A (2010) Uncovering the complexity of transcriptomes with RNA-Seq. J Biomed Biotechnol 2010:853916
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL et al (2007) High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS ONE 2:e219
Floyd SK, Zalewski CS, Bowman JL (2006) Evolution of class III homeodomain-leucine zipper genes in streptophytes. Genetics 173:373–388
Fu HJ, Zhu J, Yang M, Zhang ZY, Tie Y, Jiang H, Sun ZX, Zheng XF (2006) A novel method to monitor the expression of microRNAs. Mol Biotechnol 32:197–204
He XF, Fang YY, Feng L, Guo HS (2008) Characterization of conserved and novel microRNAs and their targets, including a TuMV-induced TIR–NBS–LRR class R gene-derived novel miRNA in Brassica. FEBS Lett 582(16):2445–2452
Herr AJ (2005) Pathways through the small RNA world of plants. FEBS Lett 579:5879–5888
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Li R, Li Y, Kristiansen K, Wang J (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24(5):713–714
Lippman Z, Martienssen R (2004) The role of RNA interference in heterochromatic silencing. Nature 431:364–370
Llave C, Kasschau KD, Rector MA, Carrington JC (2002) Endogenous and silencing-associated small RNAs in plants. Plant Cell 14:1605–1619
Lu C, Tej SS, Luo S, Haudenschild CD, Meyers BC, Green PJ (2005) Elucidation of the small RNA component of the transcriptome. Science 309:1567–1569
Mallory AC, Vaucheret H (2004) MicroRNAs: something important between the genes. Curr Opin Plant Biol 7:120–125
Morin RD, Aksay G, Dolgosheina E, Ebhardt HA, Magrini V, Mardis ER, Sahinalp SC, Unrau PJ (2008) Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res 18:571–584
Morozova O, Marra MA (2008) Applications of next-generation sequencing technologies in functional genomics. Genomics 92:255–264
Moxon S, Jing R, Szittya G, Schwach F, Rusholme Pilcher RL, Moulton V, Dalmay T (2008) Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res 18:1602–1609
Nag A, King S, Jack T (2009) miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. PNAS 106(52):22534–22539
Palatnik JF, Allen E, Wu X, Schommer C, Schwab R, Carrington JC, Weigel D (2003) Control of leaf morphogenesis by microRNAs. Nature 425(6955):257–263
Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA (2010) Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics 11:180
Park W, Li J, Song R, Messing J, Chen X (2002) CARPEL FACTORY, a dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol 12:1484–1495
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20:3407–3425
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403:901–906
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP (2002) Prediction of plant microRNA targets. Cell 110:513–520
Schommer C, Palatnik JF, Aggarwal P, Chételat A, Cubas P, Farmer EE, Nath U, Weigel D (2008) Control of jasmonate biosynthesis and senescence by miR319 targets. PLoS Biol 6(9):e230
Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D (2005) Specific effects of microRNAs on the plant transcriptome. Dev Cell 8:517–527
Song C, Wang C, Zhang C, Korir NK, Yu H, Ma Z, Fang J (2010) Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics 11:431
Sun C, Li Y, Wu Q, Luo H, Sun Y, Song J, Lui EM, Chen S (2010) De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genomics 11:262
Sunkar R, Girke T, Jain PK, Zhu JK (2005) Cloning and characterization of MicroRNAs from rice. Plant Cell 17:1397–1411
Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK (2008) Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol 8:25
Szittya G, Moxon S, Santos DM, Jing R, Fevereiro MP, Moulton V, Dalmay T (2008) High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics 9:593
Vazquez F (2006) Arabidopsis endogenous small RNAs: highways and byways. Trends Plant Sci 11:460–468
Wang J, Hou X, Yang X (2011a) Identification of conserved microRNAs and their targets in Chinese cabbage (Brassica rapa subsp. pekinensis). Genome 54:1029–1040
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Bai Y, Mun JH, Bancroft I, Cheng F et al (2011b) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039
Wu G, Poethig RS (2006) Temporal regulation of shoot development in Arabidopsis thaliana by mir156 and its target SPL3. Development 133(18):3539–3547
Xie Z, Allen E, Fahlgren N, Calamar A, Givan SA, James C, Carrington JC (2005) Expression of Arabidopsis MIRNA genes. Plant Physiol 138:2145–2154
Xie K, Wu C, Xiong L (2006) Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol 142:280–293
Zhang BH, Pan XP, Wang QL, Cobb GP, Anderson TA (2005) Identification and characterization of new plant microRNAs using EST analysis. Cell Res 15:336–360
Zhang BH, Pan XP, Cannon CH, Cobb GP, Anderson TA (2006) Conservation and divergence of plant microRNA genes. Plant J 46:243–259
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgments
This work was supported by the Prospect of Shandong Seed Project, China (2011lzgcshucaizy); National Nature Science Foundation of China (31101553); Modern Agricultural Industrial Technology System Funding of Shandong Province, China (2010sdxdcyjstxshucai) and the Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Province, China (BS2010SW027).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
F. Wang and L. Li contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Wang, F., Li, L., Liu, L. et al. High-throughput sequencing discovery of conserved and novel microRNAs in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Mol Genet Genomics 287, 555–563 (2012). https://doi.org/10.1007/s00438-012-0699-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-012-0699-3