
Abstract
Radopholus similis is an important nematode pest on fruit crops in the tropics. Unraveling the transcriptome of this migratory plant-parasitic nematode can provide insight in the parasitism process and lead to more efficient control measures. For the first high throughput molecular characterization of this devastating nematode, 5,853 expressed sequence tags from a mixed stage population were generated. Adding 1,154 tags from the EST division of GenBank for subsequent analysis, resulted in a total of 7,007 ESTs, which represent approximately 3,200 genes. The mean G + C content of the nucleotides at the third codon position (GC3%) was calculated to be as high as 64.8%, the highest for nematodes reported to date. BLAST-searches resulted in about 70% of the clustered ESTs having homology to (DNA and protein) sequences from the GenBank database, whereas one-third of them did not match to any known sequence. Roughly 40% of these latter sequences are predicted to be coding, representing putative novel protein coding genes. Functional annotation of the sequences by GO annotation revealed the abundance of genes involved in reproduction and development, which reflects the nematode population biology. Genes with a role in the parasitism process are identified, as well as genes essential for nematode survival, providing information useful for parasite control. No evidence was found for the presence of trans-spliced leader sequences commonly occurring in nematodes, despite the use of various approaches. In conclusion, we found three different sources for the EST sequences: the majority has a nuclear origin, approximately 1% of the EST sequences are derived from the mitochondrial transcriptome, and interestingly, 1% of the tags are with high probability derived from Wolbachia, providing the first molecular indication for the presence of this endosymbiont in a plant-parasitic nematode.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To investigate the transcriptome of the plant-parasite Radopholus similis (the burrowing nematode), we explored newly generated and existing expressed sequence tags (ESTs). Generation of ESTs is a cost-effective method to generate large amounts of sequences, and is widely used as a first step in obtaining molecular data of a certain species. However, these sequences are typically of relatively low quality as numerous biases can be introduced along the whole process. Artifacts introduced in the first step, the construction of a complementary DNA (cDNA) library, include low occurrence of full length transcripts, exclusion of very short and very long transcripts, generation of chimeric constructs, and the inclusion of contaminating DNA or rRNA sequences (Nagaraj et al. 2007a). Furthermore, estimates of internal priming lies around 2–3% (Aaronson et al. 1996). The sequencing step starts with a random selection of clones from the cDNA library, followed by one-time sequencing of the inserts. The resulting EST sequences are contaminated with fragments of the vector and/or adaptors, and can contain up to 3% erroneous bases (Nishikawa and Nagai 1996). Contaminating sequences are usually removed before submission or additional analyses. Nowadays, a plethora of bioinformatic tools for EST analysis exists (Nagaraj et al. 2007b). Accompanied by a thorough understanding of the generation of EST sequences, these tools can deliver valuable information, which can serve different purposes: obtaining a first impression of the molecular composition of species (McCarter et al. 2003), identifying (tissue, developmental stage or organism specific) genes (Chen et al. 2006; Dubreuil et al. 2007), estimating the level of gene expression (digital northern) (Liu and Graber 2006; Munoz et al. 2004), annotating genome sequences (Blumenthal et al. 2002), and facilitating proteome analysis (Liu et al. 2006).
Radopholus similis is an obligate migratory plant-parasitic nematode (PPN), mainly occurring in subtropical regions. It parasitizes the roots of over 365 host plants, of which banana, plantain and citrus are economically the most important (O’Bannon 1977; Sarah et al. 1996). The nematode remains mobile throughout the whole life cycle (hence the adjective “migratory”), and every mobile stage can infect new roots, except the males which make up about 5% of a normal population. With the help of secreted proteins, originating from large pharyngeal gland cells and secreted through a hard hollow spear-like structure in the head (the stylet), the nematode penetrates the root and digests the cortex cells, resulting in large necrotic lesions in which secondary infections rapidly take place, mainly by Fusarium oxysporum and Rhizoctonia solani. The result of this nematode infection is stunting and wilting of the host plant or in severe cases even toppling due to the weakened stem base (known as the “blackhead-toppling disease” in bananas). These effects can cause massive losses in crop production ranging from 5 to 75% (O’Bannon 1977; Price 2006; Sarah et al. 1996). Once established in the field, it is very hard to nearly impossible to eradicate R. similis. Chemical control is extremely hazardous for the environment and most “nematicides” (e.g. methyl bromide) are banned for this reason (United Nations Environment Programme 1995). Since only very few resistant varieties are yet available, new sources of resistance are being sought (Elsen et al. 2004; Stoffelen et al. 2000; Wuyts et al. 2007). Recently, transgenic banana expressing cystatin was proven to posses some level of resistance to R. similis infection (Atkinson et al. 2004). To support the on-going research on this devastating plant-parasitic nematode, we present the generation and analysis of EST sequences from mixed stages of R. similis to gain a first insight into the transcriptome.
Materials and methods
Laboratory experiments
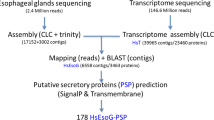
Radopholus similis was cultured at 25°C on carrot disks in parafilm sealed small petridishes (Jacob et al. 2007). Approximately 5,000 nematodes of mixed stages were collected in sterile demineralized water. After grinding of these nematodes in liquid nitrogen, RNA was extracted with TRIzol® Reagent (Invitrogen, Carlsbad, USA), precipitated with isopropanol and washed with 70% ethanol. The pellet was redissolved in diethylpyrocarbonate(DEPC)-treated demineralized water. Integrity of the RNA was checked by electrophoresis on a 0.5× TAE 1% agarose gel. Concentration was determined with the ND-1000 spectrophotometer (Nanodrop, Wilmington, DE, USA). This RNA served as a basis for cDNA library construction using the SMART™ cDNA Library Construction Kit, following the manufacturer’s instructions (Clontech, Mountain View, USA). The resulting fragments were directionally cloned in the pDNR-Lib vector provided by the kit. The R. similis mixed stage cDNA library contained over 105 primary transformants. Clones were sequenced using the M13 forward or reverse primer at the Genome Sequencing Center (GSC, Washington University, St Louis, USA). Sequences and quality files can be found on Nematode.net (Wylie et al. 2004), and sequences were submitted to the EST division of GenBank (dbEST, Boguski et al. 1993).
Cleaning and clustering
The sequences were cleaned using Seqclean (http://www.tigr.org) with a locally downloaded vector database and default parameter settings, to remove vector, poly(A) and short (<100 nt) sequences. Next, the dataset was clustered using TIGR Gene Indices Clustering Tool (TGICL) (Pertea et al. 2003), and assembled sequences were constructed by CAP3 (Huang and Madan 1999) using default settings, generating contigs (clustered ESTs) and singletons (non-clustered ESTs), commonly referred to as “unigenes” (ESTs used for clustering can be found in additional material file A5). Based on the clustering results, ESTstat was used to estimate the degree of fragmentation (Wang et al. 2006a). To gather ESTs with poly(A) sequences (and thus containing 3′ untranslated regions), cleaning by Seqclean was redone without poly(A) screening (option -A). The sequences differently cleaned compared to the first cleaning contain predicted poly(A) sequences.
BLAST-searches
The basic local alignment search tool (BLAST) analyses (Altschul et al. 1990) were performed both locally and via netblast. The BLASTx-results were parsed by an in-house perl script: for each hit, the species and phylogenetic classification was obtained from its GenBank file and used for subsequent classification of the unigene query (as nematode-, animal-, eukaryote-specific, etc.). R. similis unigenes were also used for BLASTx against C. elegans sequences (E value cut-off of 1e-05), and the top-hit sequences were used to estimate the degree of fragmentation (Mitreva et al. 2004). For this estimation, 384 of 1,632 unigenes with C. elegans hits share the same top-hit with one or more other unigenes. Of these unigenes, 221 were “redundant” as the C. elegans top-hit was already detected by another unigene(s), from which the fragmentation can be estimated. Further, all available nematode EST sequences were downloaded (June 2007) and searched locally with tBLASTx for homology to the unigenes of Radopholus similis. Of those tBLASTx hits, the developmental stage and nematode species used for the cDNA library construction were parsed from its GenBank file. A tBLASTx-search was performed with the R. similis unigenes as query against the coding mitochondrial sequences of all nematode species available in GenBank (September 2007). To address the nature of cluster 1, coding and non-coding classification was done for this cluster by ESTScan (Iseli et al. 1999) and RNAz (Washietl et al. 2005).
Translation
The FrameD gene prediction and translation program (Schiex et al. 2003) was trained by manually selected full length coding open reading frames (ORFs) from the set of unigenes, based on the BLASTx-results and Clustal W alignment with the corresponding most homologous sequences (Thompson et al. 1994) (ORFs of cluster 4, 5, 8, 11, 12, 13, 15, 18, 23, 25, 28, 31, 35, 41, 42 and 43). This set was extended with 7 full-length coding sequences of R. similis yet in GenBank (accession numbers AM691117.1, AM691118.1, EU190885, EF693940, EF693941, EF693942, EF693943), resulting in a total of 15,069 coding nucleotides. Using these sequences, an N-resistant Markov model was build on the website of FrameD (http://bioinfo.genopole-toulouse.prd.fr/apps/FrameD/FDM.pl). Predictions were analyzed for the occurrence and position of the coding sequence part of the unigene. Other unigenes which were predicted to be non-coding by FrameD, but with homologous proteins in other species according to BLASTx, were translated using Prot4EST trained with H. glycines sequences (Wasmuth and Blaxter 2004). Signal peptide prediction on this set of translations was done by SignalP 3.0 (Emanuelsson et al. 2007) and a signal peptide was only assigned if both the neural network and the hidden Markov model predicted a signal peptide. The signal peptide was cleaved from the translated sequence and subsequent transmembrane domain prediction was performed by TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) and SOSUI (Hirokawa et al. 1998). A protein was assigned to reside in the cell membrane, if both programs predicted a transmembrane domain.
Trans-spliced leader search
Searches were based on 70 spliced leader (SL) sequences reported in Guiliano and Blaxter (2006). In a first approach, a BLASTn search was set up with the SL sequences as query, a minimum matching length of 20, and a cut-off E value of 0.1. Using these parameters, a search was done in the R. similis unigene dataset. Since SLs have been shown to occur in the plant-parasitic nematode Meloidogyne incognita, a locally downloaded set of EST sequences of this nematode was used as a control (Guiliano and Blaxter 2006; McCarter et al. 2003). To minimize influence of a technical nature, we selected an EST set of M. incognita of a similar size as ours and generated by the SMART technology. The chosen control EST set has 3,098 ESTs sequenced from a SMART cDNA library, constructed from females of M. incognita (library “Meloidogyne incognita female SMART pGEM”). Since the BLASTn search yielded no results for the R. similis unigenes, a second approach was applied using a perl regular expression pattern, based on common features extracted from the SL sequences and the 5′ position of the SL in the unigene. The resulting pattern /^[AGCT]{0,30}GGT[^CG]{4,9}CCC[^C]\w{5,9}AG/ was used to search the SL sequences set, the R. similis unigenes (both strands) and the M. incognita ESTs (both strands). As a confirmation of the SL sequences found, the sequences of M. incognita containing a trans-spliced leader sequence, were used for a tBLASTx-search as a query (E value cut-off of 1e-35), simultaneously against C. elegans EST sequences (from dbEST) and against R. similis unigenes, containing full-length coding sequences and sequences with at least a 5′ UTR sequence part (based on the translation prediction, see “Translation”, and on the presence of a small piece (GGCCGGG) of the 5′ SMART primer). When highly similar ESTs (ranging from 60 to 93% identity on the protein level) in all datasets were found, the corresponding ESTs were aligned (on the DNA level) using Clustal W.
Gene ontology and KEGG biochemical pathway annotation
To map and annotate gene ontology (GO) terms, BLAST2GO was used (Conesa et al. 2005), with default parameters, except for an E value cut-off of 1e-05, maximum number of 30 BLAST hits, E value hit filter for annotation of 1e-05, the conversion of the annotation to GOSlim view, and a node scoring filter in the GO graph of 50 for biological process, 20 for molecular function and 20 for cellular component. Further, KOBAS was used to annotate KEGG biochemical pathways to the unigenes (Mao et al. 2005).
Annotating RNAi data to the unigenes
Using the RNAi data available of numerous C. elegans genes, we tried to assign an RNAi phenotype to the R. similis unigenes. A BLASTx-search revealed the top-hit C. elegans sequence for a unigene (using E value cut-off of 1e-05). Subsequently, the RNAi phenotype and GO terms (only of the C. elegans top-hits with observed RNAi phenotypes) were retrieved via WormMart (Schwarz et al. 2006) and the GO terms analyzed and visualized with WEGO (Ye et al. 2006).
Results
Dataset characteristics
A total of 5,853 new EST sequences were generated, having a slightly higher average sequence length compared to the ESTs of R. similis already deposited in the dbEST division of GenBank (Table 1). Analytical processing of both sets combined (removal of vector sequences, poly(A) tails and sequences <100 nt) resulted in 6,800 ESTs, and subsequent clustering (merging overlapping sequences together into “contigs”) established a 13% increase in sequence length. This final set of unigenes contains 1,008 contigs, grouped into 989 clusters (enclosing sequences with minor sequence variations), and 2,659 “singletons” (non-overlapping EST sequences). With growing cluster size (i.e. the number of ESTs contained in a cluster), the number of clusters decreases logarithmically (Fig. 1). A certain degree of “fragmentation”—also called underclustering—could be expected in our final dataset and was estimated by ESTstat to be as high as 15.8% (Wang et al. 2006a). Another method described by Mitreva et al. (2004), resulted in a comparable estimation of 12.9%. Due to this fragmentation error, our dataset represents at most 3,194 genes, which is approximately 16% of the total gene number, if assumed similar as in Caenorhabditis (Stein et al. 2003).

Graphical representation of the cluster size distribution. Y axis The number of clusters. X axis cluster size (i.e. the number of ESTs represented by the cluster)
BLASTx analysis
A BLASTx-search against the GenBank non-redundant protein sequences resulted in hits for 2,130 unigenes (58.1% of the total unigene set). Of these, 1,710 (46.6%) had an E value lower than 1e-05 (more significant match), whereas the remaining 420 sequences (11.5%) were only retained with higher E values (between 1e-05 and 1e-01). Ribosomal proteins were of the most abundant top-hits (n = 176 or 4.8% of the unigenes). 535 unigenes (14.6%) matched sequences originating from both eukaryotes and prokaryotes, and 622 unigenes (17.0%) matched solely to sequences from all major eukaryotic lineages. The wide occurrence of these unigenes suggests a role in basal cell metabolism. Surprisingly, 14 unigenes gave a plant-specific hit. Since R. similis was cultured on carrot disks (Daucus carota), the presence of contaminating plant tissue can explain these sequences, although the top-hit sequences originated from different plant species (with an E value range between 1e-10 and 1e-01). Of the remaining unigenes with a BLASTx-hit, 428 exclusively matched animal sequences (11.8%), of which 328 (8.9%) were nematode-specific (see Fig. 2). Seven of the nematode-specific unigenes were found to match exclusively sequences of plant-parasitic nematodes, and 8 matched both plant- and animal-parasitic nematode sequences (see Table 2). Special attention was paid to the largest clusters, as they correspond most likely to highly expressed genes in R. similis. The BLASTx-results of the largest clusters are reported in Table 3: commonly known highly expressed genes are found (such as actin, sec-2), but some pioneer sequences are present as well. A considerable subset of our unigenes (n = 1,537; 41.9%) gave no BLASTx-hits with an E value cut-off set as high as 1e-01. One striking feature of these unigenes is their shorter average sequence length (354 ± 166 nt) compared to the unigenes with hits (518 ± 185 nt) (P value two sample t test <0.001) (Fig. 4). For many of the unigenes the short sequence length is the cause for missing BLASTx hits, as their E values will not reach the preset threshold.

Overview of the EST analysis (colors are visible in the on-line version). a Graph of density lines of the G + C content of different unigene subsets; b Length distributions of different subsets of unigenes, that are represented in c; c Classification of unigenes with BLASTx homology (E value cut-off of 1e-1; blue), with indication of the portion of unigenes with nematode-specific hits (“nema specific”, matches to nematode proteins only), animal-specific hits (matches to animal proteins only), plant-specific hit (“plant sp.”; matches to plant proteins only), eukaryote specific hits (matches to eukaryote proteins only), prokaryote-specific hits (“prokaryote sp.”; matches to prokaryote proteins only) and unigenes with hits to pro- and eukaryote sequences (“general”). The orange part marked with “tBLASTx” indicates unigenes without BLASTx-hits, but with tBLASTx-hits in nematode ESTs. The gray part marked with “orphans” indicates the subset of unigenes without BLASTx- or tBLASTx-homology. The inner black and white circle correspond to coding (black) and non-coding (white) prediction based on FrameD and BLASTx-homology. The small portion of mitochondrial and Wolbachia-derived sequences are indicated in the circle respectively as violet and dark-blue
Homologues in nematode EST sequences
To find homologues in the transcriptional data of other nematodes (ESTs of all nematodes except R. similis), a tBLASTx-search was performed with the unigenes (E value cut-off of 1e-05). The E value cut-off for the tBLASTx-search was set lower than the E value cut-off for the BLASTx-search (i.e. 1e-05, compared to 1e-01), since for the majority of the unigenes a consistently lower top-hit E value with tBLASTx (i.e. more significant) was found compared to BLASTx (see additional material figure A1). This tBLASTx-search reported 2,305 hits to nematode EST sequence, of which 560 unigenes (15.3%) with homologous EST sequences exclusively in plant-parasitic nematodes (PPN), 106 unigenes (2.9%) exclusively in animal-parasitic nematodes (APN), and 147 (4.0%) unigenes exclusively in both APN and PPN. As seen for the sequences with and without BLASTx-hit, a similar difference in sequence length could still be observed between unigenes with and without tBLASTx-hit: unigenes having homologues in the nematode ESTs are generally longer (408 ± 169 nt), compared to those without homologous counterparts (344 ± 163 nt). The persistence of this difference in sequence length, points to an important influence of sequence length in finding homologues based on BLAST-searches, arguing for a thorough quality check of the used cDNA library. Furthermore, the tBLASTx-search revealed a large portion of the unigenes (n = 408 or 11.1%) without BLASTx homology to known proteins, but with homology to EST sequences of other nematodes (Fig. 2). Notably the majority of the unigenes with homology to PPN EST sequences lack a BLASTx-hit (367 of 560 unigenes, or 65.5%). Despite the efforts to identify unigenes on basis of homology using BLAST-searches, unigenes without hits (either BLASTx or tBLASTx; the so called “orphans”) constitute still a large portion (n = 1,128 or 30.8%, see Fig. 2).
Annotation of the unigenes
Annotation of “gene ontology” (GO) terms helps to categorize unigenes based on their putative function. We used the user-friendly BLAST2GO program to explore the R. similis unigene data set (Conesa et al. 2005). This annotation method is based on sequence homology determined by BLAST-searches. For 1,920 unigenes BLAST2GO could not find a homologous sequence and no mapping could be retrieved for 259 sequences with BLAST homology. Finally, after mapping a total of 5,501 GO terms to the unigenes, 812 sequences (22%) were successfully annotated, with a higher success rate as the sequences get longer (see Fig. 3). Analyzing the main GO category “biological process” and its child terms, revealed that “embryonic development”, “growth” and “reproduction” GO terms are the most represented, followed by terms involved in basal cell metabolism based on annotation scores assigned by BLAST2GO (see Fig. 3). This can be a reflection of the high reproductive rate of R. similis and the high percentage of females (and developing eggs) in the population used for cDNA library construction. In the main GO category “molecular function”, the “protein binding” term is most represented (∼38% of the terms), followed by “structural molecule activity” and “RNA binding”. Many unigenes encoding ribosomal proteins are assigned to the “protein binding” term, and also highly expressed genes coding for structural molecules (such as actin) and regulatory molecules (such as transcription factors). Since those unigenes are abundantly present in the dataset, this causes the overrepresentation of the “protein binding” term. Regarding the main GO category “cellular component”, the term “ribosome” is most represented, constituting together with the term “cytosol” almost half of the total terms. Since the nematode secretes a cocktail of proteins into the plant to control the parasitism process, interesting sequences are supposed to be found under the term “extracellular region”. However, the GO terms are assigned using homology to known annotated sequences and scarcely any parasitism gene of plant-parasitic nematodes has GO terms assigned to date; consequently most parasitism genes are likely not annotated. Furthermore, the “extracellular region” term encompasses also abundantly expressed genes coding for proteins secreted by the gut, epidermis and the nervous system. The answer to parasitism gene annotation may come from a more fine-tuned GO classification adapted to parasitism, since many parasitism genes are not easily classified in the present classification (Berriman et al. 2001). In summary, the GO annotation of the unigenes is a representation of the biology of R. similis and the characteristics of the cDNA library, and is rather unsuited to detect gene expression correlated with parasitism.

Summary of the Gene ontology annotation, results by Blast2GO; a Comparison of length distribution of all unigenes (black line) with successfully GO annotated unigenes (gray surface); b Most represented GO terms (based on annotation score) of the main category “biological process”; c Most represented GO terms of the main category “cellular component”; d Most represented GO terms of the main category “molecular function”
Assigning RNAi phenotypes
Exploring the RNAi phenotypes in the R. similis unigene dataset can lead to potential control strategies based on disrupting gene expression. We made use of the RNAi phenotypic data available for C. elegans. Using BLASTx (E value cut-off of 1e-05), 1,638 unigenes were found to have a homologous C. elegans gene. Of those C. elegans genes, 659 have a detectable RNAi phenotype. Comparing the GO term distribution of genes with RNAi phenotypes to the complete GO term distribution (see “Annotation of unigenes”) revealed marked changes. Genes involved in “biological regulation” (GO:0065007, main GO category “biological process”), are enriched from 1% in the complete GO annotation to 10% in the GO annotation of genes with RNAi phenotypes, at the cost of general biological processes, such as metabolic (GO:0008152) and cellular process terms (GO:0009987) (from 19 to 10% and 18 to 12% respectively). Similarly, in the “cellular component” GO terms, an increase is seen in “macromolecular complex” (GO:0032991) (from 7 to 13%) and “organelle part” (GO:0044422) (from 3 to 8%), while the largest decreases are for “organelle” (GO:0043226) (from 22 to 18%) and “extracellular region” (GO: GO:0005576) (from 2 to 0.5%). The GO term distribution of “molecular function” does not show any remarkable difference. Three quarter of the RNAi phenotypes (n = 506, 76.8%) report a lethal effect. Compared to the total gene set with RNAi phenotypes, the genes with lethal RNAi phenotypes are significantly enriched in the GO terms “macromolecular complex” (GO:0032991), “developmental process” (GO:0032502), “growth” (GO:0040007) and “multicellular organismal process” (GO:0032501) (see Fig. 4). On the other hand, some GO terms, such as “enzyme regulator” (GO:0030234), “molecular transducer” (GO:0060089), and “cell surface receptor linked signal transduction” (GO:0007166), are depleted in the unigenes with lethal RNAi phenotypes. While disruption of gene expression of these genes is expected to have a less profound influence on nematode survival, the strongest effects on nematode survival are expected when targeting genes involved in developmental processes.

GO terms of the genes with predicted RNAi phenotypes. Black bars represent the total set; gray bars represent the subset with predicted lethal RNAi phenotypes. Only the GO categories (up to level 5) which differ significantly between the two sets (Pearson Chi square test P value <0.05) are represented (analysis and output by WEGO)
Translation of unigenes
It was reported before that coding DNA sequences of R. similis are GC-rich. This is mainly the result of the high mean G + C content of the nucleotides at the third position of each codon (GC3%), which has been previously estimated around 63% (Cutter et al. 2006; Haegeman et al. 2008). This feature poses a potential problem for translation of the DNA sequences using standard sequence translation rules. This is due to the fact that every stop codon starts with a uracil (U) (UAG, UGA and UAA). A high GC3% means that most of the nucleotides at the last position of the codons are G or C. Hence the reverse complementary codons start mostly with G or C, reducing the likelihood to encounter a stop codon. It was noticed that translation based on the longest open reading frame (ORF) will therefore frequently result in translating the wrong strand. The translation prediction program FrameD (which uses a Markov model) is especially build for dealing with a high GC3%: it predicts coding regions and corrects frame-shifts to obtain reliable translations, based on a set of training sequences (Schiex et al. 2003). We trained FrameD with 15 kb of coding nucleotides from manually collected full length ORFs based on BLASTx-results and cloned R. similis genes submitted to GenBank. Testing the performance of FrameD, it classified all 226 unigenes containing 3′ untranslated regions (selected by the presence of a poly(A) tail and polyadenylation signal) as non-coding, indicative of a low false positive error. Consequently, the false negative error is rather high, since FrameD classified only 77% unigenes (1,317 of 1,710) with BLASTx-hits as coding. To correct for this error, the unigenes with at least one BLASTx-hit, but lacking a FrameD translation, were translated with the Prot4EST translation pipeline (Wasmuth and Blaxter 2004). On the total unigene set, FrameD classified 2,245 (61.2%) unigenes as coding, with detection and correction of frame shifts in 6.4% of the translations. Of the remaining unigenes, 552 sequences had at least one BLASTx hit, and were subsequently translated by Prot4EST. In this way, the total number of protein coding unigenes reaches 2,797, or 76.3% of the unigene dataset (Table 1). Calculation of the G + C content of the coding part of the unigenes resulted in an overall GC-percentage of 53.7%, a GC1% of 55.5%, a GC2% of 40.7% and a GC3% of 64.8% (see additional material figure A2). Searching signal peptides for secretion in the translations revealed that 216 of the 2,755 translations (7.8%) were predicted to contain a signal peptide, of which 156 (5.6% of the translations) lacked a transmembrane region. Based on these predictions, we identified 4.3% of the unigenes coding for secreted proteins. Remarkably, the translation prediction showed that 447 orphan sequences were predicted to be coding (39.6% of the orphans). These interesting unigenes are in all probability novel protein coding genes, without any known homologue in the database to date. But on the other hand and equally remarkable, a large fraction of the unigene sequences (at most 682 sequences or 18.6%) is predicted not to be protein-coding, pointing to the existence of EST sequences derived from non-coding RNA.
Trans-spliced leaders
One of the major eccentricities in the molecular biology of nematodes is the widespread occurrence of operons and trans-spliced leader sequences, frequently found at the start of transcripts (Guiliano and Blaxter 2006). To investigate the occurrence of the presently known trans-spliced leader sequences in our unigenes, searches were performed based on common features of a reference set of 70 spliced leader (SL) sequences reported in Guiliano and Blaxter (2006). Surprisingly, in the first approach using BLASTn-searches, none of the R. similis unigenes in our dataset matched to any SL sequence, whereas in a control dataset of Meloidogyne incognita ESTs (in which trans-splicing is known to occur), 293 sequences matched to a total of nine different SL sequences of our reference set. After this negative result, a second approach was applied using a pattern, which was able to match 76% of the reference SL sequences. But likewise, this pattern was unable to match any R. similis unigene. On the contrary, this pattern found motifs in 12.2% of the control data set of M. incognita ESTs (376 out of 3,098 ESTs). To exclude the possibility that by chance none of our unigenes belongs to a nematode gene family which is trans-spliced, we searched very strong homologues of the trans-spliced M. incognita ESTs (detected by the pattern search) in the R. similis unigenes and in C. elegans ESTs. Five R. similis unigenes with complete 5′ UTRs (CL13, CL18, CL25, CL112 and CL929) were found having very strong homology to trans-spliced M. incognita ESTs (ranging from 60 to 93% identity on a protein level). For each of the five cases, the unigene and the trans-spliced M. incognita EST were aligned (on a DNA level), together with the strongest C. elegans homologue. In all the five cases, this alignment revealed the occurrence of trans-spliced leader sequences on both the M. incognita EST and C. elegans gene, but absence of such sequences in the 5′ UTR region of the R. similis unigene (see additional material figure A3). Closer inspection of the 5′ UTR sequences of the five R. similis unigenes could not reveal any sequence similarity, neither with the M. incognita or C. elegans homologues, nor with each other.
Occurrence of mitochondrial ESTs
Depending on the cDNA library construction method, a remarkably high fraction of ESTs can be of mitochondrial origin and these EST can even be used as a guideline for the sequencing of the mitochondrial genome (Gissi and Pesole 2003). We searched our unigene dataset for ESTs most likely derived from the mitochondrial genome, and found five unigenes with significant similarity to various nematode mitochondrial genes (see Table 4). The G + C content of those sequences was 16.4%, very low compared to the mean G + C content of the total unigene set (48.8%; see Fig. 1). This low G + C content is a major characteristic of mitochondrial genomes, in most cases between 20 and 30% (He et al. 2005). The different G + C content of the nuclear unigenes compared to the mitochondrial unigenes could therefore be used to predict the mitochondrial origin of the EST. The G + C content density line of the total unigene dataset follows a normal distribution, pointing to a similar source of the unigenes (see additional material figure A4). However, a bias from the normal distribution is observed at one end of the curve, caused by 21 unigenes with a lower G + C content (from 10 to 26%), most likely all originating from the mitochondrial genome (summarized in Table 4). To refute the possibility of dealing with numts [nuclear insertions of mitochondrial sequences into the nuclear genome (Richly and Leister 2004)], and as part of an ongoing project to sequence the mitochondrial genome of R. similis, primers were designed on two putative mitochondrial unigenes. One primer was based on the nad5 homologue (CL86) and another on coxI (CL429). Via long distance PCR we succeeded in amplifying a piece of 3,396 nt of the mitochondrial genome of R. similis (data not shown). The average G + C content of the amplicon was 14.40% and contained the complete sequence of another mitochondrial unigene (CL21, a coxII homologue). The results of the complete sequencing will be written down in another manuscript. Interestingly, these results will probably shed light onto the origin of the largest cluster in our dataset (cluster 1). This cluster represents a disproportionate large amount (n = 575, or 8%) of the total number of ESTs. Despite this, it shows no significant homology to any known sequence using various BLAST approaches against various databases, and is also predicted to be not coding by different coding sequence prediction programs. Using cluster 1 specific primers, we succeeded in amplifying this unigene from a cDNA pool constructed from DNAse treated RNA, making genomic DNA contamination very unlikely (data not shown). In addition, genomic contamination (such as numts) is supposed to appear as singletons and not as a cluster. The only feature of this sequence that can give us a clue about its origin, is its low G + C content (16.3%, see Table 4). Based on the available mitochondrial genomic data, it is possible that cluster 1 is derived from the mitochondrial genome. Furthermore, in clustered EST datasets of other plant-parasitic nematodes (Bursaphelenchus and Pratylenchus), the largest clusters also represent a disproportionate large part of the EST dataset (Kikuchi et al. 2007; Mitreva et al. 2004). Notably, in these cases homology was found to mitochondrial genes, but certainty for cluster 1 of R. similis will only be achieved when the complete sequence of the R. similis mitochondrial genome is known.
Unigenes with similarity to Wolbachia sequences
A subset of 43 unigenes (2%) had homology exclusively to prokaryotic sequences. Although the possibility exists that some of these sequences are the result of contamination, 18 significant matches to genes of the endosymbiotic Wolbachia species are found. Further investigation of all the unigenes (having homologues not limited to prokaryotic species) revealed another 12 unigenes with BLASTx top-hits to Wolbachia (see Table 5). The mean G + C content for these 30 sequences is 36.9% (±4.2%), similar to previously reported G + C percentages of Wolbachia sequences (Foster et al. 2005). The high similarity to known Wolbachia genes indicates that the corresponding unigenes are genuine Wolbachia derived transcripts. Consequently, the discovery of these sequences suggests an endosymbiotic presence of Wolbachia within R. similis.
Unigenes putatively involved in parasitism
Some unigenes are involved in the parasitism process, based on homology to genes of other parasitic species (see Table 2). Two unigenes coding for plant cell wall degrading enzymes were found: an endoglucanase (which was cloned and characterized by Haegeman et al. (2008)) and a xylanase. Both enzymes soften the plant tissue to facilitate the intracellular migration of the nematode. Those cell-wall degrading enzymes have been identified in numerous plant-parasitic nematodes (Ledger et al. 2006; Smant et al. 1998) and these were extensively studied in the light of parasitism. Other unigenes putatively encode enzymes that can neutralize reactive oxygen (ROS) species, produced by the host as a defense mechanism in response to infection by the nematode (Dubreuil et al. 2007; Jones et al. 2004; Robertson et al. 2000). Further, three unigenes show homology to fatty acid- and retinoid-binding proteins of parasitic nematodes. Fatty acids are compounds that play a role in the host defense-signaling pathway (Kennedy et al. 1995; Prior et al. 2001). As a consequence, nematode proteins that bind such compounds could modulate the host defense to facilitate parasitism. One unigene of this group resembles a gene of the animal-parasitic nematode Brugia malayi (Kennedy et al. 1995). The other two unigenes (CL12contig1 and CL70contig1) show highest homology to SEC-2 proteins (also called FAR, fatty-acid and retinol-binding) of the PPN Globodera pallida. However, where CL12contig1 shows also homology to a (hypothetical) protein of the free-living nematode C. elegans, CL70contig1 has only homology to SEC-2 proteins of parasitic nematodes. This could point to the existence of functionally distinct SEC-2 proteins, one with a general function and another with a function related to parasitism, as hypothesized previously (Garofalo et al. 2003; Prior et al. 2001). Two unigenes have homology to SXP/RAL-2 genes, whose gene products most likely play role in host localization (Prior et al. 2001; Tytgat et al. 2005). In addition to those functionally known genes, some unigenes showed homology to putative parasitism genes without known function. To further identify additional unknown parasitism gene candidates, we searched in the 447 orphan sequences predicted to be protein coding for homology exclusively to ESTs of plant-parasitic nematodes (PPN). A total of 212 such unigenes (5.8%) were retrieved, of which 18 (1.3%) were predicted to encode secreted proteins. Five of them (CL26, CL546, CL793, 92h12, 23g06) were assigned good candidate parasitism genes, since the homologous EST sequences originated exclusively from the parasitic stages of PPN (i.e. second stage juvenile to adults). Future experiments could confirm their putative role in the parasitism process.
Discussion
With the generation of thousands of new EST sequences from mixed stages of the plant-parasitic nematode R. similis, interesting research topics are introduced. Based on our analysis, the R. similis ESTs are derived from three different sources. The majority of the ESTs are derived from the nuclear genome. A small fraction (∼0.6%) has most likely a mitochondrial origin, corresponding to sequences with a very low G + C content (∼16% G + C). Finally, a third subset (∼1%) seems to be derived from a Wolbachia species. To our knowledge, this obligate intracellular endosymbiont is only reported in arthropod species and a few filarial nematode species (Hise et al. 2004; Kramer et al. 2003; Taylor et al. 1999). In these nematode species, Wolbachia seems to be required for successful molting as well as for reproduction of the nematode. In only three genera of plant-parasitic nematodes (Heterodera, Globodera and Xiphinema), bacteria-like endosymbionts—other than Wolbachia—have been found (Noel and Atibalentja 2006; Vandekerckhove et al. 2002). On the other hand, we can not exclude the possibility of an insertion of Wolbachia genes into the genome of R. similis, as recently has been shown that these inserts can be transcriptionally active (Hotopp et al. 2007).
The majority of the unigenes are derived from the nuclear genome of R. similis. Approximately one-third of the unigenes code for proteins involved in general metabolic pathways. Other classifications based on BLASTx-results can be found in Fig. 2, but can slightly change in the future as more sequence data become available. Besides the unigenes with clear homology, a relatively large part of our unigene dataset (30.8%) lacked homology to any sequence in the database to date (called “orphan” sequences). Multiple explanations can be found for these orphans: (1) the most “preferred” one is that the unigene represents a genuine novel protein-coding gene (estimated to be 12.1% of the unigenes). However, (2) the length of the unigene also plays a role, as a correlation exists between the length of a unigene and its homology significance level. Thus significant homology can simply not be detected if the sequence is too short. Alternatively, (3) unigenes containing mainly untranslated region (UTR) will most likely lack homology, as UTRs are the most diverse regions of transcripts (McCarter et al. 2003). Moreover, (4) for unigenes derived from (non-coding) contaminating DNA, most likely no significant homology will be detected. Finally it is possible (5) that some unigenes of the orphans correspond to regulatory non-coding RNAs rather than mRNA, since evidence is accumulating on the ubiquitous role of these non-coding RNAs on translational regulation. Recent estimates in humans state that at least 20% of the genes are regulated by over 1,000 miRNAs (Bentwich et al. 2005; Lim et al. 2005) and in the model nematode C. elegans, 112 miRNA genes have been identified so far (Ruby et al. 2006). One of the most intriguing unigenes lacking homology to any known sequence is notably the largest cluster in our dataset, representing about 8% of the ESTs. In fact it is frequently reported in the literature that the largest clusters in EST analyses contain a disproportionate large number of ESTs [e.g. the largest cluster contained 10% of the ESTs in Mitreva et al. (2004), and 4.7% in Ranganathan et al. (2007)]. Often these oversized clusters do not show homology to any known sequence (Dubreuil et al. 2007; Ranganathan et al. 2007). Unfortunately, not many attempts have been undertaken to clarify this. Based on preliminary sequence data of mitochondrial genome of R. similis, we suggest that cluster 1 most likely has a mitochondrial origin. If so, it should be a part of a transcriptionally active region with a high expression level.
The translation of the unigenes revealed a very high GC3 percentage of 63.4%, while the overall G + C content of the unigenes was approximately 54%. Analysis of genome sequences lead to the thermodynamic stability hypothesis to explain observed differences in G + C content. It states that G + C content is correlated with the optimal growth temperature of the organism (in case of bacteria) or the optimal body temperature (in case of vertebrates) (Jabbari and Bernardi 2004). However, this hypothesis could not be confirmed by different other studies, pointing other unknown more complex grounds for the different G + C content between organisms (Basak and Ghosh 2005; Belle et al. 2002; Wang et al. 2006b). Although at first sight the thermodynamic stability hypothesis could apply to the G + C content of R. similis unigenes, other tropical nematodes (such as Meloidogyne species) have a clearly lower G + C content compared to nematodes occurring in moderate climates such as Heterodera and Caenorhabditis (Mitreva et al. 2006, Table 6).
Another remarkable result from this analysis is the impossibility—despite the use of various approaches—to extract sequence fragments from the unigene dataset that resemble trans-spliced leader sequences, known to occur in other nematode species, such as M. incognita and C. elegans. This could indicate that R. similis makes no use of trans-splicing. This is in conflict with the statement that trans-splicing widely occurs throughout the phylum Nematoda. Therefore it is possible that R. similis makes use of a different set of spliced leader sequences, although our attempts to detect these sequences were unsuccessful. The lack of known trans-spliced leader sequences could also explain the difficulties encountered when constructing an oligo(dT)-SL1 PCR based cDNA library of R. similis. This library turned out to have a rather low number of primary transformants, which can be due to the unsuccessful amplification step. Remarkably, a preliminary search for known trans-spliced leaders in the EST data from Heterodera glycines also gave a negative result (data not shown), arguing for a thorough investigation to validate the systematical occurrence of trans-spliced leader sequences (known and unknown) throughout the phylum Nematoda. Some comparisons between R. similis and three other nematode species can be found in Table 6.
Because of the importance of R. similis as a major pest, the EST data can deliver information on the parasitism process as well as on potential control strategies based on disrupting gene expression. Unigenes involved in the plant-parasitic life style of R. similis were found through homology with genes of parasitic species, with known and unknown function. Searching for unigenes coding for secreted proteins has proven a useful approach to identify parasitism genes used by the nematode. It is assumed that the majority of the parasitism proteins are secreted into the host to modulate the nematode’s environment. Most of them originate from the pharyngeal glands and are injected in the plant tissue through the stylet, a hollow needle-like structure in the head of the nematode. A better understanding of the parasitism process will come from identification and characterization of the parasitism genes. In this respect, it is remarkable that the majority of the R. similis unigenes with homology exclusively to PPN EST sequences lack a BLASTx-hit, indicative of a high potential for discovery of novel genes in PPN EST sequences. Besides the elucidation of the parasitism process as step-stone to parasite control, GO and RNAi-phenotype data analysis suggest that suitable targets for controlling R. similis may also be found among genes involved in the regulation of developmental processes. Experiments with RNAi show that viability of nematodes can be severely affected when essential nematode genes are silenced (Kamath et al. 2003). A promising technique in this respect is the in planta generation of nematode specific double stranded RNA, leading to a decreased viability of the nematode when it ingests the inferring RNA molecules (Bakhetia et al. 2005; Gheysen and Vanholme 2007). Therefore, the sequences delivered by this EST project can aid in various ways to establish efficient parasite control.
Abbreviations
- cDNA:
-
Complementary DNA
- EST:
-
Expressed sequence tag
- ORF:
-
Open reading frame
- GO:
-
Gene ontology
- mRNA:
-
Messenger RNA
- aa:
-
Amino acid
- PPN:
-
Plant-parasitic nematode
- APN:
-
Animal-parasitic nematode
- FLN:
-
Free-living nematode
References
Aaronson JS, Eckman B, Blevins RA, Borkowski JA, Myerson J, Imran S, Elliston KO (1996) Toward the development of a gene index to the human genome: an assessment of the nature of high-throughput EST sequence data. Genome Res 6:829–845
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Atkinson H, Grimwood S, Johnston K, Green J (2004) Prototype demonstration of transgenic resistance to the nematode Radopholus similis conferred on banana by a cystatin. Transgenic Res 13:135–142
Bakhetia M, Charlton WL, Urwin PE, McPherson MJ, Atkinson HJ (2005) RNA interference and plant parasitic nematodes. Trends Plant Sci 10:362–367
Basak S, Ghosh TC (2005) On the origin of genomic adaptation at high temperature for prokaryotic organisms. Biochem Biophys Res Commun 330:629–632
Belle EM, Smith N, Eyre-Walker A (2002) Analysis of the phylogenetic distribution of isochores in vertebrates and a test of the thermal stability hypothesis. J Mol Evol 55:256–363
Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z (2005) Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 37:766–770
Berriman M, Aslett M, Hall N, Ivens A (2001) Parasites are GO. Trends Parasitol 17:463–464
Blumenthal T, Evans D, Link CD, Guffanti A, Lawson D, Thierry-Mieg J, Thierry-Mieg D, Chiu WL, Duke K, Kiraly M, Kim SK (2002) A global analysis of Caenorhabditis elegans operons. Nature 417:851–854
Boguski MS, Lowe TMJ, Tolstoshev CM (1993) dbEST - database for “expressed sequence tags”. Nat Genet 4:332–333
Chen W-H, Wang X-X, Lin W, He X-W, Wu Z-Q, Lin Y, Hu S-N, Wang X-N (2006) Analysis of 10,000 ESTs from lymphocytes of the cynomolgus monkey to improve our understanding of its immune system. BMC Genomics 7:82. doi:10.1186/1471-2164-7-82
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Cutter AD, Wasmuth JD, Blaxter ML (2006) The evolution of biased codon and amino acid usage in nematode genomes. Mol Biol Evol 23:2303–2315
Dubreuil G, Magliano M, Deleury E, Abad P, Rosso MN (2007) Transcriptome analysis of root-knot nematode functions induced in the early stages of parasitism. New Phytol 176:426–436
Elsen A, Jain SM, Swennen R, De Waele D (2004) Recent developments in early in vitro screening for resistance against migratory endoparasitic nematodes in Musa. Banana improvement: cellular, molecular biology, and induced mutations. Proceedings of a meeting held in Leuven, Belgium, 24–28 September 2001, Science publishers, Inc
Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP, and related tools. Nat Protoc 2:953–971
Foster J, Ganatra M, Kama I, Ware J, Makarova K, Ivanova N, Bhattacharyya A, Kapatral V, Kumar S, Posfai J, Vincze T, Ingram J, Moran L, Lapidus A, Omelchenko M, Kyrpides N, Ghedin E, Wang S, Goltsman E, Joukov V, Ostrovskaya O, Tsukerman K, Mazur M, Comb D, Koonin E, Slatko B (2005) The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLOS Biol 3:0599–0614
Garofalo A, Kennedy MW, Bradley JE (2003) The FAR proteins of parasitic nematodes: their possible involvement in the pathogenesis of infection and the use of Caenorhabditis elegans as a model system to evaluate their function. Med Microbiol Immunol 192:47–52
Gheysen G, Vanholme B (2007) RNAi from plants to nematodes. Trends Biotechnol 25:89–92
Gissi C, Pesole G (2003) Transcript mapping and genome annotation of Ascidian mtDNA using EST data. Genome Res 13:2203–2212
Guiliano DB, Blaxter ML (2006) Operon conservation and the evolution of trans-splicing in the phylum Nematoda. PLoS Genet 2:e198. doi:10.1371/journal.pgen.0020198
Haegeman A, Jacob J, Vanholme B, Kyndt T, Gheysen G (2008) A family of GHF5 endo-1,4-beta-glucanases in the migratory plant-parasitic nematode Radopholus similis. Plant Pathol (in press). doi:10.1111/j.1365-3059.2007.01814.x
Hammond MP, Bianco AE (1992) Genes and genomes of parasitic nematodes. Parasitol Today 8:299–305
He Y, Jones J, Armstrong M, Lamberti F, Moens M (2005) The mitochondrial genome of Xiphinema americanum sensu stricto (Nematoda: Enoplea): considerable economization in the length and structural features of encoded genes. J Mol Evol 61:819–833
Hirokawa T, Boon-Chieng S, Mitaku S (1998) SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379
Hise AG, Gillette-Ferguson I, Pearlman E (2004) The role of endosymbiotic Wolbachia bacteria in filarial disease. Cell Microbiol 6:97–104
Hotopp JCD, Clark ME, Oliveira DCSG, Foster JM, Fischer P, Torres MCM, Giebel JD, Kumar N, Ishmael N, Wang S, Ingram J, Nene RV, Shepard J, Tomkins J, Richards S, Spiro DJ, Ghedin E, Slatko BE, Tettelin H, Werren JH (2007) Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science 317:1753–1756
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9:868–877
Iseli C, Jongeneel V, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol 138–148
Jabbari K, Bernardi G (2004) Body temperature and evolutionary genomics of vertebrates: a lesson from the genomes of Takifugu rubripes and Tetraodon nigroviridis. Gene 333:179–181
Jacob J, Vanholme B, Haegeman A, Gheysen G (2007) Four transthyretin-like genes of the migratory plant-parasitic nematode Radopholus similis: members of an extensive nematode-specific family. Gene 402:9–19
Jones JT, Reavy B, Smant G, Prior AE (2004) Glutathione peroxidases of the potato cyst nematode Globodera rostochiensis. Gene 324:47–54
Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421:231–237
Kennedy MW, Allen JE, Wright AS, McCruden AB, Cooper A (1995) The gp15/400 polyprotein antigen of Brugia malayi binds fatty acids and retinoids. Mol Biochem Parasitol 71:41–50
Kikuchi T, Aikawa T, Kosaka H, Pritchard L, Ogura N, Jones JT (2007) Expressed sequence tag (EST) analysis of the pine wood nematode Bursaphelenchus xylophilus and B. mucronatus. Mol Biochem Parasitol 115:9–17
Kramer L, Passeri B, Corona S, Simoncini L, Casiraghi M (2003) Immunohistochemical/immunogold detection and distribution of the endosymbiont Wolbachia of Dirofilaria immitis and Brugia pahangi using a polyclonal antiserum raised against WSP (Wolbachia surface protein). Parasitol Res 89:381–386
Ledger TN, Jaubert S, Bosselut N, Abad P, Rosso M-N (2006) Characterization of a new beta-1,4-endoglucanase gene from the root-knot nematode Meloidogyne incognita and evolutionary scheme for phytonematode family 5 glycosyl hydrolases. Gene 382:121–128
Leroy S, Bouamer S, Morand S, Fargette M (2007) Genome size of plant-parasitic nematodes. Nematology 9:449–450
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM (2005) Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433:769–773
Liu D, Graber J (2006) Quantitative comparison of EST libraries requires compensation for systematic biases in cDNA generation. BMC Bioinformatics 7:77. doi:10.1186/1471-2105-7-77
Liu F, Lu J, Hu W, Wang SY, Cui SJ, Chi M, Yan Q, Wang XR, Song HD, Xu XN, Wang JJ, Zhang XL, Zhang X, Wang ZQ, Xue CL, Brindley PJ, McManus DP, Yang PY, Feng Z, Chen Z, Han ZG (2006) New perspectives on host-parasite interplay by comparative transcriptomic and proteomic analyses of Schistosoma japonicum. PLoS Pathog 2:e29. doi:10.1371/journal.ppat.0020029
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21:3787–3793
McCarter JP, Mitreva MD, Martin J, Dante M, Wylie T, Rao U, Pape D, Bowers Y, Theising B, Murphy CV, Kloek AP, Chiapelli BJ, Clifton SW, Bird DM, Waterston RH (2003) Analysis and functional classification of transcripts from the nematode Meloidogyne incognita. Genome Biol 4:R26. doi:10.1186/gb-2003-4-4-r26
Mitreva M, Elling AA, Dante M, Kloek AP, Kalyanaraman A, Aluru S, Clifton SW, Bird DM, Baum TJ, McCarter JP (2004) A survey of SL1-spliced transcipts from the root-lesion nematode Pratylenchus penetrans. Mol Genet Genomics 272:138–148
Mitreva M, Wendl MC, Martin J, Wylie T, Yin Y, Larson A, Parkinson J, Waterston RH, McCarter JP (2006) Codon usage patterns in Nematoda: analysis based on over 25 million codons in thirty-two species. Genome Biol 7:R75. doi:10.1186/gb-2006-7-8-r75
Munoz E, Bogarad L, Deem M (2004) Microarray and EST database estimates of mRNA expression levels differ: the protein length versus expression curve for C. elegans. BMC Genomics 5:30–30
Nagaraj SH, Deshpande N, Gasser RB, Ranganathan S (2007a) ESTExplorer: an expressed sequence tag (EST) assembly and annotation platform. Nucleic Acids Res 35:W143–W147. doi:10.1093/nar/gkm378
Nagaraj SH, Gasser RB, Ranganathan S (2007b) A hitchhiker’s guide to expressed sequence tag (EST) analysis. Brief Bioinform 8:6–21
Nishikawa T, Nagai K (1996) EST error analysis in a large-scale GenBank search of ESTs using rapid-identity searching program for DNA sequences. In: Genome mapping and sequencing, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Noel GR, Atibalentja N (2006) “Candidatus Paenicardinium endonii”, an endosymbiont of the plant-parasitic nematode Heterodera glycines (Nemata: Tylenchida), affiliated to the phylum Bacteroidetes. Int J Syst Evol Microbiol 56:1697–1702
O’Bannon JH (1977) Worldwide dissemination of Radopholus similis and its importance in crop production. J Nematol 9:16–25
Opperman CH, Bird DM (1998) The soybean cyst nematode, Heterodera glycines: a genetic model system for the study of plant-parasitic nematodes. Current Opin Plant Biol 1:342–346
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J (2003) TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19:651–652
Price NS (2006) The banana burrowing nematode, Radopholus similis (Cobb) Thorne, in the lake Victoria region of East Africa: its introduction, spread and impact. Nematology 8:801–817
Prior A, Jones JT, Blok VC, Beauchamp J, McDermott L, Cooper A, Kennedy MW (2001) A surface-associated retinol- and fatty acid-binding protein (Gp-FAR-1) from the potato cyst nematode Globodera pallida: lipid binding activities, structural analysis and expression pattern. Biochem J 356:387–394
Ranganathan S, Nagaraj SH, Hu M, Strube C, Schnieder T, Gasser RB (2007) A transcriptomic analysis of the adult stage of the bovine lungworm, Dityocaulus viviparus. BMC Genomics 8:311. doi:10.1186/1471-2164-8-311
Richly E, Leister D (2004) NUMTs in sequenced eukaryotic genomes. Mol Biol Evol 21:1081–1084
Robertson L, Robertson WM, Sobczak M, Helder J, Tetaud E, Ariyanayagam MR, Ferguson MAJ, Fairlamb A, Jones JT (2000) Cloning, expression and functional characterisation of a peroxiredoxin from the potato cyst nematode Globodera rostochiensis. Mol Biochem Parasitol 111:41–49
Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP (2006) Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell 127:1193–1207
Sarah JL, Pinochet J, Stanton J (1996) The burrowing nematode of bananas, Radopholus similis Cobb, 1913. International network for the improvement of banana and plantain, Montpellier, France
Schiex T, Gouzy J, Moisan A, de Oliveira Y (2003) FrameD: a flexible program for quality check and gene prediction in prokaryotic genomes and noisy matured eukaryotic sequences. Nucleic Acids Res 31:3738–3741
Schwarz EM, Antoshechkin I, Bastiani C, Bieri T, Blasiar D, Canaran P, Chan J, Chen N, Chen WJ, Davis P, Fiedler TJ, Girard L, Harris TW, Kenny EE, Kishore R, Lawson D, Lee R, Muller HM, Nakamura C, Ozersky P, Petcherski A, Rogers A, Spooner W, Tuli MA, Van Auken K, Wang D, Durbin R, Spieth J, Stein LD, Sternberg PW (2006) WormBase: better software, richer content. Nucleic Acids Res 34:D475–D478
Smant G, Stokkermans JPWG, Yan YT, de Boer JM, Baum TJ, Wang XH, Hussey RS, Gommers FJ, Henrissat B, Davis EL, Helder J, Schots A, Bakker J (1998) Endogenous cellulases in animals: isolation of beta-1,4-endoglucanase genes from two species of plant-parasitic cyst nematodes. PNAS 95:4906–4911
Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, Coulson A, D’Eustachio P, Fitch DHA, Fulton LA, Fulton RE, Griffiths-Jones S, Harris TW, Hillier LW, Kamath R, Kuwabara PE, Mardis ER, Marra MA, Miner TL, Minx P, Mullikin JC, Plumb RW, Rogers J, Schein JE, Sohrmann M, Spieth J, Stajich JE, Wei C, Willey D, Wilson RK, Durbin R, Waterston RH (2003) The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLOS Biol 1:e45–e45
Stoffelen R, Verlinden R, Pinochet J, Swennen RL, De Waele D (2000) Host plant response of Fusarium wilt resistant Musa genotypes to Radopholus similis and Pratylenchus coffeae. Int J Pest Manag 46:289–293
Taylor MJ, Bilo K, Cross HF, Archer JP, Underwood AP (1999) 16S rDNA phylogeny and ultrastructural characterization of Wolbachia intracellular bacteria of the filarial nematodes Brugia malayi, B. pahangi, and Wuchereria bancrofti. Exp Parasitol 91:356–361
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tytgat T, Vercauteren I, Vanholme B, De Meutter J, Vanhoutte I, Gheysen G, Borgonie G, Coomans A, Gheysen G (2005) An SXP/RAL-2 protein produced by the subventral pharyngeal glands in the plant parasitic root-knot nematode Meloidogyne incognita. Parasitol Res 95:50–54
United Nations Environment Programme (1995) Report of the methyl bromide technical options committee. Montreal protocol on substances that deplete the ozone layer
Vandekerckhove TTM, Coomans A, Cornelis K, Baert P, Gillis M (2002) Use of the Verrucomicrobia-specific probe EUB338-III and fluorescent in situ hybridization for detection of “Candidatus Xiphinematobacter” cells in nematode hosts. Appl Environ Microbiol 68:3121–3125
Wang J-PZ, Lindsay BG, Cui L, Wall PK, Marion J, Zhang J, dePamphilis CW (2006a) Gene capture prediction and overlap estimation in EST sequencing from one or multiple libraries. BMC Bioinformatics 6:300
Wang HC, Susko E, Roger AJ (2006b) On the correlation between genomic G + C content and optimal growth temperature in prokaryotes: data quality and confounding factors. Biochem Biophys Res Commun 342:681–684
Washietl S, Hofacker IL, Stadler PF (2005) Fast and reliable prediction of noncoding RNAs. PNAS 102:2454–2459
Wasmuth JD, Blaxter ML (2004) Prot4EST: translating expressed sequence tags from neglected genomes. BMC Bioinformatics 5:187
Wuyts N, Lognay G, Verscheure M, Marlier M, De Waele D, Swennen R (2007) Potential physical and chemical barriers to infection by the burrowing nematode Radopholus similis in roots of susceptible and resistant banana (Musa spp.). Plant Pathol 56:878–890
Wylie T, Martin JC, Dante M, Mitreva MD, Clifton SW, Chinwalla A, Waterston RH, Wilson RK, McCarter JP (2004) Nematode.net: a tool for navigating sequences from parasitic and free-living nematodes. Nucleic Acids Res 32:D423–D426
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:W293–W297
Acknowledgments
J.J. has a Ph. D grant funded by Ghent University (BOF) and B.V. has a postdoctoral grant from Ghent University (BOF). Work at Washington University School of Medicine was supported by NIH-NIAID research grant AI 46593.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2008_340_MOESM1_ESM.pdf
Figure A1: Comparison of the E-values obtained by BLASTx-search (Y-axis) and tBLASTx-search (X-axis). Every dot represents one unigene. Unigenes without hits in either the two searches are not represented.(76.8 KB)
438_2008_340_MOESM2_ESM.pdf
Figure A2: Density lines of the calculated total G+C content (blue line), GC1 (red), GC2 (green) and GC3 (orange) content of the coding sequence part of the unigenes, based on FrameD translation predictions.(174 KB)
438_2008_340_MOESM3_ESM.pdf
Figure A3: Alignment of five R. similis unigenes (first sequence) with the M. incognita EST with strongest homology on protein level (second sequence) and C. elegans EST with strongest homology on protein level (third sequence). Conserved sequence features of the trans-spliced leader sequences are indicated with black boxes, the start ATG codon is shaded in gray. (40.6 KB)
438_2008_340_MOESM4_ESM.pdf
Figure A4: Detection of unigenes with a G+C content of 16%, compared to the average G+C content of the majority of the unigenes. A. The density line shows normal distribution, except for a bias at the left handed side, indicated by the dashed line. B. This bias, indicated by the dashed line, can also be observed in the ‘QQ-normal plot’ (line indicates the normal distribution). C. When the total unigene set is split up into the outliers with lower G+C content (1) and the remaining unigenes (2), this bias from the normal distribution is removed. D. The absence of any bias from the normal distribution can also be observed when the QQ-normal plot is constructed for both subsets, i.e. the outliers with lower G+C content (1) and the remaining unigenes (2). (210 KB)
Rights and permissions
About this article
Cite this article
Jacob, J., Mitreva, M., Vanholme, B. et al. Exploring the transcriptome of the burrowing nematode Radopholus similis . Mol Genet Genomics 280, 1–17 (2008). https://doi.org/10.1007/s00438-008-0340-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-008-0340-7