
Abstract
The widespread occurrence of epigenetic alterations in allopolyploid species deserves scrutiny that DNA methylation systems may be perturbed by interspecies hybridization and polyploidization. Here we studied the genes involved in DNA methylation in Nicotiana tabacum (tobacco) allotetraploid containing S and T genomes inherited from Nicotiana sylvestris and Nicotiana tomentosiformis progenitors. To determine the inheritance of DNA methyltransferase genes and their expression patterns we examined three major DNA methyltransferase families (MET1, CMT3 and DRM) from tobacco and the progenitor species. Using Southern blot hybridization and PCR-based methods (genomic CAPS), we found that the parental loci of these gene families are retained in tobacco. Homoeologous expression was found in all tissues examined (leaf, root, flower) suggesting that DNA methyltransferase genes were probably not themselves targets of uniparental epigenetic silencing for over thousands of generations of allotetraploid evolution. The level of CG and CHG methylation of selected high-copy repeated sequences was similar and high in tobacco and its diploid progenitors. We speculate that natural selection might favor additive expression of parental DNA methyltransferase genes maintaining high levels of DNA methylation in tobacco, which has a repeat-rich heterochromatic genome.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Allopolyploidy, defined as interspecific hybridization and genome multiplication, has been considered a major force in the evolution of angiosperm species. Experiments with synthetic and natural allopolyploids showed that parental genomes evolve in a complex manner, involving numerous genetic and epigenetic changes (Soltis and Soltis 1995; Leitch and Leitch 2008). Of the epigenetic changes, the most important in plants is cytosine methylation at symmetrical (CG, CHG) and non-symmetrical (CHH) sequence motifs (Ingelbrecht et al. 1994; Meyer et al. 1994; Vanyushin 2006). DNA methylation plays a pivotal role in silencing of genes, transposons and maintenance of chromosome integrity (Vaillant and Paszkowski 2007). Changes in gene expression, transposon activation and karyological instability have been frequently observed in allopolyploid plants and DNA methylation is likely to be involved in harmonizing genome structure and expression after the genome merger. Changes in DNA methylation patterns occur more frequently in synthetic allotetraploids than their parents (Madlung et al. 2002), perhaps related to the silencing of “redundant” genes in a doubled genome. The molecular basis underlying these allopolyploid-induced epigenetic changes is largely unsolved, but one possibility is that there is altered expression of parental alleles or genes encoding DNA methyltransferase activities.
Three major families (MET1, CMT and DRM) involved in DNA methylation have been identified in many monocot and dicot plant species (for reviews see Finnegan and Kovac 2000; Pavlopoulou and Kossida 2007), and homologous sequences can be found in conifers and mosses (http://genome.jgi-psf.org/euk_home.html). Methylation at CG dinucleotides is maintained by the MET1 family of enzymes that is common to higher eukaryotes. The second family, called Domains Rearranged Methyltransferase (DRM) has characteristic rearrangement of conserved motifs in the catalytic domain (Cao et al. 2000), and probably catalyzes methylation of naive DNA (Wada et al. 2003). The chromomethylases (CMT) are unique in higher plants (Henikoff and Comai 1998). These enzymes maintain methylation of CHG trinucleotides (Lindroth et al. 2001; Papa et al. 2001).
Nicotiana tabacum is an important model plant for genetic, biochemical and evolutionary studies. Its allotetraploid genome (2n = 4x = 48) is thought to be formed by interspecific hybridization between diploid progenitors close to modern Nicotiana sylvestris (2n = 24) and Nicotiana tomentosiformis (2n = 24). Numerous coding genes and repeated sequences have been characterized in detail and recently a gene space library has been constructed (Rushton et al. 2008) making tobacco a convenient plant system for genetic manipulation. Tobacco genes encoding MET1 and DRM activities have been isolated and characterized at the biochemical level (Nakano et al. 2000; Wada et al. 2003). NtMET1 is a putative major tobacco maintenance enzyme expressed in dividing cells (Nakano et al. 2000). Tobacco transgenic lines carrying antisense NtMET1 construct showed hypomethylation of DNA in CG sequences (Nakano et al. 2000). However, NtMET1 peculiar features are a complete lack of in vitro activity, a hypo- rather hypermethylation effect on DNA when overexpressed in vivo, and interaction with a GTPa-ase protein (Kim et al. 2007). The NtDRM enzyme shows strong preference towards non-CG motifs (Wada et al. 2003) and may be involved in RNA-directed DNA methylation reactions in tobacco (Mette et al. 2000; Fojtova et al. 2006).
The focus of this study was to determine: (1) inheritance of parental DNA methyltransferase loci in tobacco allotetraploid; (2) expression patterns of homoeologous methyltransferase genes; (3) DNA methylation levels of selected high-copy sequences in both S and T compartments of tobacco’s genome.
Materials and methods
Plant material
Nicotiana sylvestris Speg. & Comes (accession number ITB626) was obtained from SEITA, Institut du Tabac, Bergerac, France. Nicotiana tomentosiformis Goodsp. (NIC 479/84) was obtained from the Institute of Plant Genetics and Crop Plant Research, Gatersleben, Germany. Nicotiana tabacum L. cv. Vielblättriger (00246) originally obtained from the Tobacco research Institute, Báb, Slovakia has been cultivated at the Institute of Biphysics for at least 20 years (line T3). The synthetic allotetraploid of N. sylvestris and N. tomentosiformis called TR1A was described elsewhere (Lim et al. 2006b). Seeds were surface sterilised and germinated for 6–8 days in sterile water, seedlings were then transferred to soil, and plants were grown in a greenhouse under standard conditions. Calli were established from leaf explants by hormonal treatment according to standard procedures (Koukalova et al. 2005).
DNA and RNA isolation
DNA was isolated from fresh young leaves by a CTAB method (Saghai-Maroof et al. 1984). Total RNA was isolated from 100 mg of fresh 6–8 days old seedlings, young, fully developed and old leaves, root tips, flower buds, and calli using RNeasy Plant Mini Kit (Qiagen) or TRIzol Reagent (Invitrogen). Oligotex mRNA Mini Kit (Qiagen) was used to isolate mRNA from total RNA. Possible contaminating DNA in RNA preparations was degraded by a DNase treatment (DNA-Free Turbo, Ambion). The quantity and quality of RNA and DNA preparations were checked by absorbance at 260/280 nm and by agarose gel electrophoresis.
Molecular cloning
For cloning of DNA methyltransferase cDNAs from N. sylvestris and N. tomentosiformis we designed several forward and reverse COnserved-DEgenerated Hybrid Oligonucleotide Primers (CODEHOP, Rose et al. 1998) according to the conserved motifs of the catalytic domains of MET1, CMT3 and DRM genes in Arabidopsis thaliana and Zea mays. Sequences of primers are given in Supplementary Table S1. These primers were used in subsequent RT-PCR reactions (CODEHOP, 3′, 5′ RACE and gene-specific). Chronology of experimental amplification methods used, primers, the lengths of PCR products and the clone names are given in Table S2. Cloning procedures are described in detail in Supplementary methods. Sequence data were submitted to the EMBL Nucleotide Sequence Database. Nicotiana sylvestris cDNA molecules with complete coding sequences: NsMET1 clone 10—accession AM946605; NsCMT3 clones ch991, ch031, ch034, chm2—accessions AM946608–AM946611; NsDRM clones d1991 = d10311, d2992, d2036, d2m10—accessions AM946617–AM946620. N. tomentosiformis cDNA molecules with partial coding sequences: NtoMET1 clone mt1—accession FM872474; NtoCMT3 clone ct2—accession FM872475; NtoDRM clone dt1—accession FM872476.
Southern blot analysis
Genomic DNA was digested with 10 U μg−1 of restriction endonucleases and the fragments were electrophoretically separated in a 0.8% agarose gel. The DNA fragments were alkali blotted onto Hybond XL membrane (GE-Healthcare) and hybridized with 32P-labeled DNA probes (>108 dpm μg−1 DNA, DekaLabel DNA Labeling Kit, MBI Fermentas). The blots were hybridized with full-length cDNAs derived from NsMET1, NsCMT3 and NsDRM clones (Table 1). The repetitive probes involved cloned HRS60 (Matyasek et al. 1989) and NTRS (Matyasek et al. 1997) satellites, 5S_T-genome sub-region (Fulnecek et al. 2002a), 5S_S-spec (ClaI–BstUI fragment of the intergenic spacer AJ131164), endogenous NsEPRV pararetroviruses (Gregor et al. 2004) and endogenous geminiviral GRD5 (Ashby et al. 1997). Southern blot hybridization was carried out under high-stringency conditions using standard procedures (Sambrook and Russell 2001) in a 0.25 M sodium phosphate buffer (pH 7.0) supplemented with 7% w/v sodium dodecyl sulfate (SDS) at 65°C for 16 h. Probes were washed with 2× SSC (20× SSC = 3 M NaCl and 0.3 M sodium citrate, pH 7.0), 0.1% SDS (twice for 5 min) then with 0.2× SSC and 0.1% SDS (twice for 15 min at 65°C). The membranes were exposed to Storage Phosphor Screen (Storm, GE-Healthcare) for 2 days.
Northern blot analysis
Ten μg of total RNA per lane were separated in 1% agarose gel and transferred to nylon membranes (Hybond XL, GE-Healthcare) in 10× SSPE [20× SSPE = 3.6 M NaCl, 0.2 M (NaH2PO4/Na2HPO4, pH = 7.0) and 0.02 M EDTA]. The RNAs were hybridized with the antisense MET1, CMT3 and DRM-specific riboprobes derived from inserts of 10, ch991 and d2036 N. sylvestris clones. The probes were prepared by incorporation of [alpha-32P]UTP (10 mCi ml−1, 3,000 Ci mmol−1, MP Biomedicals) in a RNA polymerase reaction (RNAMaxx High Yield Transcription Kit, Stratagene). The membranes were prehybridized and hybridized in UltraHyb Ultrasensitive Hybridization Buffer (Ambion) at 68°C for 2 h and at 68°C for 24 h, respectively. The blots were washed twice with 2× SSC, 0.1% (w/v) SDS at 68°C for 5 min each, then twice with 0.1× SSC, 0.1% (w/v) SDS at 68°C for 15 min each, and finally high salt wash [5× SSC, 0.5% (w/v) SDS at 68°C for 15 min each] was used. The membranes were exposed to a Storage Phosphor Screen (Storm, GE-Healthcare) for 2 days.
Cleaved amplified polymorphic sequence assay
The genomic cleaved amplified polymorphic sequence (CAPS) was used to analyze species-specific polymorphisms within the MET1, CMT3 and DRM genes. The selected coding regions were as follows: 72–4,549 nt of NsMET1, 28–1,892 nt of NsCMT3 and 1–1,816 nt of NsDRM. For the sequences of primers, see Tables S1, S2. PCR conditions: initial denaturation at 95°C for 1 min was followed by 35 cycles of 95°C for 20 s, annealing step for 30 s and 72°C for 6 min. Annealing temperatures were 52.5, 50 and 55°C for MET1, CMT3 and DRM, respectively. All amplifications were performed using PfuUltra II Fusion HS DNA polymerase (Stratagene). After PCR the products were digested with selective restriction enzymes and separated on agarose gels.
The cDNA-CAPS was carried out using cDNA samples prepared from total RNA by random-primed reverse transcription. The amplified regions involved 72–2,420 nt of NsMET1, 38–1,336 nt of NsCMT3 and 580–1,266 nt of NsDRM coding sequences. PCR conditions were as follows: initial denaturation at 95°C for 1 min was followed by 35 cycles (40 cycles for CMT3) of 95°C for 20 s, 50°C (MET1, DRM) or 55°C (CMT3) annealing step for 30 s and 72°C for 2 min (MET1, CMT3) or 1 min (DRM).
Phylogenetic analysis
Amino acid sequences of the C-terminal catalytic domains were aligned using a PILEUP program (Wisconsin Package Version 10.3). Sequences used in distance analysis are given in Table S3. The gaps were deleted from alignments prior to distance analysis. In order to root the CMT3 and DRM trees the sequences were treated as follows: chromodomains in CMT3 orthologues were deleted and catalytic motifs of the DRM proteins were rearranged at their point of rearrangement. The evolutionary relationships were inferred using Phylip programs (Felsenstein 1989; http://mobyle.pasteur.fr/cgi-bin/MobylePortal/portal.py). The distance data were obtained by running a PROTDIST program according to the Jones–Taylor–Thornton distance model (Jones et al. 1992) and bootstrap resampling of 100 replicates. The distance trees were constructed from distance data (randomized input order) employing a Neighbor-Joining algorithm (Saitou and Nei 1987) implemented within the NEIGHBOR program. Finally, consensus distance trees including bootstrap values were plotted using CONSENSE and DRAWTREE programs. The divergence (d) was defined as a number of sites with mismatches/total sites.
Bioinformatic analysis of tobacco genomic database
We have used N. sylvestris methyltransferase cDNA sequences as queries to search a tobacco (cultivar Hicks Broadleaf) gene space library using BLASTN program. The library data sets were generated by the Tobacco Genome Initiative (TGI, http://www.tobaccogenome.org/) using methylation filtration technology. To the date of analysis (3 January 2008) the nucleotide database contained 1,271,256 gene-space sequence reads (GSRs). The GSR hits (about 50) for each methyltransferase family were transferred to the SeqLab interface of Wisconsin Package Version 10.3. Each GSR was then aligned (pairwise, BESTFIT) to cDNA sequences of N. sylvestris and N. tabacum and adjusted manually. Intron–exon boundaries were determined using a GT–AG rule. The number of sequences was narrowed by discarding false positives. Finally, the GSRs were assembled to genomic contigs (Supplementary files MET1_assembly, CMT3_assembly, DRM_assembly).
Results
Cloning of DNA methyltransferase cDNAs from tobacco diploid progenitors
Based on homologies to known plant DNA methyltransferase sequences we have cloned and sequenced cDNAs of MET1, CMT3 and DRM genes from N. sylvestris and N. tomentosiformis. The clones obtained are listed in Table 1.
The MET1 family was represented by a single cDNA clone in each species that we called NsMET1 and NtoMET1, respectively.
The CMT3 family was represented by four cDNA clones of N. sylvestris (NsCMT3) and a single clone of N. tomentosiformis (NtoCMT3). Nicotiana sylvestris clones ch991 and chm2 had identical coding sequences while clones ch031 and ch034 differed from the consensus by one and two silent point mutations, respectively. We do not know whether the sequence polymorphisms reflect cloning/sequencing errors, heterozygosity or more gene copies/variants in the genome. Some variability in the length of the 3′ untranslated region (3′UTR) could originate from alternatively polyadenylated molecules.
Two DRM-specific RT-PCR products (d1 and d2) were obtained from N. sylvestris. Sequencing revealed that the d1 clones were about 200 bp shorter in 3′UTR than the d2 clones. Since the coding region (1,827 bp) was identical between the clones and the variation in 3′UTR probably arose from alternative splicing we therefore assign these cDNA clones to one NsDRM gene. A single NtoDRM clone was isolated from N. tomentosiformis.
Since conventional RT-PCR cannot reveal the structure of the 5′ mRNA terminus we applied 5′ RACE. Through this method we identified 5′ untranslated regions (5′UTR) for transcripts corresponding to all three gene families in N. sylvestris. In frame stop codons preceding the first AUG codon delimitated 5′UTR in NsMET1 and NsDRM whose lengths were 198 and 283 nt, respectively. There were no in frame stop codons in the NsCMT3 clones. However, a motif at +389 similar to a Kozak consensus sequence (AACAATGGC, Lütcke et al. 1987) suggested translation initiation within that region.
Phylogenetic analysis
The phylogenetic trees of DNA methyltransferase catalytic domains of the MET1, CMT3 and DRM families were constructed using amino acid sequences deduced from plant DNA methyltransferase cDNAs (Figs. 1, 2, 3; Table S3). The trees yielded several well-supported branches reflecting phylogenetic relationships between the species. As expected the Nicotiana genes fell to the same branch as those of tomato (Solanaceae). All three trees showed tight clustering of N. sylvestris and N. tomentosiformis sequences with those of N. tabacum. In general the topologies were congruent with those previously published (Pavlopoulou and Kossida 2007). At the amino acid level, the similarities between obtained Nicotiana orthologs were as follows: 98.2% NsMET1 versus NtoMET1, 97.8% NsCMT3 versus NtoCMT3 and 99.2% between NsDRM and NtoDRM. At the nucleotide level, sequence identities were 98.8, 97.1, 97.9% for MET1, CMT3 and DRM gene families, respectively.

Phylogenetic relationships between members of the MET1 family. a Schematic drawing of a domain structure. Translated region is in gray, conserved amino acid motifs in N-terminal regulatory domains are in dark gray and black boxes with roman numerals indicate conserved motifs in C-terminal catalytic domain (Kumar et al. 1994). Region used for phylogenetic analysis in b is underlined. S-stretch multiple serine residues; E-rich region glutamic acid residue rich region; GK repeat repeated glycine lysine residues; BAH bromo adjacent homology domain. b A phylogenetic tree constructed based on amino acid sequences of C-terminal catalytic domains. Clustering of Solanaceae species is highlighted. Distance data were calculated using a Neighbor-Joining method (Saitou and Nei 1987) and the tree was constructed in Phylip programs (Felsenstein 1989; http://mobyle.pasteur.fr/). Accession numbers and references are given in Table S3. The bootstrap values are indicated at the nodes

Phylogenetic relationships between members of the CMT3 family. a Domain structure. A unique chromodomain is indicated. b A phylogenetic tree comprising CMT3 sequences. The tree was rooted to AtMET1, OsMET1–2, ZmMET1 sequences. The methods and symbols used are as described in the legend of Fig. 1

Phylogenetic relationships between members of the DRM family. a Domain structure. UBA Ubiquitin-associated domain; nls nuclear localization signal. The “###” symbol denotes point of motif rearrangement. b A phylogenetic tree comprising DRM sequences. The methods and symbols used are as described in the legend of Fig. 1. The tree was rooted to Mus musculus MmDnmt3a, MmDnmt3b (GenBank: AAC40177, AAC40178) as an outgroup
Inheritance of DNA methyltransferase loci in tobacco
Next we determined the ancestral origin of tobacco loci using Southern blot hybridization and genomic CAPS (Fig. 4). In the Southern blot hybridization Fig. 4a, the probes derived from NsMET1 cDNA hybridized to three fragments (2.1, 3.1 and 4.2 kb) in N. sylvestris and two fragments (2.1, 3.7 kb) in N. tomentosiformis. In N. tabacum all four parental bands were detected. Similar hybridization band-additivity was observed with the NsCMT3 and NsDRM cDNA probes. Also genomic CAPS profiles (Fig. 4b, S2) were additive for all three families of DNA methyltransferases. These results indicated that both parental MET1, CMT3 and DRM methyltransferase loci remained intact in tobacco.

Inheritance of parental MET1, CMT3 and DRM loci in tobacco genome. a Southern blot hybridization of restricted genomic DNA with the NsMET1, NsCMT3 and NsDRM cDNA probes. b Genomic CAPS showing additivity of progenitor restriction fragments in tobacco samples. Regions used for genomic CAPS are depicted under panels. Short vertical bars represent positions of restriction enzymes. N. sylvestris (SY), N. tomentosiformis (TO) and N. tabacum (TA) species. M Size markers (MBI Fermentas)
Reconstitution of genomic structure of tobacco DNA methyltransferase loci
To further characterize tobacco DNA methyltransferase genes we queried the tobacco gene space database for sequences homologous to N. sylvestris cDNAs. The gene-space sequence reads (GSRs) were aligned with cDNA sequences of each methyltransferase family (for the alignments see supplementary files MET1_assembly, CMT3_assembly, DRM_assembly). The GSR hits split into two groups matching N. sylvestris sequences and N. tabacum sequences, likely inherited from the N. tomentosiformis progenitor. For each locus, we designed a tentative map of its genomic organization (Fig. S3). The contigs assembled from individual GSRs involved genic regions, promoters, introns and 3′ adjacent sequences. The size and position of introns were similar between homoeologous loci. Moreover, the tobacco MET1 and CMT3 orthologs had the same number of introns as those of Arabidopsis indicating a conserved genomic structure among dicots. The DRM loci were more divergent having 11 introns in tobacco and 9 introns in Arabidopsis. A prominent feature of tobacco MET1 was a microsatellite-like sequence in the eighth intron; in the N. sylvestris derived homoeolog it was (AT)9(GT)27, and in the N. tomentosiformis derived homoeolog it was (GTAT)4(GT)17.
Expression analysis
To reveal the size of respective DNA methyltransferase transcripts we carried out Northern blot hybridization of N. sylvestris RNA (Fig. 5a). The hybridization signals of NsMET1, NsCMT3 and NsDRM probes were detected as single bands in regions of 5, 3.2 and 2.8 kb respectively, which correlated with the predicted sizes of RNA species based on cloning experiments.

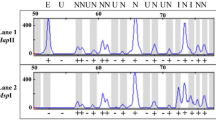
Expression analysis of MET1, CMT3 and DRM genes in Nicotiana. a Northern blot hybridization of N. sylvestris root tip total RNA probed with riboprobes derived from NsMET1, NsCMT3 and NsDRM cDNAs (left lanes). The exposure times of blots were identical. 18S and 25S rDNA bands are shown in the right lanes as loading controls (etbr). b cDNA-CAPS experiments were carried out using total RNA isolated from N. sylvestris (SY), N. tomentosiformis (TO) and N. tabacum (TA). The RNA samples were from young (lane 1), fully developed (2), and old (3) leaves, seedlings (4, SY, TO), roots (5), flower buds (6) and calli (7, 8)
To examine expression of parental gene families in allotetraploid tobacco we carried out cDNA-CAPS analysis of RNA samples isolated from tobacco leaves, roots, flowers, and dedifferentiated calli (Fig. 5b). The PCR products obtained from amplification of each methyltransferase family were similar between the species in accord with conserved lengths of amplified subregions. Upon restriction, the MET1 fragment (Fig. 5b, upper panel) remained undigested in N. tomentosiformis while it was digested into two bands in N. sylvestris. The CMT3 (middle panel) and DRM (bottom panel) fragments were digested in N. tomentosiformis while they were undigested in N. sylvestris. In N. tabacum, all three parental fragments were visible in all tissues examined. There were little or no differences in restriction profiles between the tobacco tissues, suggesting that both parental homoeologs were expressed. The ratio of the signal of DRM homoeologs varied slightly among the RNAs isolated from two independent calli (Fig. 5b, bottom panel, MspI digestion, lanes 7, 8).
Methylation analysis of repeated sequences in tobacco and progenitor genomes
We next determined methylation status of retroelements (NsEPRV), endogenous viruses (GRD5), 5S rDNA and satellites in tobacco, its progenitors and synthetic doubled F1 hybrid (Fig. 6). The total sequence analyzed accounts for approximately 10% of tobacco genome (Skalicka et al. 2005). The genomic DNAs were digested with methylation-insensitive (MboI, MvaI) and methylation-sensitive (HpaII, MspI, Sau3AI, EcoRII, ScrFI) restriction enzymes. MvaI (cuts at CCWGG) is isoschizomeric, or nearly so, with EcoRII (CCWGG) and ScrFI (CCNGG), respectively. Both EcoRII and ScrFI are sensitive to CHG methylation. MboI (GATC) is isoschizomeric with Sau3AI and the context of methylation-sensitivity is dependent on overlapping sequence. HpaII is sensitive to methylation of both Cs within the CCGG while MspI (CCGG) is sensitive to methylation of outer C only. Methylation sensitivities of individual restriction enzymes were taken from a REBASE website (http://rebase.neb.co).

DNA methylation of repeated sequences in N. tabacum and diploid parental species. Restricted genomic DNA of N. sylvestris (SY), N. tomentosiformis (TO), N. tabacum (TA) and doubled F1 hybrid of N. sylvestris and N. tomentosiformis (F1) were hybridized on blots with the S- and T- genome-specific probes. About 5–10 μg of restricted DNA was loaded into each lane. a Endogenous pararetrovirus family NsEPRV; b, c Geminivirus-related GRD5 family; d, f 5S rDNA intergenic spacer from N. sylvestris; e, g 5S rDNA intergenic spacer from N. tomentosiformis; h HRS 60 subtelomeric satellite; i NTRS subcentromeric satellite. Restriction enzymes were: MvaI (Mv), EcoRII (Ec), ScrfI (Sc), MboI (Mb), Sau3AI (Sa), MspI (Ms) and HpaII (Hp)
The NsEPRV elements belong to the family of endogenous pararetroviruses (Staginnus and Richert-Poggeler 2006) known to be highly methylated in tobacco and inherited from the N. sylvestris progenitor (Gregor et al. 2004). Consistently, the methylation-sensitive EcoRII and ScrFI enzymes failed to significantly digest NsEPRV repeats (most material migrated as undigested, unfractionated high molecular weight DNA) in tobacco and N. sylvestris, while they were digested with the methylation-insensitive enzyme MvaI, forming 2.2 kb bands (Fig. 6a). A similar result was obtained with a probe derived from N. tomentosiformis pararetroviral sequences (not shown). The GRD5 family of geminiviruses, inherited from N. tomentosiformis progenitor (Murad et al. 2002), was also highly resistant to enzymes sensitive to CHG methylation (Fig. 6b). The pattern of GRD5 bands revealed with Sau3AI digestion of genomic DNA (Fig. 6c) indicates multiple target sites within the 1.2 kb units, some apparently lacking methylation. It is likely that the undermethylated Cs are located in a non-symmetrical context since the CHH methylation is far less frequent than that of CG and CHG in tobacco (Fulnecek et al. 1998). In conclusion the methylation status of endogenous viruses has not been materially changed since the allopolyploidy event.
There is a single 5S locus in each of the progenitor species, N. tomentosiformis and N. sylvestris, and both are found in tobacco (Fulnecek et al. 2002a). The methylation status of the 5S rRNA genes (Fig. 6d–g) was studied using locus-specific probes derived from intergenic spacers. In general there was little (if any) digestion of 5S rDNA with the methylation-sensitive enzymes, consistent with high levels of methylation of 5S repeats in plant genomes (Fulnecek et al. 2002b; Vaillant et al. 2007).
Most of the HRS60 satellite was digested with MboI into monomeric bands, indicating conserved GATC sites within the units (Fig. 6h). In contrast a ladder of bands was formed after the Sau3AI digestion in both N. sylvestris and tobacco samples. The sequence context of the restriction site (GATC) is GATCCG in HRS60 (Kovarik et al. 2000), and the ladder-like pattern indicates that there might be relatively little methylation of the cytosine. The NTRS sequence was analyzed with methylation-insensitive MvaI and isoschizomeric methylation-sensitive ScrFI and EcoRII (Fig. 6i). There was almost no digestion of NTRS repeats with EcoRII and ScrFI in both diploid and allotetraploid species while there was extensive digestion with MvaI. The faint signal in tobacco is consistent with the fewer numbers of repeats in tobacco (Skalicka et al. 2005). The unit loss, however, was not associated with material change in methylation status of the NTRS locus. These results showed that methylation levels of repetitive DNA are comparable in the genomes of tobacco and the diploid progenitors.
Discussion
DNA methyltransferase genes are highly conserved among Nicotiana diploid species
Nicotiana sylvestris and N. tomentosiformis are thought to have diverged about 70 million years ago (Goodspeed 1954). Our data indicate that despite a considerable period of independent evolution, relatively few changes have occurred to coding sequences of the three main DNA methyltranferase families, MET1, CMT3 and DRM. Of these, the MET1 family appears to be most conserved (divergence, d = 0.02) while the CMT and DRM families are slightly more diverged (d = 0.04). In tobacco homoeologs, the introns displayed somewhat higher variation than the coding sequences and a conserved microsatellite-like motif was discovered in the eighth introns of MET1 genes. The microsatellite sequences differed, adding significantly to genetic variability between homoeologous sequences. The presence of the microsatellite could be unique to Nicotiana genes since there is no such microsatellite in Arabidopsis. Microsatellites appear to be suitable markers for phylogenic studies in Nicotiana (Moon et al. 2008) and it will be interesting to investigate distribution of MET1 intron-linked microsatellites in other species.
The divergence values of coding sequences are in good agreement with values reported for other sequences isolated from N. sylvestris and N. tomentosiformis, including GTP-binding proteins (Takumi et al. 2002) and putrescin N-methytransferases (Riechers and Timko 1999). Divergence of internal transcribed spacer (ITS) of rDNA, believed to be generally under lower selection pressures (Nieto Feliner and Rossello 2007) than protein coding sequences, was in a similar range (d = ~0.04, Clarkson et al. 2004). Perhaps, mutation rates of transcribed gene families are similar irrespective to their copy number. However, several genomic in situ hybridization studies showed clear differences between the genomes of diploid species in Nicotiana (Lim et al. 2000b; Lim et al. 2006a). This is probably due to a rapidly evolving repetitive fraction (Lim et al. 2007) that may account up to 90% of plant genomes (Schmidt and Heslop-Harrison 1998). Indeed, many species-specific satellites (Koukalova et al. 1989; Matyasek et al. 1997; Jakowitsch et al. 1998), retroelements (Petit et al. 2007), endogenous viruses (Gregor et al. 2004; Murad et al. 2004) and intergenic spacer elements (Lim et al. 2004) were isolated from N. sylvestris and N. tomentosiformis (Fig. 6, and for review see Hemleben et al. 2007). Recent whole genomic comparison of species of Solanum (including tomato), diverging >70 million years ago, confirmed near identical coding sequences and significant differences in the repetitive fraction (Zhu et al. 2008). Together, it seems that there is little divergence of genic sequences while there is massive divergence of repeated DNA in Solanaceae genomes.
Mendelian inheritance of parental DNA methyltransferase loci in tobacco
Several single nucleotide polymorphisms allowed us to identify progenitor DNA methyltransferase genes in tobacco. We found that both homoeologs of all three families (MET1, CMT3 and DRM) were transmitted from diploid species and faithfully inherited in tobacco. A complete additivity of hybridization bands (Fig. 4a) also supports the view that the structure of flanking sequences was not significantly influenced by allopolyploidy. In tobacco, Mendelian inheritance of parental loci has also been reported for pleiotropic drug resistance NtPDR1 gene (Schenke et al. 2003), putrescin N-methyltransferases (Riechers and Timko 1999), a family of small GTP-binding proteins (Takumi et al. 2002), lignin forming peroxidase genes (Matassi et al. 1991), nitrate reductase genes (Matassi et al. 1991) and glutamine synthase (Matassi et al. 1991). The only exception seems to be a family of tobacco glucan endo-1,3-beta-glucosidase genes that appear to be a recombinant between both ancestral sequences (Sperisen et al. 1991). Together most parental low-copy coding sequences seem to be unchanged in the tobacco supporting the more recent origin of its genome (perhaps <2.105 years, Clarkson et al. 2005) than previously inferred from flower morphology and DNA reassociation experiments (6.106 years, Goodspeed 1954; Okamuro and Goldberg 1985).
The intactness of low-copy sequences contrasts with rapid elimination of some non-coding repeats (Matzke et al. 2004; Melayah et al. 2004) and 35S rDNA (Volkov et al. 1999; Lim et al. 2000a) seen in natural tobacco and some synthetic lines (Skalicka et al. 2003, 2005). There may be several explanations. First, the repeated fraction of genome could be more sensitive than low-copy genes to the genomic shock associated with allopolyploidy, e.g. Plohl et al. 2008 report intrinsic instability of tandems of inverted repeats. It is also possible that protein coding genes are maintained in allopolyploid genomes by natural selection, while repeated DNA sequences tend to evolve more rapidly probably due to less stringent functional constrains. Finally, the relatively young age of tobacco needs to be considered. For example, N. quadrivalvis and N. clevelandii allotetraploids have protease inhibitor genes from one progenitor donor (N. obtusifolia) while those of the partner genome (N. attenuata) have been lost (Wu et al. 2006). However, both N. quadrivalvis and N. clevelandii are more ancient (~106 years old) allotetraploids than tobacco and their chromosomes have major molecular reconstructions (Leitch et al. 2008). Perhaps, in older polyploids with genome downsizing, duplicate copies of DNA methyltransferases could be lost. Based on these observations we can tentatively write a tempo of structural changes in Nicotiana allopolyploids as: 35S rDNA = some transposons > satellites = 5S rDNA > low-copy genic sequences.
Additive expression of parental DNA methyltransferase loci in different plant organs
Copies of duplicate genes may be expected to be retained only if both copies have a selective function. Selection may arise from a necessity to maintain gene product dosage, in relative or absolute terms in a duplicated genome (see below), or through neofunctionalization or subfunctionalization of duplicate copies (Adams and Wendel 2005). Subfunctionalization can apparently occur rapidly since synthetic allopolyploid lines showed 5–6% transcriptome divergence from the parental values in Arabidopsis (Wang et al. 2006). We tested the possibility that paternal/maternal DNA methyltransferase genes are differentially expressed in various organs. We found that in leaves, roots and flowers all three families of genes were equally expressed from both S and T loci, suggesting absence of uniparental silencing. The MET1 and CMT3 signals in mature and old leaves were rather weak compared to other tissues confirming limited expression of “maintenance” enzymes in non-dividing cells (Nakano et al. 2000). However, the expression patterns were similar among diploid and tetraploid species indicating that relatively low MET1 and CMT3 levels in developed leaves were not related to allopolyploidy. It is therefore likely that little (if any) epigenetic silencing was imposed on parental DNA methyltransferase genes during evolution of tobacco.
Additive expression of DNA methyltransferase loci correlates with inheritance of dense cytosine methylation at repeated loci
Tobacco possesses a highly repeat-rich allotetraploid genome in which ~28% of cytosines appear to be methylated (Messeguer et al. 1991). In animals, interspecific hybridization can trigger drastic hypomethylation of the genome (O’Neill et al. 1998). We found that satellites, 5S rDNA and endogenous viruses were equally methylated at both CG and non-CG motifs in progenitor species, natural tobacco and synthetic F1 hybrids (Fig. 6). Although we cannot exclude the possibility of methylation changes at other sequences (medium and low copy) not analyzed in this study, the data are consistent with Mendelian inheritance of DNA methylation patterns in large fraction of parental genomes. Thus, in contrast to animal hybrids, there is no evidence of dramatic methylation changes in tobacco. Endoreduplication (up to 64C) of cells during tomato fruit ripening was not accompanied by material changes in global methylation levels (Teyssier et al. 2008), suggesting that methylation patterns may be rapidly established on dramatically multiplied chromosomes. Methylation of repeated sequences could be important for their stability in cell nucleus (Peng and Karpen 2008) decreasing their recombination frequency (Maloisel and Rossignol 1998) and preventing intergenomic homogenization (Dadejova et al. 2007). Silencing of DNA methyltransferase genes (or their physical elimination) might be more deleterious in allopolyploids combining highly heterochromatic and highly methylated genomes than in systems with less DNA repeats and heterochromatin. Certainly some repeated sequences were lost or reduced in copy number in both natural and synthetic tobacco (Skalicka et al. 2005). Perhaps, DNA methyltransferase loci and their expression could be under strong selection constraints in plant allopolyploid genomes. In this context artificial inhibition of DNA methyltransferase activities was associated with severe developmental defects in tobacco (Vyskot et al. 1995) and other allopolyploids (Madlung et al. 2002). Further, RNAi experiments in Arabidopsis indicate that reduced DNA methylation disrupts regulation of a subset of transposons and heterochromatic genes (Wang et al. 2006). Considering that DNA methylation is a complex process requiring a number of auxiliary factors in addition to DNA methyltransferases, including short RNAs (Rangwala and Richards 2004), we can even speculate that some factors could be genome-specific, and that each compartment could be methylated by its own methylation system.
The maintenance of parental DNA methyltransferase gene families in a relatively young tobacco allotetraploid genome, their active expression status opens the opportunity for subneofunctionalization and neofunctionalization on longer evolutionary scales. In this context, it will be interesting to analyze these genes in ancient (>1 million years old) Nicotiana allotetraploids that have experienced deep chromosomal reconstructions (Lim et al. 2007).
References
Adams KL, Wendel JF (2005) Polyploidy and genome evolution in plants. Curr Opin Plant Biol 8:135–141
Ashby MK, Warry A, Bejarano ER, Khashoggi A, Burrell M, Lichtenstein CP (1997) Analysis of multiple copies of geminiviral DNA in the genome of four closely related Nicotiana species suggest a unique integration event. Plant Mol Biol 35:313–321
Cao X, Springer NM, Muszynski MG, Phillips RL, Kaeppler S, Jacobsen SE (2000) Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc Natl Acad Sci USA 97:4979–4984
Clarkson JJ, Knapp S, Garcia VF, Olmstead RG, Leitch AR, Chase MW (2004) Phylogenetic relationships in Nicotiana (Solanaceae) inferred from multiple plastid DNA regions. Mol Phylogenet Evol 33:75–90
Clarkson JJ, Lim KY, Kovarik A, Chase MW, Knapp S, Leitch AR (2005) Long-term genome diploidization in allopolyploid Nicotiana section Repandae (Solanaceae). New Phytol 168:241–252
Dadejova M, Lim Y, Souckova-Skalicka K, Matyasek R, Leitch AR, Grandbastien M-A, Kovarik A (2007) Transcription activity of rRNA genes correlates with a tendency towards intergenomic homogenization in Nicotiana allotetraploids. New Phytol 174(3):658–668
Felsenstein J (1989) PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 5:164–166
Finnegan EJ, Kovac KA (2000) Plant DNA methyltransferases. Plant Mol Biol 43:189–201
Fojtova M, Bleys A, Bedrichova J, Van Houdt H, Krizova K, Depicker A, Kovarik A (2006) The trans-silencing capacity of invertedly repeated transgenes depends on their epigenetic state in tobacco. Nucleic Acids Res 34:2280–2293
Fulnecek J, Matyasek R, Kovarik A, Bezdek M (1998) Mapping of 5-methylcytosine residues in Nicotiana tabacum 5S rRNA genes by genomic sequencing. Mol Gen Genet 259:133–141
Fulnecek J, Lim KY, Leitch AR, Kovarik A, Matyasek R (2002a) Evolution and structure of 5S rDNA loci in allotetraploid Nicotiana tabacum and its putative parental species. Heredity 88:19–25
Fulnecek J, Matyasek R, Kovarik A (2002b) Distribution of 5-methylcytosine residues in 5S rRNA genes in Arabidopsis thaliana and Secale cereale. Mol Genet Genomics 268:510–517
Goodspeed TH (1954) The genus Nicotiana. Waltham, Massachusetts
Gregor W, Mette MF, Staginnus C, Matzke MA, Matzke AJ (2004) A distinct endogenous pararetrovirus family in Nicotiana tomentosiformis, a diploid progenitor of polyploid tobacco. Plant Physiol 134:1191–1199
Hemleben V, Kovarik A, Torres RA, Volkov RA, Beridze T (2007) Plant highly repeated satellite DNA: molecular evolution, distribution and use for identification of hybrids. Syst Biodivers 5:277–289
Henikoff S, Comai L (1998) A DNA methyltransferase homolog with a chromodomain exists in multiple polymorphic forms in Arabidopsis. Genetics 149:307–318
Ingelbrecht I, Van Houdt H, Van Montagu M, Depicker A (1994) Posttranscriptional silencing of reporter transgenes in tobacco correlates with DNA methylation. Proc Natl Acad Sci USA 91:10502–10506
Jakowitsch J, Papp I, Matzke MA, Matzke AJ (1998) Identification of a new family of highly repetitive DNA, NTS9, that is located predominantly on the S9 chromosome of tobacco. Chromosom Res 6:649–651
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Kim HJ, Yano A, Wada Y, Sano H (2007) Properties of a tobacco DNA methyltransferase, NtMET1 and its involvement in chromatin movement during cell division. Ann Bot (Lond) 99:845–856
Koukalova B, Reich J, Matyasek R, Kuhrova V, Bezdek M (1989) A BamHI family of highly repeated DNA sequences of Nicotiana tabacum. Theor Appl Genet 78:77–80
Koukalova B, Fojtova M, Lim KY, Fulnecek J, Leitch AR, Kovarik A (2005) Dedifferentiation of tobacco cells is associated with ribosomal RNA gene hypomethylation, increased transcription, and chromatin alterations. Plant Physiol 139:275–286
Kovarik A, Van Houdt H, Holy A, Depicker A (2000) Drug-induced hypomethylation of a posttranscriptionally silenced transgene locus of tobacco leads to partial release of silencing. FEBS Lett 467:47–51
Kumar S, Cheng X, Klimasauskas S, Mi S, Posfai J, Roberts RJ, Wilson GG (1994) The DNA (cytosine-5) methyltransferases. Nucleic Acids Res 22:1–10
Leitch AR, Leitch IJ (2008) Genomic plasticity and the diversity of polyploid plants. Science 320:481–483
Leitch IJ, Hanson L, Lim KY, Kovarik A, Chase MW, Clarkson JJ, Leitch AR (2008) The ups and downs of genome size evolution in polyploid species of Nicotiana (Solanaceae). Ann Bot (Lond) 101:805–814
Lim KY, Kovarik A, Matyasek R, Bezdek M, Lichtenstein CP, Leitch AR (2000a) Gene conversion of ribosomal DNA in Nicotiana tabacum is associated with undermethylated, decondensed and probably active gene units. Chromosoma 109:161–172
Lim KY, Matyasek R, Lichtenstein CP, Leitch AR (2000b) Molecular cytogenetic analyses and phylogenetic studies in the Nicotiana section Tomentosae. Chromosoma 109:245–258
Lim KY, Skalicka K, Koukalova B, Volkov RA, Matyasek R, Hemleben V, Leitch AR, Kovarik A (2004) Dynamic changes in the distribution of a satellite homologous to intergenic 26-18S rDNA spacer in the evolution of Nicotiana. Genetics 166:1935–1946
Lim KY, Kovarik A, Matyasek R, Chase MW, Knapp S, McCarthy E, Clarkson JJ, Leitch AR (2006a) Comparative genomics and repetitive sequence divergence in the species of diploid Nicotiana section Alatae. Plant J 48:907–919
Lim KY, Souckova-Skalicka K, Sarasan V, Clarkson JJ, Chase MW, Kovařík A, Leitch A (2006b) A genetic appraisal of a new synthetic Nicotiana tabacum (Solanaceae) and the Kostoff synthetic tobacco. Am J Bot 93:875–883
Lim KY, Kovarik A, Matyasek R, Chase MW, Clarkson JJ, Grandbastien MA, Leitch AR (2007) Sequence of events leading to near-complete genome turnover in allopolyploid Nicotiana within five million years. New Phytol 175:756–763
Lindroth AM, Cao X, Jackson JP, Zilberman D, McCallum CM, Henikoff S, Jacobsen SE (2001) Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292:2077–2080
Lütcke HA, Chow KC, Mickel FS, Moss KA, Kern HF, Scheele GA (1987) Selection of AUG initiation codons differs in plants and animals. Embo J 6:43–48
Madlung A, Masuelli RW, Watson B, Reynolds SH, Davison J, Comai L (2002) Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiol 129:733–746
Maloisel L, Rossignol J-L (1998) Suppression of crossing-over by DNA methylation in Ascobolus. Genes Dev 12:1381–1389
Matassi G, Melis R, Macaya G, Bernardi G (1991) Compositional bimodality of the nuclear genome of tobacco. Nucleic Acids Res 19:5561–5567
Matyasek R, Koukalova B, Reich J (1989) Isolation and sequencing of HRS60dim1, a dimeric member of the HRS60-family of a tobacco DNA repeat. Nucleic Acids Res 17:4377
Matyasek R, Gazdova B, Fajkus J, Bezdek M (1997) NTRS, a new family of highly repetitive DNAs specific for the T1 chromosome of tobacco. Chromosoma 106:369–379
Matzke M, Gregor W, Mette MF, Aufsatz W, Kanno T, Jakowitsch J, Matzke AJM (2004) Endogenous pararetroviruses of allotetraploid Nicotiana tabacum and its diploid progenitors, N. sylvestris and N. tomentosiformis. Biol J Linn Soc 82:627–638
Melayah D, Lim KY, Bonnivard B, Chalhoub B, Dorlac de Borne F, Mhiri CARL, Grandbastien MA (2004) Distribution of the Tnt1 retrotransposon family in the amphidiploid tobacco (Nicotiana tabacum) and its wild Nicotiana relatives. Biol J Linn Soc 82:639–649
Messeguer R, Ganal MW, Steffens JC, Tanksley SD (1991) Characterization of the level, target sites and inheritance of cytosine methylation in tomato nuclear DNA. Plant Mol Biol 16:753–770
Mette MF, Aufsatz W, van der Winden J, Matzke MA, Matzke AJM (2000) Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J 19:5194–5201
Meyer P, Niedenhof I, ten Lohuis M (1994) Evidence for cytosine methylation of non-symmetrical sequences in transgenic Petunia hybrida. EMBO J 13:2084–2088
Moon HS, Nicholson JS, Lewis RS (2008) Use of transferable Nicotiana tabacum L. microsatellite markers for investigating genetic diversity in the genus Nicotiana. Genome 51:547–559
Murad L, Lim KY, Christopodulou V, Matyasek R, Lichtenstein CP, Kovarik A, Leitch AR (2002) The origin of tobacco’s T genome is traced to a particular lineage within Nicotiana tomentosiformis (Solanaceae). Am J Bot 89:921–928
Murad L, Bielawski JP, Matyasek R, Kovarik A, Nichols RA, Leitch AR, Lichtenstein CP (2004) The origin and evolution of geminivirus-related DNA sequences in Nicotiana. Heredity 92:352–358
Nakano Y, Steward N, Sekine M, Kusano T, Sano H (2000) A tobacco NtMET1 cDNA encoding a DNA methyltransferase: molecular characterization and abnormal phenotypes of transgenic tobacco plants. Plant Cell Physiol 41:448–457
Nieto Feliner G, Rossello JA (2007) Better the devil you know? Guidelines for insightful utilization of nrDNA ITS in species-level evolutionary studies in plants. Mol Phylogenet Evol 44:911–919
O’Neill RJ, O’Neill MJ, Graves JA (1998) Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393:68–72
Okamuro JK, Goldberg RB (1985) Tobacco single-copy DNA is highly homologous to sequences present in the genomes of its diploid progenitors. Mol Gen Genet 198:290–298
Papa CM, Springer NM, Muszynski MG, Meeley R, Kaeppler SM (2001) Maize chromomethylase Zea methyltransferase2 is required for CpNpG methylation. Plant Cell 13:1919–1928
Pavlopoulou A, Kossida S (2007) Plant cytosine-5 DNA methyltransferases: structure, function, and molecular evolution. Genomics 90:530–541
Peng JC, Karpen GH (2008) Epigenetic regulation of heterochromatic DNA stability. Curr Opin Genet Dev 18:204–211
Petit M, Lim KY, Julio E, Poncet C, Dorlhac de Borne F, Kovarik A, Leitch AR, Grandbastien MA, Mhiri C (2007) Differential impact of retrotransposon populations on the genome of allotetraploid tobacco (Nicotiana tabacum). Mol Genet Genomics 278:1–15
Plohl M, Luchetti A, Mestrovic N, Mantovani B (2008) Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene 409:72–82
Rangwala SH, Richards EJ (2004) The value-added genome: building and maintaining genomic cytosine methylation landscapes. Curr Opin Genet Dev 14:686–691
Riechers DE, Timko MP (1999) Structure and expression of the gene family encoding putrescine N-methyltransferase in Nicotiana tabacum: new clues to the evolutionary origin of cultivated tobacco. Plant Mol Biol 41:387–401
Rose TM, Schultz ER, Henikoff JG, Pietrokovski S, McCallum CM, Henikoff S (1998) Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res 26:1628–1635
Rushton PJ, Bokowiec MT, Laudeman TW, Brannock JF, Chen X, Timko MP (2008) TOBFAC: the database of tobacco transcription factors. BMC Bioinformatics 9:53
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schenke D, Sasabe M, Toyoda K, Inagaki Y, Shiraishi T, Ichinose Y (2003) Genomic structure of the NtPDR1 gene, harboring the two miniature inverted-repeat transposable elements, NtToya1 and NtStowaway101. Genes Genet Syst 78:409–418
Schmidt T, Heslop-Harrison JS (1998) Genomes, genes and junk: the large-scale organisation of plant chromosomes. Trends Plant Sci 3:195–199
Skalicka K, Lim KY, Matyasek R, Koukalova B, Leitch AR, Kovarik A (2003) Rapid evolution of parental rDNA in a synthetic tobacco allotetraploid line. Am J Bot 90:988–996
Skalicka K, Lim KY, Matyasek R, Matzke M, Leitch AR, Kovarik A (2005) Preferential elimination of repeated DNA sequences from the paternal, Nicotiana tomentosiformis genome donor of a synthetic, allotetraploid tobacco. New Phytol 166:291–303
Soltis DE, Soltis PS (1995) The dynamic nature of polyploid genomes. Proc Natl Acad Sci USA 92:8089–8091
Sperisen C, Ryals J, Meins F (1991) Comparison of cloned genes provides evidence for intergenomic exchange of DNA in the evolution of a tobacco glucan endo-1, 3-beta-glucosidase gene family. Proc Natl Acad Sci USA 88:1820–1824
Staginnus C, Richert-Poggeler KR (2006) Endogenous pararetroviruses: two-faced travelers in the plant genome. Trends Plant Sci 11:485–491
Takumi S, Ida M, Haisa Y, Ando S, Nakamura C (2002) Genomic structure and homoeologous relationship of the two alpha-subunit genes of a heterotrimeric GTP-binding protein in tobacco. Genome 45:626–633
Teyssier E, Bernacchia G, Maury S, How Kit A, Stammitti-Bert L, Rolin D, Gallusci P (2008) Tissue dependent variations of DNA methylation and endoreduplication levels during tomato fruit development and ripening. Planta 228:391–399
Vaillant I, Paszkowski J (2007) Role of histone and DNA methylation in gene regulation. Curr Opin Plant Biol 10:528–533
Vaillant I, Tutois S, Cuvillier C, Schubert I, Tourmente S (2007) Regulation of Arabidopsis thaliana 5S rRNA Genes. Plant Cell Physiol 48:745–752
Vanyushin BF (2006) DNA methylation in plants. Curr Top Microbiol Immunol 301:67–122
Volkov RA, Borisjuk NV, Panchuk II, Schweizer D, Hemleben V (1999) Elimination and rearrangement of parental rDNA in the allotetraploid Nicotiana tabacum. Mol Biol Evol 16:311–320
Vyskot B, Koukalova B, Kovarik A, Sachambula L, Reynolds D, Bezdek M (1995) Meiotic transmission of a hypomethylated repetitive DNA family in tobacco. Theor Appl Genet 91:659–664
Wada Y, Ohya H, Yamaguchi Y, Koizumi N, Sano H (2003) Preferential de novo methylation of cytosine residues in non-CpG sequences by a domains rearranged DNA methyltransferase from tobacco plants. J Biol Chem 278:42386–42393
Wang J, Tian L, Lee HS, Wei NE, Jiang H, Watson B, Madlung A, Osborn TC, Doerge RW, Comai L, Chen ZJ (2006) Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172:507–517
Wu J, Hettenhausen C, Bladwin IT (2006) Evolution of protease inhibitor defenses in North American allopolyploid species of Nicotiana. Planta 224:750–760
Zhu W, Ouyang S, Iovene M, O’Brien K, Vuong H, Jiang J, Buell CR (2008) Analysis of 90 Mb of the potato genome reveals conservation of gene structures and order with tomato but divergence in repetitive sequence composition. BMC Genomics 9:286
Acknowledgments
The authors wish to thank Professor Andrew Leitch (University of London, UK) for reading the manuscript and valuable comments. This research was funded by the Grant Agency of the Czech Republic (204/06/1432, 521/07/0116) and the Academy of Sciences of the Czech Republic (AVOZ50040507 and AVOZ50040702).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G. Reuter.
Nucleotide sequence data reported are available in the DDBJ/EMBL/GenBank databases under the accession numbers AM946602–AM946620 and FM872474–FM872476.
Electronic supplementary material
Alignment data sets: MET1_assembly, CMT3_assembly, DRM_assembly: Multiple alignments of tobacco gene-space sequence reads, cDNA and protein sequences of N. sylvestris and N. tabacum MET1 genes (MET1_assembly), CMT3 genes (CMT3_assembly) and DRM genes (DRM_assembly). Files are in fasta format (tfa file extension) and in GCG format (rsf extension).
These files are unfortunately not in the Publisher's archive anymore:
-
Supplementary material 1 (RSF 157 kb)
-
Supplementary material 2 (TFA 148 kb)
-
Supplementary material 3 (RSF 109 kb)
-
Supplementary material 4 (TFA 104 kb)
-
Supplementary material 5 (RSF 276 kb)
-
Supplementary material 6 (TFA 262 kb)
Rights and permissions
About this article
Cite this article
Fulneček, J., Matyášek, R. & Kovařík, A. Faithful inheritance of cytosine methylation patterns in repeated sequences of the allotetraploid tobacco correlates with the expression of DNA methyltransferase gene families from both parental genomes. Mol Genet Genomics 281, 407–420 (2009). https://doi.org/10.1007/s00438-008-0420-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-008-0420-8