
Abstract
Our goal in this work was to develop a method to minimize the chromosomes of Aspergillus oryzae, to arrive at a deeper understanding of essential gene functions that will help create more efficient industrially useful strains. In a previous study, we successfully constructed a highly reduced chromosome 7 using multiple large-scale chromosomal deletions (Jin et al. in Mol Genet Genomics 283:1–12, 2010). Here, we have created a further reduced chromosome A. oryzae mutant harboring a reduced chromosome 7 and a reduced chromosome 8 both of which contain a large number of non-syntenic blocks. These are the smallest A. oryzae chromosomes that have been reported. Protoplast fusion between the two distinct chromosome-reduced mutants produced a vigorous and stable fusant which was isolated. PCR and flow cytometry confirmed that two kinds of nuclei, derived from the parent strains, existed in this fusant and that the chromosome DNA per nucleus was doubled, suggesting that the fusant was a heterozygous diploid strain. By treating the cell with 1 μg/ml benomyl, cell nuclei haploidization was induced in the stable diploid strain. Array comparative genomic hybridization and pulsed-field gel electrophoresis confirmed that the reduced chromosomes 7 and 8 co-existed in the haploid fusant and that no other chromosomal modifications had occurred. This method provides a useful tool for chromosome engineering in A. oryzae to construct an industry-useful strain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The filamentous fungus Aspergillus oryzae is well known for its capacity to secrete large amounts of hydrolytic enzymes. For more than a thousand years, it has been safely used as an industrially important koji mold in manufacturing traditional oriental fermented foods, such as soy sauce, miso, and sake. A. oryzae has also been used in the enzyme industry for numerous native and heterologous enzymes production. Considering the industrial importance of A. oryzae, it is desirable to create industrially more efficient and useful strain that, by eliminating nonessential regions of the genome, would greatly benefit its industrial applications. For example, although the A. oryzae is considered to be safe, it still remained some nonessential and even unfavorable regions within its genome, such as imperfect clustered genes related to cyclopiazonic acid production as well as aflatoxin biosynthesis. Therefore, the elimination of the potential risk caused by these detrimental genes will further improve the safety of the industrially important koji mold, A. oryzae. At the same time, the elimination of unnecessary regions coding various dispensable functions implies that the strain may have more energy available for growth and/or more useful products.
However, because A. oryzae is resistant to antifungal antibiotics, has multiple nuclei, and no report indicated the presence of a sexual life cycle, although in A. flavus, a close relative, has been proven to be capable of sexual recombination (Horn et al. 2009), gene manipulation, such as gene disruption, is difficult. Recently, by disrupting the genes involved in the non-homologous end-joining pathway (Jones et al. 2001; DeFazio et al. 2002), researchers have improved the gene targeting frequency (Ninomiya et al. 2004; Takahashi et al. 2006). In addition, loop-out and replacement-type recombination methods that could be used to efficiently construct large-scale chromosomal deletion mutants have been developed in A. oryzae (Takahashi et al. 2008, 2009). The replacement-type recombination method is useful for single deletions, while the loop-out type recombination [5-fluoroorotic acid (5-FOA) selection] method enables the construction of multiple deletions in a single strain by pyrG marker recycling.
These large-scale chromosomal deletion methods together with the recent completion of the A. oryzae genomic sequencing project (Machida et al. 2005; Kobayashi et al. 2007) allowed us to analyze the whole genome and made it possible for us to create an industrially versatile strain of A. oryzae by experimentally reducing the genome size to eliminate nonessential regions (Jin et al. 2010). Mutants with significantly reduced genomes have been constructed in E. coli (Kolisnychenko et al. 2002; Yu et al. 2002; Goryshin et al. 2003; Hashimoto et al. 2005; Kato and Hashimoto 2007) and S. cerevisiae (Murakami et al. 2007). Constructing a highly reduced A. oryzae genomic strain is more challenging because the gene manipulation in A. oryzae is more difficult than in E. coli.
Protoplast fusion techniques, first described by Anné and Peberdy (1975, 1976) and Ferenczy et al. (1975, 1976) in fungi, have proved to be valuable for interspecific hybridization. Fusion of protoplasts resulted in interspecific heterokaryons as primary fusion products, and these heterokaryons can lead to hybrid formation following nuclear fusion, as reported previously (Kevei and Peberdy 1977, 1979). Similarly, protoplast fusion between different Aspergillus species has successfully produced heterokaryons. However, this was successful in part because A. nidulans has a sexual life cycle that facilitates the exchange of chromosomes; A. oryzae lacks a sexual life cycle, making the chromosomal exchange by nuclear fusion extremely difficult. Recently, the discovery of sexual cycle by crossing the strains of opposite mating types in A. flavus and A. fumigatus (Horn et al. 2009; O’Gorman et al. 2009), which were long considered to be asexual, opened up a new perspective for other supposed ‘asexuals’, especially for biotechnologically important fungi such as A. oryzae.
In a previous study, we successfully deleted multiple large chromosomal segments and constructed a significantly reduced chromosome 7 strain using the loop-out type recombination method (Jin et al. 2010). However, it is extremely difficult to completely delete the nonessential regions on all A. oryzae chromosomes using only this method. In the present study, we used the protoplast fusion method to create a new A. oryzae hybrid haploid fusant harboring both a reduced chromosome 7 and a reduced chromosome 8. The new chromosome-reduced fusant was confirmed by microarray-based comparative genomic hybridization (CGH) and pulsed-field gel electrophoresis (PFGE).
Materials and methods
Strains, media, and transformation of A. oryzae
The A. oryzae wild type strain RIB40 was used as a DNA donor, and the RkuAF7B strain (Jin et al. 2010) was used for further deletion of the AO090011000096–AO090011000104 region of chromosome 7 to acquire an RkuAF8B mutant. The PNDR T6-1 strain, with a reduced chromosome 8 and a niaD disruption, was derived from pT470 k (Takahashi et al. 2009) and was used for protoplast fusion with the RkuAF8B mutant. E. coli DH5α was used for DNA manipulation. Czapek-Dox (CD) minimal agar medium (0.2% NaNO3, 0.05% KCl, 0.05% NaCl, 0.1% KH2PO4, 0.05% MgSO4·7H2O, 0.002% FeSO4·7H2O, 2% glucose, and 2% agar, pH 5.5) was used for selecting A. oryzae transformants and fusants. CD minimal medium plates supplemented with 15 mM uridine and 5-fluoroorotic acid (5-FOA) (2 mg/ml; Sigma, St. Louis, MO, USA) were used for the positive selection of pyrG-deficient strains. Malt medium (80 g of malt extract, 2.0 mg of CuSO4, 0.04 mg of Na2B4O7, 0.87 mg of FePO4, 0.95 mg of MnSO4, 0.8 mg of Na2MoO4, 8.0 mg of ZnSO4, and 15 g of agar per liter; pH 6.0) was used as the complete medium. DPY medium (2% dextrin, 1% polypeptone, 0.5% yeast extract, 0.5% KH2PO4, and 0.05% MgSO4·7H2O) was used for the liquid cultivation of A. oryzae strains.
Construction of the RkuAF8B mutant
The AO090011000096–AO090011000104 region of chromosome 7 was deleted from the RkuAF7B strain. The strategy for construction of the AO090011000096–AO090011000104 region deletion is described by Jin et al. (2010). To construct the deletion plasmid, we first amplified an approximately 2.9-kb fragment from a target gene AO090011000096 by PCR with Ex-Taq polymerase (TaKaRa, Kyoto, Japan) using primer pair No. 1/No. 2 (all primer pairs used in this study are listed in Table 1). The amplified PCR product was inserted into the pCR2.1-TOPO vector, using a TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA, USA). The generated plasmid was linearized by PCR with primer pair No. 7/No. 8. A 1.5-kb fragment of gene AO090011000104 was amplified with primer pair No. 9/No. 10 and was combined with the linearized cloning vector using the In-Fusion cloning enzyme. The newly generated plasmid was linearized again by PCR with primer pair No. 3/No. 4. A 2.2-kb pyrG marker was amplified with primer pair No. 5/No. 6 and was fused to the linearized cloning vector using the In-Fusion cloning enzyme. Finally, the plasmid for deleting genes AO090011000096–AO090011000104 was constructed and was used to transform the RkuAF7B strain. The transformants that were obtained were plated on CD medium containing 5-FOA and uridine. Deletants were obtained by homologous recombination between direct repeats using the 5-FOA treatment described earlier (Jin et al. 2010). The targeted region of AO090011000096–AO090011000104 containing the pyrG marker was eliminated. The obtained strain was named RkuAF8B.
Construction of a niaD gene deletion cassette
Using the genomic DNA from the PNDR T6-1 strain (a niaD gene disruptant) as a template, PCR was performed with primer pair No. 11/No. 12. A 2.8-kb PCR product, consisting of a 1.5-kb upstream region and a 1.3-kb downstream region of the niaD deletion region, was amplified and used to disrupt the niaD gene.
Disruption of the niaD gene located in chromosome 4 of the RkuAF8B strain
The RkuAF8B was transformed by the niaD gene disruption cassette and was grown on a non-selective CD minimal medium containing 10 mM glutamate as a sole nitrogen source, 10 mM uracil, and 1.2 M sorbitol. The obtained conidia were suspended in 10 ml of 0.01% Tween 80 and were filtered through a 70-μm cell strainer (BD Falcon, USA) to remove the mycelia matter. To select the transformants, separated conidia were spread-cultured on CD minimal medium containing 10 mM glutamate, 10 mM uracil, and 470 mM KClO3. The obtained chlorate-resistant strain was confirmed by PCR with primer pairs No. 11/No. 12 and No. 13/No. 14, and was designated 8BDelN_1.
Generation of the niaD insertion cassette for chromosome 7 of the 8BDelN_1 strain by fusion PCR
The niaD insertion cassette was constructed using fusion PCR (Yu et al. 2004). The genomic DNA of A. oryzae RIB40 was used as a template. A 5.3-kb niaD gene with primer pair No. 11/No. 12 and two 1.5-kb DNA fragments outside the inserted region (upstream region with primer pair No. 1/No. 15, and downstream region with primer pair No. 16/No. 10 listed) were amplified by PCR using a GeneAmp 9600 system (Applied Biosystems, Foster City, CA) with KOD-plus DNA polymerase (Toyobo, Osaka, Japan). The sequences from the upstream and downstream regions were designed to overlap the ends of the niaD gene. The three amplified DNA fragments were mixed, and fusion PCR was performed using the primer pair No. 1/No. 17. The resultant insertion cassette was used to transform the 8BDelN_1 strain. The successful transformants were confirmed by PCR with primer pairs No. 18/No. 19, and No. 20/No. 21. The confirmed niaD insertion strain was designated 8BCh7N_3.
Protoplast fusion
The chromosome 7-reduced strain, 8BCh7N_3, and chromosome 8-reduced strain, PNDR T6-1, were used for protoplast fusion. Protoplasts were prepared from mycelial cultures by Yatalase treatment (Takara, Kyoto, Japan) for 3 h at 30°C according to the A. oryzae transformation method previously described (Takahashi et al. 2004) and were then suspended in SolB solution (1.2 M sorbitol, 50 mM CaCl2, and 10 mM Tris–HCl, pH 7.5). Equal quantities of protoplasts were mixed and added to the PEG solution (50% PEG3350, 50 mM CaCl2, and 10 mM Tris–HCl, pH 7.5). The mixture was placed at room temperature for 20 min to allow protoplast fusion. After fusion treatment, the protoplasts were washed with SolB solution and grown on CD minimal agar medium containing 1.2 M sorbitol.
Flow cytometry
Strains were spot-cultivated on minimal medium for several days. The conidia were harvested with 0.01% Tween 80, fixed with 70% ethanol at 4°C for overnight, and then washed twice with Tween solution. The conidia were incubated with 1 mg/ml RNase A at 37°C for 2 h to remove the RNA. After filtering through a 35-μm cell strainer (BD Falcon, USA), the separated conidia were stained with 25 μg/ml propidium iodide (PI) and were analyzed by a flow cytometer (FACScan, Becton–Dickinson). The intensity of fluorescence, which reflects DNA content, was measured for each of the 20,000 conidia.
Array comparative genomic hybridization (aCGH)
The obtained haploid candidate strain was confirmed by array comparative genomic hybridization (aCGH). DNA microarrays were purchased from Agilent Technologies. Genomic DNAs were extracted and sheared by sonication (UCD-250, Cosmo Bio). They were then fluorescently labeled with Cy3/Cy5-dUTP and hybridized according to the Agilent array CGH protocol. After hybridization for 24 h at 65°C, the array slide was washed at 37°C. Finally, the DNA microarray slide was scanned using an Agilent scanner (Agilent Technologies, Santa Clara, CA). Data were extracted using Agilent Feature Extraction Software.
Pulsed-field gel electrophoresis
Conidia from each strain were cultivated in 10 ml of DPY liquid medium for approximately 22–24 h in a 50-ml Falcon tube. Protoplasts were prepared from the mycelial cultures by Yatalase treatment for 3 h at 30°C. The protoplasts were embedded in agarose gel and treated with NDS buffer [0.45 M EDTA, 0.01 M Tris–HCl (pH 7.5), 1% SDS and 1 mg/ml proteinase K] at 50°C. Separation of the A. oryzae chromosomes was performed by pulsed-field gel electrophoresis (PFGE) using a CHEF-DRIII system (Bio-Rad). The separations were performed at 14°C using 0.8% Certified Megabase agarose (Bio-Rad) in TAE buffer [40 mM Tris-base, 40 mM acetate, and 2.0 mM EDTA (pH 8.0)]. The total electrophoresis time was 96 h and the PFGE programs were as follows: the first stage of electrophoresis was performed for 24 h at 2 V/cm with a pulse time of 1,200 s at an angle of 96o; the second stage was performed for 24 h at 2 V/cm with a pulse time of 1,500 s at an angle of 100o; and the final stage of electrophoresis was performed for 48 h at 2 V/cm with a pulse time of 1,800 s at an angle of 106o. Chromosomes of S. pombe were used as high-molecular-mass DNA standards. Gels were stained by Gel-Red (Wako).
Southern blot hybridization
Southern hybridization was performed according to a previously described method (Takahashi et al. 2004). Digoxigenin (DIG)-labeled probes were constructed using a DIG PCR labeling kit (Roche Diagnostics, Mannheim, Germany). Hybridization and signal detection were performed using a DIG system according to the manufacturer’s instructions (Roche Diagnostics). Primer pairs No. 30/No. 31 and No. 32/No. 33 were used to obtain DIG-labeled probes for the detection of chromosome 7 and chromosome 8, respectively.
Results and discussion
Construction of strains for protoplast fusion
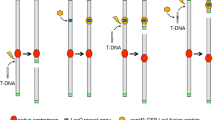
Earlier, we constructed a chromosome 7-reduced strain (Jin et al. 2010) and a chromosome 8-reduced strain (Takahashi et al. 2009). The chromosome 7-reduced strain, RkuAF7B, was successfully constructed through multiple large-scale chromosomal deletions using a recursive pyrG-mediated transformation system. Here, the AO090011000096–AO090011000104 region of the reduced chromosome 7 was deleted from the RkuAF7B strain and the obtained strain, RkuAF8B, was used for trial of further chromosome minimization. In this study, the protoplast fusion method was used to create the further reduced chromosome mutant. The strategy that was used to construct the strain with both the reduced chromosome 7 and chromosome 8 is described in Fig. 1. To select the fusant after protoplast fusion, the auxotrophic genetic markers, niaD and pyrG, were deleted from their original chromosomal locations and reintroduced into the reduced chromosome 7 and 8, respectively. This method can be simply described as follows: first, the niaD gene that was original located in chromosome 4 of the RkuAF8B strain harboring a largely reduced chromosome 7 and pyrG deficiency was disrupted by selecting a chlorate-resistant strain; then, the cassette for the reinsertion of the niaD gene into chromosome 7 was generated by fusion PCR and used to transform the chromosome 7-reduced strain; finally, the new strain (named 8BCh7N_3) containing the translocated niaD gene was used directly for protoplast fusion with the chromosome 8-reduced strain, PNDR T6-1, deleting the original niaD and pyrG genes and reinserting the pyrG gene into chromosome 8 (Fig. 1).

Strategy for constructing a strain with reduced chromosomes 7 and 8. Chromosomes 1 to 6 are shown as one line here
Acquisition of the heterozygous diploid strain by protoplast fusion
The new chromosome 7-reduced strain, 8BCh7N_3, and the chromosome 8-reduced strain, PNDR T6-1 strain, were used for protoplast fusion. The fusion method is described in the “Materials and methods” section. The protoplasts of fusion treatment were plated on CD minimal agar medium containing 1.2 M sorbitol. After 5 days of incubation, conidia were harvested from the plate, diluted, and spread-cultured on CD minimal agar medium with 500 conidia per plate. Approximately 100 conidia per plate grew into colonies. However, almost all the colonies formed segregated sectors and displayed unstable growth phenotypes (Fig. 2, left). These colonies were most likely heterokaryotic strains. Conidia were re-collected, diluted, and again spread-cultured onto CD minimal agar medium. This procedure was repeated 3 times until a vigorous and stable colony appeared. The isolated strain that we named 8BT6_6 was assumed to be a heterozygous diploid candidate strain (Fig. 2, right).

Growth of fusants on Czapek-Dox (CD) minimal medium plate. Initially, all colonies formed a segregated sector of colonies and displayed an unstable growth phenotype (left). After selecting 3 times on CD minimal medium, a vigorous and stable colony was acquired. This strain was considered to be a potential candidate heterozygous diploid strain (right). The circles indicate nuclei
The acquired 8BT6_6 strain was confirmed by PCR (Fig. 3a). The primer pairs No. 22/No. 23 for detection of reduced chromosome 7, No. 24/No. 25 for detection of normal chromosome 7, No. 26/No. 27 for detection of reduced chromosome 8, and No. 28/No. 29 for detection of normal chromosome 8 were used for PCR amplification. Two bands, representing reduced chromosome and normal chromosome, were detected for chromosomes 7 and 8 of the 8BT6_6 fusant (Fig. 3a). However, this pattern was also observed for the heterokaryons. Although this result confirmed that the 8BT6_6 candidate strain had two kinds of nuclei derived from the distinct parent strains, the method could not identify whether or not the strain was a heterozygous diploid.

Confirmation of a heterozygous diploid. a PCR analysis. Primer pairs No. 22/No. 23 for detection of reduced chromosome 7, No. 24/No. 25 for detection of normal chromosome 7, No. 26/No. 27 for detection of reduced chromosome 8, and No. 28/No. 29 for detection of normal chromosome 8 were used for PCR amplification. The PCR amplification pattern in the candidate strain was the same as in the heterokaryons. 8BCh7N_3, chromosome 7-reduced strain; PNDR T6-1, chromosome 8-reduced strain; 8BT6_6, candidate for heterozygous diploid. b Chromosomal ploidy analysis by flow cytometry. Conidia were collected, fixed, stained with propidium iodide (PI), and analyzed in a flow cytometer. RIB40, A. oryzae wild type strain, 8BT6_6, candidate for heterozygous diploid
The 8BT6_6 strain was further analyzed by flow cytometry and the intensity of fluorescence (FL) was measured. A typical FL histogram is shown in Fig. 3b. In the A. oryzae RIB40 strain, four major peaks at an interval of intensity that reflects the distribution of the nuclear number in the conidia (Hara et al. 2002) were obtained. Compared with the RIB40 strain, the 8BT6_6 fusant only displayed the even number nuclear peaks (2 and 4 nuclei), implying that the quantity of chromosomal DNA per nucleus was doubled in the 8BT6_6 fusant and confirming that 8BT6_6 is a heterozygous diploid strain. For the heterokaryon, although the separation of the peaks was indistinct, the peak pattern showed that this strain contained anywhere from 1 nucleus to 4 nuclei proving that the primary fusion colonies were indeed heterokaryons. The prolonged cultivation of the heterokaryons under selective conditions resulted in the formation of hybrid diploid nuclei as a consequence of nuclear fusion. However, the frequency of nuclear fusion was very low because, after fusion treatment, the formed conidia that were cultivated on 5 plates of CD minimal medium with 500 conidia per plate, finally, produced only one vigorous and stable heterozygous diploid. When this experiment was repeated, the same result was obtained.
Isolation of haploid fusant by treatment with benomyl
The 8BT6_6 strain was spot-cultivated on malt agar medium supplemented with 1 μg/ml benomyl, to induce the cell nuclei haploidization. After the strain formed a giant colony, the 6 different sectors that formed were individually cultivated on CD minimal agar medium. Four of these sectors were further investigated by PCR and only one strain that contained both reduced chromosome 7 and chromosome 8 was found (Fig. 4). The method used here is the same as the method described in the legend to Fig. 3a. In the identified candidate strain, a 1.3-kb band was detected for the reduced chromosome 7 using primer pair No. 22/No. 23; however, no band was detected for the normal chromosome 7 using primer pair No. 24/No. 25. This result suggested that haploidization of the diploid strain had occurred and the reduced chromosome 7 existed only in the candidate strain. The PCR amplification for chromosome 8 produced the same results. This isolated strain named 1112_4_12 was identified as a candidate haploid fusant and was further investigated. The objective strain could not be produced without benomyl treatment (data not shown).

Confirmation of the haploidization of heterozygous diploid strain by PCR. The primer pairs No. 22/No. 23 for detection of reduced chromosome 7, No. 24/No. 25 for detection of normal chromosome 7, No. 26/No. 27 for detection of reduced chromosome 8, and No. 28/No. 29 for detection of normal chromosome 8 were used for PCR amplification. 8BCh7N_3, chromosome 7-reduced strain; PNDR T6-1, chromosome 8-reduced strain; 8BT6_6, heterozygous diploid; 1112_4_12, haploid fusant with reduced chromosomes 7 and 8
Confirmation of the haploid fusant by aCGH and pulsed-field gel electrophoresis (PFGE)
Further analysis was performed using aCGH (Fig. 5) and PFGE (Fig. 6) to verify whether the reduced chromosomes 7 and 8 co-existed in the candidate strain, 1112_4_12. In the aCGH experiments, we found that the ratio of fluorescence intensity of the strains 1112_4_12 to RIB40 decreased to nearly zero in some of the regions of chromosome 7 that are identical to regions in the reduced chromosome 7 in the 8BCh7N_3 strain (Fig. 5b). Because, the same regions were missing in both strains, it appeared likely that chromosome 7 in the 1112_4_12 strain was derived from the 8BCh7N_3 strain. In the heterozygous diploid strain, the ratio of fluorescence intensity relative to the RIB40 strain decreased to around 0.5 in some of the regions that were completely deleted in 8BCh7N_3 strain, implying that the numbers of reduced chromosome 7 (derived from 8BCh7N_3 strain) and normal chromosome 7 (derived from PNDR T6-1 strain) were the same in both strains (Fig. 5b). Similarly, a histogram analysis of chromosome 8 showed that the regions of chromosome 8 that were lost in the 1112_4_12 strain are identical to those that are missing in the chromosome 8-reduced strain, PNDR T6-1, suggesting that chromosome 8 in the 1112_4_12 strain was derived from the PNDR T6-1 strain (Fig. 5c). The aCGH results clearly showed that the two types of nuclei derived from the two distinct parent strains are in equal numbers in the heterozygous diploid strain, while in the 1112_4_12 strain in which chromosomal segregation and rearrangement had occurred, and the expected regions of chromosome 7 and chromosome 8 were simultaneously removed without other significant modifications in other chromosomes (data not shown).

Analysis of the haploid fusant by array comparative genomic hybridization (aCGH). a aCGH analysis. The spots represent the signal intensity ratio. b Data analysis of aCGH in SC011 of chromosome 7. Chromosome 7 consists of two supercontigs (SC), SC011 and SC206 and the deletion regions of chromosome 7-reduced strain mainly exist in SC011 of chromosome 7. c Histogram of the aCGH in SC103 of chromosome 8. The deletion region of chromosome 8-reduced strain exists in SC103. Each row represents the ratio of normalized signal intensity for the genes from each of the strains to the wild type, RIB40. 8BCh7N_3, chromosome 7-reduced strain; PNDR T6-1, chromosome 8-reduced strain; 8BT6_6, heterozygous diploid; 1112_4_12, haploid fusant with both reduced chromosomes 7 and 8

Pulsed-field gel electrophoresis (PFGE) analysis. a Separation of chromosomes by PFGE. DNA was visualized by GelRed staining and UV irradiation. Chromosomes from S. pombe were used as molecular weight markers to calculate the size of the A. oryzae chromosomes. b Southern hybridization analysis of chromosome 7 and chromosome 8. Chromosomes 7 and 8 were hybridized with 0.5 kb probes from gene AO090011000577 located on chromosome 7 and gene AO090010000491 located on chromosome 8. The hybridization signals indicate that chromosome 7 and chromosome 8 were largely reduced in the obtained haploid fusant. 8BCh7N_3, chromosome 7-reduced strain; PNDR T6-1, chromosome 8-reduced strain; 8BT6_6, heterozygous diploid; 1112_4_12, haploid fusant with reduced chromosomes 7 and 8. Ch7 normal chromosome 7, Ch7Δ reduced chromosome 7, Ch8 normal chromosome 8, Ch8Δ reduced chromosome 8
The PFGE analysis also demonstrated that the chromosomal pattern of the 1112_4_12 strain was altered compared with that of the RIB40 strain (Fig. 6a). To further validate whether the changed pattern occurred simultaneously in chromosomes 7 and 8, Southern blot analysis was performed (Fig. 6b). Approximately, 0.5 kb of DIG-labeled probes were designed and constructed to target undeleted regions of chromosomes 7 and 8. The gel mobility shift showed simultaneous reduction of chromosome 7 and chromosome 8 in the candidate strain. Interestingly, the reduced chromosome 7 in the heterozygous diploid strain moved faster than that from both the 8BCh7N_3 strain and the 1112_4_12 fusant. In chromosome 7 of heterozygous diploid strain, no other deletions were detected using aCGH (Fig. 5b); therefore, the reason why it moved faster still remains unclear. These results confirmed that the 1112_4_12 strain was an objective haploid fusant harboring the two reduced chromosomes, 7 and 8.
Phenotype observation of the new haploid strain
The growth phenotype of the new haploid fusant was observed (Fig. 7). The new haploid fusant 1112_4_12, the heterozygous diploid strain, and the wild type strain RIB40 were separately cultivated on malt agar medium for 5 days. Except that growth was slower, the morphogenesis of the heterozygous diploid strain was most similar to that of the wild type strain. On the other hand, although the new haploid fusant harbored various combinations of parental genetic markers and showed parental morphology, the phenotype of the haploid fusant resembled that of the chromosome 8-reduced strain, PNDR T6-1, rather than that of the chromosome 7-reduced strain, 8BCh7N_3. In addition, because the parent strains, 8BCh7N_3 and PNDR T6-1, are auxotrophic strains carrying the pyrG and niaD deletion, respectively (Fig. 1), they displayed slower growth rates than did the 1112_4_12 strain (Fig. 7).

Growth phenotype of the new haploid fusant. 1112_4_12, the new haploid fusant, 8BT6_6, the heterozygous diploid strain, and the wild type strain RIB40 were cultivated on malt agar medium at 37°C for 5 days. The phenotype of the haploid fusant resembled that of the chromosome 8-reduced strain, PNDR T6-1, rather than the chromosome 7-reduced strain, 8BCh7N_3
Conclusions
In this study, we successfully created a new chromosome-reduced strain that contained the reduced chromosomes 7 and 8 using a protoplast fusion method. This method provides a useful tool for large-scale chromosome manipulation and further chromosomal reduction. Combining the protoplast fusion method with the loop-out method, which is useful for multiple large-scale chromosomal deletions (Takahashi et al. 2009; Jin et al. 2010), may eventually help minimize the whole A. oryzae genome and lead to the construction of a safer, more efficient industrially useful A. oryzae strain.
References
Anné J, Peberdy JF (1975) Conditions for induced fusion of fungal protoplasts in polyethylene glycol solutions. Arch Microbiol 105:201–205
Anné J, Peberdy JF (1976) Induced fusion of fungal protoplasts following treatment with polyethylene glycol. J Gen Microbiol 92:413–417
DeFazio LG, Stansel RM, Griffith JD, Chu G (2002) Synapsis of DNA sends by DNA-dependent protein kinase. EMBO J 21:3192–3200
Ferenczy L, Kevei F, Szegedi M (1975) High frequency fusion of fungal protoplasts. Experientia 31:1028–1030
Ferenczy L, Kevei F, Szegedi M, Franko A, Rojik I (1976) Factors affecting high frequency fungal protoplast fusion. Experientia 32:1156–1158
Goryshin IY, Naumann TA, Apodaca J, Reznikoff WS (2003) Chromosomal deletion formation system based on Tn5 double transposition: use for making minimal genomes and essential gene analysis. Genome Res 13:644–653
Hara S, Tsuji RF, Hatamoto O, Masuda T (2002) A simple method for enrichment of uninucleate conidia of Aspergillus oryzae. Biosci Biotechnol Biochem 66:693–696
Hashimoto M, Ichimura T, Mizoguchi H, Tanaka K, Fujimitsu K, Keyamura K, Ote T, Yamakawa T, Yamazaki Y, Mori H, Katayama T, Kato J (2005) Cell size and nucleoid organization of engineered Escherichia coli cells with a reduced genome. Mol Microbiol 55:137–149
Horn BW, Moore GG, Carbone I (2009) Sexual reproduction in Aspergillus flavus. Mycologia 101:423–429
Jin FJ, Takahashi T, Utsushikawa M, Furukido T, Nishida M, Ogawa M, Tokuoka M, Koyama Y (2010) A trial of minimization of chromosome 7 in Aspergillus oryzae by multiple chromosomal deletions. Mol Genet Genomics 283:1–12
Jones JM, Gellert M, Yang W (2001) A Ku bridge over broken DNA. Structure 9:881–884
Kato J, Hashimoto M (2007) Construction of long chromosomal deletion mutants of Escherichia coli and minimization of the genome. Methods Mol Biol 416:279–294
Kevei F, Peberdy JF (1977) Interspecific hybridization between Aspergillus nidulans and Aspergillus rugulosus by fusion of somatic protoplasts. J Gen Microbiol 102:255–262
Kevei F, Peberdy JF (1979) Induced segregation in interspecific hybrids of Aspergillus nidulans and Aspergillus rugulosus obtained by protoplast fusion. Mol Gen Genet 170:213–218
Kobayashi T, Abe K, Asai K, Gomi K, Juvvadi PR, Kato M, Kitamoto K, Takeuchi M, Machida M (2007) Genomics of Aspergillus oryzae. Biosci Biotechnol Biochem 71:646–670
Kolisnychenko V, Plunkett G, Herring CD, Feher T, Posfai J, Blattner FB, Posfai G (2002) Engineering a reduced Escherichia coli genome. Genome Res 12:640–647
Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R, Iwashita K, Juvvadi PR, Kato M, Kato Y, Kin T, Kokubun A, Maeda H, Maeyama N, Maruyama J, Nagasaki H, Nakajima T, Oda K, Okada K, Paulsen I, Sakamoto K, Sawano T, Takahashi M, Takase K, Terabayashi Y, Wortman JR, Yamada O, Yamagata Y, Anazawa H, Hata Y, Koide Y, Komori T, Koyama Y, Minetoki T, Suharnan S, Tanaka A, Isono K, Kuhara S, Ogasawara N, Kikuchi H (2005) Genome sequencing and analysis of Aspergillus oryzae. Nature 438:1157–1161
Murakami K, Tao E, Ito Y, Sugiyama M, Kaneko Y, Harashima S, Sumiya T, Nakamura A, Nishizawa M (2007) Large scale deletions in the Saccharomyces cerevisiae genome create strains with altered regulation of carbon metabolism. Appl Microbiol Biotechnol 75:589–597
Ninomiya Y, Suzuki K, Ishii C, Inoue H (2004) Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc Natl Acad Sci USA 101:12248–12253
O’Gorman CM, Fuller HT, Dyer PS (2009) Discovery of a sexual cycle in the opportunistic fungal pathogen Aspergillus fumigatus. Nature 457:471–474
Takahashi T, Hatamoto O, Koyama Y, Abe K (2004) Efficient gene disruption in the koji-mold Aspergillus sojae using a novel variation of the positive–negative method. Mol Genet Genomics 272:344–352
Takahashi T, Masuda T, Koyama Y (2006) Enhanced gene targeting frequency in ku70 and ku80 disruption mutants of Aspergillus sojae and Aspergillus oryzae. Mol Genet Genomics 275:460–470
Takahashi T, Jin FJ, Sunagawa M, Machida M, Koyama Y (2008) Generation of large chromosomal deletions in koji molds Aspergillus oryzae and Aspergillus sojae via a loop-out recombination. Appl Environ Microbiol 74:7684–7693
Takahashi T, Jin FJ, Koyama Y (2009) Nonhomologous end-joining deficiency allows large chromosomal deletions to be produced by replacement-type recombination in Aspergillus strains. Fungal Genet Biol 46:815–824
Yu BJ, Sung BH, Koob MD, Lee CH, Lee JH, Lee WS et al (2002) Minimization of the Escherichia coli genome using a Tn5-targeted Cre/loxP excision system. Nat Biotechnol 20:1018–1102
Yu JH, Hamari Z, Han KH, Seo JA, Reyes-Domínguez Y, Scazzocchio C (2004) Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet Biol 41:973–981
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Perez-Martin.
S. Hara and F. J. Jin contributed equally to this work.
Rights and permissions
About this article
Cite this article
Hara, S., Jin, F.J., Takahashi, T. et al. A further study on chromosome minimization by protoplast fusion in Aspergillus oryzae . Mol Genet Genomics 287, 177–187 (2012). https://doi.org/10.1007/s00438-011-0669-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-011-0669-1