
Abstract
Heterodera glycines, the soybean cyst nematode (SCN), is a damaging agricultural pest that could be effectively managed if critical phenotypes, such as virulence and host range could be understood. While SCN is amenable to genetic analysis, lack of DNA sequence data prevents the use of such methods to study this pathogen. Fortunately, new methods of DNA sequencing that produced large amounts of data and permit whole genome comparative analyses have become available. In this study, 400 million bases of genomic DNA sequence were collected from two inbred biotypes of SCN using 454 micro-bead DNA sequencing. Comparisons to a BAC, sequenced by Sanger sequencing, showed that the micro-bead sequences could identify low and high copy number regions within the BAC. Potential single nucleotide polymorphisms (SNPs) between the two SCN biotypes were identified by comparing the two sets of sequences. Selected resequencing revealed that up to 84% of the SNPs were correct. We conclude that the quality of the micro-bead sequence data was sufficient for de novo SNP identification and should be applicable to organisms with similar genome sizes and complexities. The SNPs identified will be an important starting point in associating phenotypes with specific regions of the SCN genome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In humans and model experimental organisms such as C. elegans and Drosophila, genetic approaches to studying interesting phenotypes have been very productive because large communities of researchers were able to catalogue DNA sequence variations and map-base clone genes involved in important traits (International HapMap Consortium 2005). Such complex and expensive projects have not been an option for non-model organisms like the agricultural pest Heterodera glycines, the soybean cyst nematode (SCN), because of the expense involved in comparative genome analysis. SCN is amenable to genetic analysis of important traits such as virulence (Dong and Opperman 1997) host range and aggressiveness (Schmitt 2004). Recently an initial genetic map was generated for SCN (Atibalentja et al. 2005) and a close relative, the potato cyst nematode (Rouppe van der Voort 1999). SCN virulence is the ability of a nematode to grow on a resistant plant, the main method of SCN control. If SCN virulence could be understood at the molecular genetic level, then the nematode might be managed. The lack of DNA sequence data for SCN and other important, non-model organisms prevent the application of molecular genetic approaches to studying these organisms.
However, the recent development and commercialization of new massively parallel genome sequencing platforms promises to generate vastly more DNA sequence data at a cost that is much lower than conventional dideoxy sequencing (Mardis 2006). Such sequence data can be used to conduct polymorphism discovery at a whole genome scale for a fraction of the cost of conventional discovery methods. One DNA sequencing platform, developed by 454 Life Sciences Corporation, uses solid-phase libraries bound to micro-beads to routinely produce 20 Mbp of sequence in a 4-h sequencing run (Margulies et al. 2005). This new sequencer has been used to sequence bacterial genomes (Goldberg et al. 2006), cDNAs (Ng et al. 2006), small RNAs (Girard et al. 2006), PCR products for whole genome surveys (Thomas et al. 2006) and for sequencing ancient DNA (Green et al. 2006; Poinar et al. 2006). In addition, the large sequence depth that can be obtained with micro-bead sequencing has made it possible to conduct a number of new metagenomic projects (Angly et al. 2006; Edwards et al. 2006; Krause et al. 2006; Leininger et al. 2006; Sogin et al. 2006; Turnbaugh et al. 2006). To date, a few publications have used this micro-bead sequencing platform for large-scale single nucleotide polymorphism (SNP) or structural variation discovery in animals (Korbel et al. 2007; Malhi et al. 2007) and for mutation discovery in cancer cells (Thomas et al. 2006); however, these approaches have relied upon a completed reference genomes for sequence comparison.
In this paper we describe the use of 454 micro-bead sequence data for the identification of de novo sequence polymorphisms from two inbred SCN biotypes using the massively parallel micro-bead sequencing method developed by 454 Life Sciences Corporation. We evaluate the micro-bead sequence quality by comparing the sequences to a SCN bacterial artificial chromosome (BAC) clone and then extrapolate the analysis to the whole SCN genome. This is the first report of significant numbers of SNPs discovered in a plant parasitic nematode and the first use of micro-bead sequences to discover SNPs in the absence of a reference genome. Our experiments suggest that the micro-bead sequence data was of sufficient quality to predict SCN gene copy number and to identify sequence polymorphisms via a reference sequence or randomly in the SCN genome.
Results
454-Based genome sequencing
The 454 micro-bead sequencing method uses pyrosequencing combined with a picoliter plate to perform hundreds of thousands of sequencing reactions at once (Margulies et al. 2005). We collected sequence data from the genomes of two SCN biotypes. Both nematode biotypes were inbred by repeated sib-mating for 50+ generations and differ from each other by virulence, where the TN20 biotype is highly virulent and the TN10 biotype is avirulent. For SCN inbred TN20, the micro-bead sequence data collected totaled 327,784,600 bp of genomic sequence (average size 116 bp), while 73,755,873 bp of sequence was collected from inbred SCN TN10 (average size 111 bp). By counting the bases in the TN10 data set, it was estimated that the SCN genome has a 36% GC content.
Sequencing of a SCN BAC clone
To have a reference DNA, we sequenced a section of the SCN genome contained in a BAC derived from inbred SCN TN20. A single BAC, L19-1, that contained a single copy of a gene homologous to Hgcm-1, a chorismate mutase thought to play a role in nematode parasitism (Bekal et al. 2003), was isolated and sequenced using a shotgun sequencing strategy and conventional dideoxy sequencing chemistry to approximately a fivefold redundancy. The insert of L19-1 was 147,261 bp in length. Using a C. elegans gene model in the gene prediction program Twin Scan (Korf et al. 2001; Wei et al. 2005), we predicted that L19-1 contained 41 genes, seven of which had significant protein homology to known nematode genes in GenBank upon conducting a BLASTP search (Table 1). Of the predicted genes, six had homology to C. elegans sequences and one to a SCN gene Hg-cm-1. A more detailed analysis of the L19-1 sequence will be publishes elsewhere, but the homology of L19-1 to known nematode genes suggests the BAC is of H. glycines origin.
Estimation of sequence copy number in a SCN BAC using micro-bead DNA reads
The L19-1 sequence was compared to databases of TN20 and TN10 micro-bead sequences via BLASTN. The micro-bead sequences that matched were retrieved from the database and aligned to the L19-1 reference sequence. After assembling the TN20 sequences onto L19-1, 3,185 micro-bead sequences were found to matched the L19-1 sequence at a level of 95% sequence identity or higher. Of the 3,185 TN20 micro-bead sequences that matched, we observed 75% coverage of the L19-1 sequence with TN20 micro-bead reads. By adding the TN10 sequences to the alignment, the coverage of the BAC with micro-bead sequences increased to 88%.
Overall, the depth of the TN20 micro-bead sequence coverage on L19-1 was not even, with micro-bead sequences often clustered in certain regions of L19-1 (Fig. 1). We interpreted this increased depth of coverage as areas in the genome that are composed of repetitive DNA. To map these repetitive sequences in L19-1, the BAC sequence was sequentially fragmented in silico into 100 bp fragments. The 1,500 fragments were then compared to 660,549 micro-bead sequences from both SCN biotypes using BLASTN. The number of significant hits per 100-bp fragment was plotted for both biotypes, and the resulting graphs are shown in Fig. 1. Most of the 100 bp BAC sections had a low copy number, however; some sequences had over 100 significant matches. Interestingly, the pattern of micro-bead matches was nearly identical between the two SCN biotypes, except for a few minor differences in predicted copy number at 78,100 and 91,100 bp, suggesting the reproducibility of the sequence data is good (Fig. 1). For DNA sequence polymorphisms discovery, we were interested in the low copy regions of the SCN genome since such sequences have a greater potential for use as molecular markers than high copy regions.

SCN micro-bead DNA sequences from TN20 (top panel) and TN10 (bottom panel) aligned to the SCN BAC L19–1. The arrows indicate repetitive sequences in L19-1 that differ between the two biotypes
DNA sequence polymorphisms discovery in a SCN BAC
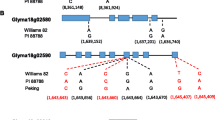
Upon comparing the low copy sequences from TN20 and TN10 to L19-1, insertion–deletion mutations (indels) and SNPs were observed. To authenticate polymorphisms, we resequenced regions of L19-1 containing micro-bead sequences containing putative SNP and indel mutations. We chose regions of L19-1 that had one or a few micro-bead matches for resequencing. To confirm the polymorphisms we designed PCR primers flanking a micro-bead sequence containing the putative SNPs/indels. The primers were used to PCR amplify the target regions from SCN genomic DNA from both biotypes. The PCR products were sequenced using conventional dideoxy sequencing. When the micro-bead sequences and the resequenced amplicons from SCN TN10 and TN20 were aligned to the L19-1 sequence, each TN10/L19-1 polymorphism could be assessed for accuracy. Out of 278 potential sequence polymorphisms assessed, 206 consisted of single base pair insertions or deletions. About half the indels were part of a homopolymer sequence. Regardless of sequence context, only 3% of these indels proved to be correct upon resequencing (Table 2). Seventy-two putative SNPs were resequenced, upon examination of the resequence data, 65% of the SNPs were confirmed to be correctly predicted by the micro-bead sequence. However, as the data analysis was being conducted, it was noticed that the incorrect SNPs often occurred at the end of a micro-bead read or associated with nearby indel mutations. This suggested that low micro-bead sequence quality might have adversely affected SNP calling accuracy. To remedy this situation, the SNPs were re-assessed using a criterion that did not allow the SNP to be within 5 bp of the 3′ end of the micro-bead sequence and could not have another sequence difference with in 10 bp on either side of the SNP. Upon applying this criterion, 84% of the SNPs were authenticated.
SNP discovery between two SCN biotypes directly from 454 sequence data
Since the SNP discovery rate on the L19-1 sequence was acceptable, we were interested in extrapolating our approach to random SNPs in the entire SCN genome. To do this, we compared all TN20 micro-bead sequences to each other using BLASTN. 193,623 TN20 micro-bead sequences that were low copy (less than ten significant matches) were retained and were then compared to all TN10 micro-bead sequences, also via BLASTN. This comparison resulted in 51,999 low-copy of TN20/TN10 matches containing sequence polymorphisms. These matching sequences were extracted and 9,748 sequences with putative single SNPs were retained, the remaining sequences contained just indel mutations and were discarded. PCR primers were designed flanking 1,536 putative SNPs, which were use to PCR amplify the selected region form genomic DNA from SCN TN10 and TN20. The resulting amplicons were sequenced using conventional dideoxy sequencing. Of the 3,072 amplicons sequenced, 1,108 amplicons yielded informative sequences for both genotypes, providing us information for SNP confirmation. Comparison of TN10 and TN20 sequences for each SNP showed that 64% of the putative SNPs (715 SNPs) had been predicted correctly from the micro-bead sequences. Of those, 239 were homozygous SNPs between TN20 and TN10 (Table 2), where 13% of the SNPs were A to C, 28% were A to G, 11% were A to T, 5% were C to G, 30% were C to T, and 12% were G to T.
Discussion
Genetic analysis requires significant genomic and/or cDNA sequence data to discover useful polymorphisms between genomes (Picoult-Newberg et al. 1999; Taillon-Miller et al. 1998). Typically, sequence data is collected from a reference genome and then cDNAs or sequence tagged sites (STS) are used to discover sequence differences (Choi et al. 2007; Hudson et al. 1995). To discover useful genetic variation one needs to avoid high copy number sequences and acquire accurate sequence data. The SNP frequency in the genome and whether the genome has extra complications such as polyploidy, are also important factors for estimating the degree of difficulty in a particular genetic analysis (Choi et al. 2007). The introduction of new massively parallel DNA sequencing technologies (Shaffer 2007) promises to allow non-model experimental organisms to be studied using comparative genomics approaches by providing large amounts of DNA sequence at a low cost. In this project we evaluated the micro-bead sequence to ascertain how well this type of sequence data allows SNP discovery in SCN. We were concerned about two issues that might have hindered SNP discovery in SCN; namely, we wanted to avoid SNPs in gene families or highly repetitive sequences and we were concerned about low DNA sequence quality that may provide many false SNP predictions.
In our initial analysis of biotypes TN10 and TN20, BLASTN of micro-bead sequence matches to the L19-1 reference sequence was used to predict putative SCN DNA copy number. It was assumed that many significant micro-bead sequence matches to a region in L19-1 were indicative of multi copy sequences. This assumption is not without merit since several publications (Macas et al. 2007; Swaminathan et al. 2007) have indicated the 454 micro-bead sequence data, due to the random shearing process used in the solid phase library construction and the lack of microbial cloning bias, represent a very good random sample of the genome. In our data, a very reproducible pattern of putative repetitive DNA between nematode biotypes was observed, supporting this assumption. Following the same logic, we assumed low numbers of micro-bead matches was indicative of low copy regions. To test this idea, we conducted resequencing of low copy regions for both the L19-1 reference sequence and for random low copy regions in the SCN genome. The resequencing was conducted by PCR amplification of putative SNPs, and flanking regions, from total SCN genomic DNA. These PCR amplicons were then used as a template for conventional dideoxy DNA sequencing. The observation that clear DNA sequence was obtained, confirming most of our predicted SNPs, suggests our low copy selection process was successful. Thus we conclude that the 454 micro-bead sequences are useful for estimating sequence copy number. This type of analysis could be applied to EST data to estimate gene copy number or it could be used to filter the SCN genome in silico to identify the low and high copy genomic DNAs to facilitate genome assembly or understand the nature of repetitive elements in the genome.
To discover DNA sequence polymorphisms the quality of the DNA sequence is critical. Four hundred and fifty-four micro-bead sequencing is a relatively new methodology, thus we were not sure it was of sufficient quality for SNP discovery at a low sequence coverage density in the absence of a SCN reference sequence (de novo SNP discovery). The raw sequence accuracy of sequence data from a Roche GS20 instrument is 96; however, removal of poor sequence data can improve the accuracy to 99.75% (Huse et al. 2007). The 454 micro-bead sequences do have quality score values associated with each base, but these values represent probability that a homopolymer length at a position is correct, not base call accuracy (Huse et al. 2007; Margulies et al. 2005). Therefore, we chose not to rely on the quality scores for selecting SNPs, but instead used an empirical approach. That is, we used raw low copy sequences and to compare them to the L19-1 BAC or randomly between the two genomes, we then assessed the frequency of correct SNP and modified our selection criteria. Our approach allowed us to remove sequences of low quality and reduced our false SNP discovery rate. We assume as the quality of 454 micro-bead sequence data improves, the de novo SNP discovery rate will also improve. Our approach of eliminating SNPs originating in sequences with multiple indels, ambiguous bases and avoiding 3′ ends of micro-bead reads significantly improved SNP discovery rate up to 84% for the BAC and up to 77% for random SNPs in the SCN genome. More sophisticated removal of obvious low quality micro-bead sequences (Huse et al. 2007) should enhance the accuracy of de novo SNP discovery.
In addition to SNP discovery, we were also interested in identifying indel polymorphisms. However, as has been noted by other researchers sequence errors associated with homopolymers are the most common types of errors (Margulies et al. 2005). The high frequency of indel artifacts is particular to pyrosequencing methods. In pyrosequencing, dNTPs are sequentially added to a sequencing reaction where, when incorporated, a flash of light is emitted when a specific nucleotide is added. The light flashes, caused by the metabolism of release pyrophosphate via the combined reaction with ATP-sulfurylase and luciferase, vary in intensity depending on the number of dNTP’s added at a particular time (Ronaghi 2001). A pyrosequencing machine must not only detect the flash of light, but the intensity of light if homopolymer runs of sequence are present in the template DNA. It was interesting that indel mutations that were not associated with homo polymers were also mostly incorrect for some unknown reason. While indel mutations are of interest, we conclude the micro-bead data is not of sufficient quality to discover indels at a frequency high enough to be useful.
We conclude micro-bead sequence data is of sufficient quality to correct SNPs between the two nematode genomes. Even though SNPs can be discovered using single or low redundancy of micro-bead sequence, it would be helpful to have a higher depth of sequence to facilitate the calling of homozygous and heterozygous SNPs for genetic analysis. One reason for the success SCN SNP discovery project is undoubtedly due to the high rate of SNPs between our two SCN biotypes. If the SNP rate was an order of magnitude lower, the error rate of the micro-bead sequence would have exceeded that of the SNP rate and thus the SNP discovery efficiency would drop dramatically. This issue may be eliminated by an increase of micro-bead sequence quantity or sequence output quality as this next generation sequencing technology matures and improves. One advantage of this micro-bead based method of de novo SNP discovery is that it identifies large number of putative SNPs. This abundance of polymorphisms allows one to choose only putative SNP that had optimal sequence context and Tm for conversion to a molecular marker assay. This preselection of putative SNPs allows a very efficient and cost effective selection of sequence polymorphisms.
In summary, we have shown 454-based pyrosequencing to be useful for whole genome SNP discovery and DNA copy number estimation. This is the first study to identify a significant number of SNPs in a plant parasitic nematode species and the first report of using 454 micro-bead sequence for de novo SNP discovery. This technology has enabled the analysis of orphan genomes, such as SCN, and shows the promise to extend genomic analysis of a wide range of important, but overlooked, organisms. The SNPs discovered in this project will form an important infrastructure for the genetic analysis of SCN traits that are vital for its long-term management. As the next generation DNA sequencing technology matures, and generates larger amounts of sequence data, SNP discovery and verification at the whole genome level will further facilitate whole genome genetic analysis for non-model organisms such as SCN.
Materials and methods
Nematode inbred line culture and characteristics
A collection of SCN inbred lines (selected by single cyst descent (half-sib matting) for over 30 generations) is maintained at 27°C in a greenhouse facility on appropriate host plants using plastic crocks as described by (Niblack 1993). TN10 was selected on tomato (cultivar Tiny Tim) and TN20 was selected on the SCN resistant soybean cultivar Hartwig. TN20 is HG type 1, 2, 3, 4, 5, 6, 7: while TN10 is Hg Type 0 (Niblack 2002).
Nematode and genomic DNA isolation
SCN eggs were harvested as described in Niblack (1993). Eggs were deposited on top of an egg hatching apparatus consisting of 41 μm mesh sieve held taut between two stacked 50 ml polypropylene beakers with there bottoms cut off. The bottom surface of the sieve cloth was carefully laid on top of a shallow layer of sterile dH2O containing 1% streptomycin sulfate solution (Sigma) in a 60 mm petri dish bottom. As the second-stage juveniles (J2s) hatched, they crawled through the mesh to the liquid in the petri dish. The clean J2s were collected from the petri dish after allowing the J2 to accumulate for 24 h. The J2s were concentrated by centrifugation until a 250 μl packed J2 pellet was obtained, then the J2’s were flash frozen in liquid nitrogen.
The frozen J2 pellet was transferred to a tissue pulverizer (Fisher Scientific) precooled in liquid nitrogen and crushed. The pulverized frozen nematode fragments were mixed with 2 ml of proteinase K solution [0.1 M Tris–Cl (pH 8.0), 1 mM EDTA, 0.15 M NaCl, 1% sodium dodecyl sulfate, 1 mM dithiothreitol, 2 mg/ml proteinase K] and were incubated for 1 h at 37°C. The DNA was phenol/chloroform-extracted, then ethanol precipitated as described in (Sambrook et al. 1989). The DNA was resuspended in 1 ml of TE (10 mM Tris–Cl pH 8, EDTA pH 8) containing RNase A at 10 μg/ml and incubated at 37°C for 1 h. The DNA was next diluted to 20 ml with column buffer (750 mM NaCl; 50 mM MOPS, pH 7; 15% isopropanol, 0.15% Triton X-100) and then further purified on a Genomic-tip 500/G as described in the manufacturers protocol (Qiagen, Valencia, CA, USA). The eluted SCN genomic DNA was concentrated by ethanol precipitation and resuspended in 200 μl of TE.
SCN BAC library construction, screening and sequencing
Two grams of frozen TN20 J2 nematode pellets were collected as described above. The frozen nematodes were shipped to Bio S&T (Montreal, Quebec) for BAC library construction and quality assessment. The insert SCN DNA was produced via partial digestion with restriction enzyme HindIII. The SCN DNA was ligated into BAC vector pIndigo-5 (Epicentre, Madison, WI, USA) and transformed into Escherichia coli DH10B. The final BAC library constructed contained 4, 608 independent clones. The average insert size was 129 kb, with 86% of the clones containing an insert of >100 kb.
The SCN BAC library was screened using a PCR-based approach. The BAC library was arrayed in 12, 384 well plates and the bacterial cultures contained in these plates were pooled in rows and columns using a hydra pipetting robot. Each plate had 40 pools and one master pool to facilitate library screening. Total BAC DNA was isolated from each pool and master pool using the Large-Construct kit (Qiagen, CA, USA). The master pools were screened using a real-time PCR assay designed to detect the SCN gene Hgcm-1 (Lambert et al. 2005). The rows and column pools for each master pool that contained a positive signal Hg-cm-1 signal were assayed for Hgcm-1.
One clone that reproducibly tested positive for the Hg-cm-1 gene, named L19-1, was selected for shotgun sequencing. The BAC clone was amplified overnight in 500 ml of LB medium + 20 μg/ml of chloramphenicol. BAC DNA was purified using the Large-Construct kit. Purified BAC DNA was sheared with a nebulizer, blunt-ended using the DNA Terminator(r) kit (Lucigen, Middleton, WI, USA). Fragments 1.6–5 kb were purified from a 0.8% low melting point agarose gel, dephosphorylated and cloned into the pCR(r)4Blunt-TOPO vector (Invitrogen, Carlsbad, CA, USA) and transformed according to the manufacturers instructions into One Shot TOP10 electrocompetent cells (Invitrogen).
Individual colonies were robotically picked from agar plates with the Genetix Q-pix and racked as glycerol stocks in 384-well plates. After overnight growth of the glycerol stocks, bacteria were inoculated into 96-well plates with LB medium + 100 μg/ml of carbenicilin and grown overnight. Plasmid DNAs were purified from the bacterial cultures with the Qiagen 8000 and Qiagen 9600 BioRobots. ABI BigDye(r) terminator chemistry was used for sequencing reactions from the 5′- and 3′-ends of the inserts using T7 and M13reverse-48 primers, respectively. Sequencing reactions were loaded into ABI 3730xl capillary systems.
Four 384 plates of randomly picked clones were sequenced from both ends using dideoxy sequencing. Low quality sequence data and vector sequences were removed and sequences were assembled using SeqManPro in the Lasergene7.1 sequence analysis software package.
Micro-bead DNA sequencing
SCN micro-bead sequencing was conducted by 454 Life Sciences Corporation (Branford, CT, USA). Ten machine runs were conducted, using a Roche-454 GS20 sequencer, for inbred SCN line TN20 and two sequence runs were preformed for TN10. Both SCN biotypes had an average micro-bead sequence length of 116 bp.
Micro-bead sequence alignment to L19-1
Micro-bead sequence coverage for BAC L19-1 was determined by comparing L19-1 to databases of all micro-bead sequences of SCN biotypes TN10 and TN20 using NCBI’s Blastall BLASTN program (Altschul et al. 1990). Micro-bead sequences that matched L19-1 were imported into the LaserGene 7.1 program SeqManPro (DNASTAR, Madison, WI, USA) for final assembly to the L19-1 reference sequence. The assembly parameters for SeqManPro, were a mismatch size of 25, a minimum match percentage of 95, a match spacing of 50, minimum sequence length of 25, a gap penalty of 0.00, a gap length penalty of 0.70 and a max mismatch of ends of 15. The micro-bead sequence coverage of L19-1 was calculated by exporting the micro-bead consensus sequences as a concatenated FASTA file and then measuring the length of the sequences using the word count feature of Microsoft word.
To identify high copy regions of the reference sequence, L19-1was fragmented in silico into sequential 100 bp fragments using the Bioperl script “split_seq.pl”. The L19-1 fragments were separately compared to databases containing 660,549 micro-bead sequences of SCN biotypes TN10 or TN20 using NCBI’s Blastall blastn program. Micro-bead sequences were considered potential matches to the L19-1 if they had over 90% identity and an E value below 10−7. The number of micro-bead sequence matches was plotted against the sequential list of 100 bp fragments using Microsoft Excel.
L19-1 SNP discovery and validation
Micro-bead sequences from TN10 that matched L19-1 were aligned to the BAC reference sequence using SeqManPro. The assembly was preformed using a minimum mismatch % of 85%, a match size of 25 bp, a minimum sequence length of 25, a gap penalty of 0.00, and a gap length penalty of 0.70 and a maximum mismatch end of 15. Low copy micro-bead reads (1–2 micro-bead sequence matches) showing sequence polymorphisms were targeted for resequencing. To resequence polymorphic L19-1 regions, the BAC sequence imported into the Primer 3 (Rozen and Skaletsky 2000) where PCR primers flanking putative SNPs and indels were designed to amplify 500 bp amplicons. All PCR primers were ordered from Invitrogen. One of the PCR primers was tagged with the M13R primer sequence (AACAGCTATGACCATG) to facilitate high-throughput sequencing of the selected regions of L19-1. PCR from TN10 and TN20 genomic DNA was conducted and the PCR products were separated on a 1.5% agarose gel to evaluate the quality of the PCR product. Successful PCR products were cleaned with ExoSAP-IT (USB Biochemicals, Cleveland, OH, USA) to remove primers and nucleotides and diluted to 2 ng/μl final template concentration. DNA sequencing was conducted at the UIUC Biotechnology Center as described above.
SNP discovery pipeline between the SCN TN20 and TN10 lines
A database of all TN20 micro-bead sequences was compared against itself using NCBI’s “BLASTALL BLASTN” program. The results were parsed to obtain low-copy sequences (fewer than ten hits to the database). In order to pull out the analogous sequence in the TN10 line, the low-copy sequences of TN20 were compared to the TN10 micro-bead sequence database using BLASTN. Hits with 90–99% homology and an E value below 10−6, were pulled out as having potential SNPs. These low copy micro-bead sequences were imported into the sequence assembly program Sequencher 4.6 (Gene Codes Corporation, Arbor, Michigan) and assembled using a parameter of 95% identity over at least 30 bp. This allowed for the contigs (containing only one TN10 and one TN20 sequence) with SNPs to be identified, selected, and exported. TN10-20 contigs containing potential SNPs were converted into Genbank SNP format using a perl script, and imported into the sequence analysis program AlleleID 4 (Premier Biosoft, Palo Alto, CA, USA), where the SNPs were compared, via a desktop BLASTN function in the program, to the TN10 and TN20 micro-bead sequences originally imported into Sequencher. SNPs were selected by examining the BLASTN alignments to choose SNPs with a good sequence context surrounding the SNP. AlleleID 4 was used to design PCR primers surrounding the SNP, again a M13R primer sequence was added to one of the PCR primers to facilitate DNA sequencing. The primer pairs were ordered from Invitrogen. PCR amplification for both TN20 and TN10 SCN DNA and the template preparation was conducted as described above. The SNPs were confirmed by assembling the resulting sequences using Sequencher 4.6 and visually confirming the SNP in the chromatogram. In order to calculate the total %G/C of the SCN, a modified version of the Bioperl.org perl script “gccontent.pls,” was used. The DNA sequence from the two runs of SCN inbred TN10 was used for the calculation. The total number of sequences analyzed was 660,649, which contained 73,755,873 nucleotides, of which 26,533,099 were G or C, indicating TN10 has a 35.97% G/C content.
Assession numbers
The SCN BAC L19-1 sequence was submitted to GenBank (accession number EU290604) and all confirmed SNPs were submitted to dbSNP at NCBI (ss# 79089825 through 79090072). All 454 micro-bead sequences and quality scores have been submitted to the NCBI short read archive (SRA000210).
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, Chan AM, Haynes M, Kelley S, Liu H, Mahaffy JM, Mueller JE, Nulton J, Olson R, Parsons R, Rayhawk S, Suttle CA, Rohwer F (2006) The marine viromes of four oceanic regions. PLoS Biol 4:e368
Atibalentja N, Bekal S, Domier LL, Niblack TL, Noel GR, Lambert KN (2005) A genetic linkage map of the soybean cyst nematode Heterodera glycines. Mol Gen Genomics 273:273–281
Bekal S, Niblack TL, Lambert KN (2003) A chorismate mutase from the soybean cyst nematode Heterodera glycines shows polymorphisms that correlate with virulence. Mol Plant Microbe Interact 16:439–446
Choi I Y, Hyten DL, Matukumalli LK, Song Q, Chaky JM, Quigley CV, Chase K, Lark KG, Reiter RS, Yoon MS, Hwang EY, Yi SI, Young ND, Shoemaker RC, van Tassell CP, Specht JE, Cregan PB (2007) A soybean transcript map: gene distribution, haplotype and SNP analysis. Genetics 176:685–696
Dong K, Opperman CH (1997) Genetic analysis of parasitism in the soybean cyst nematode Heterodera glycines. Genetics 146:1311–1318
Edwards RA, Rodriguez-Brito B, Wegley L, Haynes M, Breitbart M, Peterson DM, Saar MO, Alexander S, Alexander EC Jr, Rohwer F (2006) Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7:57
Girard A, Sachidanandam R, Hannon GJ, Carmell MA (2006) A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442:199–202
Goldberg SM, Johnson J, Busam D, Feldblyum T, Ferriera S, Friedman R, Halpern A, Khouri H, Kravitz SA, Lauro FM, Li K, Rogers YH, Strausberg R, Sutton G, Tallon L, Thomas T, Venter E, Frazier M, Venter JC (2006) A sanger/pyrosequencing hybrid approach for the generation of high-quality draft assemblies of marine microbial genomes. Proc Natl Acad Sci USA 103:11240–11245
Green RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons JF, Du L, Egholm M, Rothberg JM, Paunovic M, Paabo S (2006) Analysis of one million base pairs of Neanderthal DNA. Nature 444:330–336
Hudson TJ, Stein LD, Gerety SS, Ma J, Castle AB, Silva J, Slonim DK, Baptista R, Kruglyak L, Xu SH, Hu X, Colbert AM, Rosenberg C, Reeve-Daly MP, Rozen S, Hui L, Wu X, Vestergaard C, Wilson KM, Bae JS, Maitra S, Ganiatsas S, Evans CA, DeAngelis MM, Ingalls KA, Nahf RW, Horton LT Jr, Anderson MO, Collymore AJ, Ye W, Kouyoumjian V, Zemsteva IS, Tam J, Devine R, Courtney DF, Renaud MT, Nguyen H, O’Connor TJ, Fizames C, Faure S, Gyapay G, Dib C, Morissette J, Orlin JB, Birren BW, Goodman N, Weissenbach J, Hawkins TL, Foote S, Page DC, Lander ES (1995) An STS-based map of the human genome. Science 270:1945–1954
Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM (2007) Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol 8:R143
International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320
Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, Taillon BE, Chen Z, Tanzer A, Saunders AC, Chi J, Yang F, Carter NP, Hurles ME, Weissman SM, Harkins TT, Gerstein MB, Egholm M, Snyder M (2007) Paired-end mapping reveals extensive structural variation in the human genome. Science 318:420–426
Korf I, Flicek P, Duan D, Brent MR (2001) Integrating genomic homology into gene structure prediction. Bioinformatics 17:S140–148
Krause L, Diaz NN, Bartels D, Edwards RA, Puhler A, Rohwer F, Meyer F, Stoye J (2006) Finding novel genes in bacterial communities isolated from the environment. Bioinformatics 22:e281–e289
Lambert KN, Bekal S, Domier LL, Niblack TL, Noel GR, Smyth CA (2005) Selection of Heterodera glycines chorismate mutase-1 alleles on nematode-resistant soybean. Mol Plant Microbe Interact 18:593–601
Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW, Prosser JI, Schuster SC, Schleper C (2006) Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442:806–809
Macas J, Neumann P, Navratilova A (2007) Repetitive DNA in the pea (Pisum sativum L.) genome: comprehensive characterization using 454 sequencing and comparison to soybean and Medicago truncatula. BMC Genomics 8:427
Malhi RS, Sickler B, Lin D, Satkoski J, Tito RY, George D, Kanthaswamy S, Smith DG (2007) MamuSNP: a resource for Rhesus Macaque (Macaca mulatta) genomics. PLoS ONE 2:e438
Mardis ER (2006) Anticipating the 1,000 dollar genome. Genome Biol 7:112
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Ng P, Tan JJ, Ooi HS, Lee YL, Chiu KP, Fullwood MJ, Srinivasan KG, Perbost C, Du L, Sung WK, Wei CL, Ruan Y (2006) Multiplex sequencing of paired-end ditags (MS-PET): a strategy for the ultra-high-throughput analysis of transcriptomes and genomes. Nucleic Acids Res 34:e84
Niblack TL, Arelli PR, Noel GR, Opperman CH, Orf JH (2002) A revised classification scheme for genetically diverse populations of Heterodera glycines. J Nematology 34:279–288
Niblack TL, Heinz RD, Smith GS, Donald PA (1993) Distribution, density, and diversity of Heterodera glycines populations in Missouri. Ann Appl Nematology 25:880–886
Picoult-Newberg L, Ideker TE, Pohl MG, Taylor SL, Donaldson MA, Nickerson DA, Boyce-Jacino M (1999) Mining SNPs from EST databases. Genome Res 9:167–174
Poinar HN, Schwarz C, Qi J, Shapiro B, Macphee RD, Buigues B, Tikhonov A, Huson DH, Tomsho LP, Auch A, Rampp M, Miller W, Schuster SC (2006) Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA. Science 311:392–394
Ronaghi M (2001) Pyrosequencing sheds light on DNA sequencing. Genome Res 11:3–11
Rouppe van der Voort JNAM, van Eck HJ, van Zandvoort PM, Overmars H, Helder J, Bakker J (1999) Linkage analysis by genotyping of sibling populations: agenetic map for the potato cyst nematode contructed using “pseudo-F2” mapping strategy. Mol Gen Genet 261:1021–1031
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schmitt DP (2004). In: Schmitt DP, Wrather JA, Riggs RD (eds) Biology and management of soybean cyst nematode. Walsworth Publishing Company, Marceline, pp 1–8
Shaffer C (2007) Next-generation sequencing outpaces expectations. Nat Biotechnol 25:149
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci USA 103:12115–12120
Swaminathan K, Varala K, Hudson ME (2007) Global repeat discovery and estimation of genomic copy number in a large, complex genome using a high-throughput 454 sequence survey. BMC Genomics 8:132
Taillon-Miller P, Gu Z, Li Q, Hillier L, Kwok PY (1998) Overlapping genomic sequences: a treasure trove of single-nucleotide polymorphisms. Genome Res 8:748–754
Thomas RK, Nickerson E, Simons JF, Janne PA, Tengs T, Yuza Y, Garraway LA, LaFramboise T, Lee JC, Shah K, O’Neill K, Sasaki H, Lindeman N, Wong KK, Borras AM, Gutmann EJ, Dragnev KH, DeBiasi R, Chen TH, Glatt KA, Greulich H, Desany B, Lubeski CK, Brockman W, Alvarez P, Hutchison SK, Leamon JH, Ronan MT, Turenchalk GS, Egholm M, Sellers WR, Rothberg JM, Meyerson M (2006) Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med 12:852–855
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031
Wei C, Lamesch P, Arumugam M, Rosenberg J, Hu P, Vidal M, Brent MR (2005) Closing in on the C. elegans ORFeome by cloning TWINSCAN predictions. Genome Res 15:577–582
Acknowledgments
This work was supported by grant 2006-35604-16731 from the United States Department of Agriculture National Research Initiative Competitive Grants Program. The CFAR program at the University of Illinois at Urbana-Champaign. The Illinois Soybean Production Operating Board, The North Central Regional Integrated Pest Management, Competitive Grants Program and the United Soybean Board. We also wish to thank Kam Colgrove for his technical support. Mention of a trademark or proprietary product does not constitute a guarantee or warranty of the product by the USDA and does not imply its approval to the exclusion of other products that may also be suitable. This work has been carried out in compliance with the current laws governing genetic experimentation in the United States of America.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. Ussery.
Rights and permissions
About this article
Cite this article
Bekal, S., Craig, J.P., Hudson, M.E. et al. Genomic DNA sequence comparison between two inbred soybean cyst nematode biotypes facilitated by massively parallel 454 micro-bead sequencing. Mol Genet Genomics 279, 535–543 (2008). https://doi.org/10.1007/s00438-008-0331-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-008-0331-8