
Abstract
Diversity Arrays Technology (DArT) is a microarray-based DNA marker technique for genome-wide discovery and genotyping of genetic variation. DArT allows simultaneous scoring of hundreds of restriction site based polymorphisms between genotypes and does not require DNA sequence information or site-specific oligonucleotides. This paper demonstrates the potential of DArT for genetic mapping by validating the quality and molecular basis of the markers, using the model plant Arabidopsis thaliana. Restriction fragments from a genomic representation of the ecotype Landsberg erecta (Ler) were amplified by PCR, individualized by cloning and spotted onto glass slides. The arrays were then hybridized with labeled genomic representations of the ecotypes Columbia (Col) and Ler and of individuals from an F2 population obtained from a Col × Ler cross. The scoring of markers with specialized software was highly reproducible and 107 markers could unambiguously be ordered on a genetic linkage map. The marker order on the genetic linkage map coincided with the order on the DNA sequence map. Sequencing of the Ler markers and alignment with the available Col genome sequence confirmed that the polymorphism in DArT markers is largely a result of restriction site polymorphisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The availability of reliable molecular markers is of great importance for plant breeding. The ideal molecular marker technique should generate hundreds of molecular markers that cover the entire genome in a single, simple and reliable experiment (Luikart et al. 2003). Genome-wide molecular markers are used for germplasm characterization, assessment of genetic diversity, to accelerate introgression or backcrossing programs, and for the mapping of complex traits.
Comparison of DNA sequences from closely related organisms has revealed Single Nucleotide Polymorphisms (SNPs) as the most common feature underlying genetic variation within species (The Arabidopsis Genome Initiative 2000; Sachidanandam et al. 2001). This type of genetic variation can be screened by means of a wide variety of technologies, usually based on primer extension or on ligation of oligonucleotide ends (Kwok 2000; Jander et al. 2002; Jenkins and Gibson 2002; Peters et al. 2003). The development of these SNP scoring technologies has led to an impressive increase in throughput capacity. A general prerequisite for these technologies is DNA sequence information. Therefore most SNP assays have been developed for the human genome (Chee et al. 1996; Wang et al. 1998) and for some model organisms for which large amounts of DNA sequence information are available (Borevitz et al. 2003; Winzeler et al. 2003; Törjék et al. 2003; Van Eijk et al. 2003).
However, for the majority of organisms, including agriculturally important crops, information on SNPs is still scarce and difficult to obtain, due to limited resources or to the complex nature of polyploid genomes. For marker-assisted breeding in such crops it is rarely cost-effective to perform SNP discovery since large numbers of markers scattered throughout the genome are needed for the identification of markers that are closely linked to major genes or Quantitative Trait Loci (QTL). Furthermore, in backcrossing programs genome-wide markers are used to select the progeny with the maximum genetic contribution from the recurrent parent. For such purposes marker technologies that do not require SNP discovery based on sequence information may be preferred. The Amplified Fragment Length Polymorphism (AFLP)TM technology (Vos et al. 1995) has proven its value in this context. However, AFLP relies on electrophoretic separation of PCR products on gel. This is also true for Simple Sequence repeats (SSRs), another kind of widely used marker. This constrains the throughput capacity, although capillary based apparatus provides significant improvement. The microarray platform is better suited for high-throughput analysis of thousands of genetic markers for 100 of individuals (Syvanen 1999). Several microarray-based marker methods have been developed, but most of these still require sequence information (Pastinen et al. 2000; Cutler et al. 2001; Flavell et al. 2003; Borevitz et al. 2003; Winzeler 2003; Li et al. 2004; Ji et al. 2004).
Recently, a method called Diversity Arrays Technology (DArT)TM has been developed (Jaccoud et al. 2001; Wenzl et al. 2004). This technology does not require sequence information and can be deployed on a microarray platform. DArT uses an array of individualized clones from a genomic representation prepared from amplified restriction fragments. Labeled genomic representations of individuals to be genotyped, such as the progeny of a segregating population, are then hybridized to the arrays. The polymorphisms scored are the presence versus absence of hybridization to individual array elements. The platform allows high-throughput screening of hundreds of molecular markers in parallel, and is especially suited for the generation of genome-wide markers for e.g. genetic linkage mapping. The initial proof-of-concept was provided for a species with a relatively simple genome i.e. rice (Jaccoud et al. 2001), and later for a species with a more complex genome, i.e. barley (Wenzl et al. 2004). We used Arabidopsis thaliana as a model species (Meinke et al. 1998) to validate the quality and molecular basis of the DArT markers. The published whole-genome sequence of A. thaliana (Lukowitz et al. 2000; The Arabidopsis Genome initiative 2000) provided us with the opportunity to compare the order of the DArT markers on the genetic linkage map with the order of the same markers on the DNA sequence map. In addition, the genome sequence enabled us to validate whether the DArT markers were indeed based on SNPs and InDels in the restriction sites recognized by the endonucleases used.
Our approach in A. thaliana deviates in two aspects from the DArT methods applied in rice and barley. First, we generated genomic representations according to a modified procedure. Instead of using a single restriction enzyme and one adapter, with (Wenzl et al. 2004) or without (Jaccoud et al. 2001) the use of a co-digesting enzyme, we used two enzymes with two different adapters and a co-digesting enzyme. No adapters were ligated to the sites created by the third restriction enzyme. A similar approach has successfully been used in another fingerprinting technique called three-endonuclease (TE)-AFLP (Van der Wurff et al. 2000). In addition, the number of amplifiable fragments was further reduced by ligating an asymmetric adapter with an amino group at the 3′ end of the short strand to one of the restriction sites. The use and effectiveness of such an adapter in combination with suppression PCR has already been demonstrated (Siebert et al. 1995; Broude et al. 2000; Van der Linden et al. 2004).
A second difference relative to the previously described use of DArT is that the adapter sequences of the printed fragments were different from the adapter sequences of the fragments that were hybridized to the slides. This prevented hybridization of complementary adapter strands, common to all clones on the array.
Using these modifications, we generated DArT markers and scored them in the F2 progeny of a cross between the A. thaliana ecotypes Columbia (Col) and Landsberg erecta (Ler). This allowed us to construct a genetic linkage map of Ler. The results described in this paper focus on the quality and suitability of DArT for mapping and marker-assisted breeding. For that purpose we evaluated (1) the reproducibility of DArT scores; (2) the consistency of detected genetic differences between parents and their Mendelian segregation in the progeny; (3) the robustness of the genetic linkage map; (4) the colinearity of the genetic map with the genome sequence map; (5) the molecular basis of the DArT markers by direct sequence comparison of the two parents.
Materials and methods
In silico predictions
A number of genomic representations were simulated; using an algorithm designed in-house which models restriction enzyme digestion and amplification of fragments within a user-defined window of fragment sizes and enzyme combinations. For this purpose a locally installed version of the Col genome sequence (The Arabidopsis Genome Initiative 2000) was used. Different combinations of three restriction enzymes were tested. Adapters would be ligated to the ends of the fragments produced by two of these enzymes. The program predicted which restriction fragments would be amplified by taking into account the effect of a long asymmetrical adapter with a 3′ amino group at the end of the short strand, but it disregarded possible methylation of the restriction sites.
Based on the outcomes of these in silico predictions, the enzyme combination PstI-EcoRI was chosen, with TaqI as co-digesting enzyme. The program predicted the generation of 5237 unique PstI-EcoRI, and PstI-PstI fragments that lack a recognition site for the co-digesting enzyme TaqI and fall within the size range of 50–1000 bp.
Generation of microarrays bearing a genomic representation of Ler
Plant material and DNA isolation
Genomic DNA from Ler was isolated, according to Pereira and Aarts (1998), from flower buds of adult plants grown in the greenhouse plants, after dry grinding with tungsten carbide beads using a Mixer Mill MM300 (Retsch).
Restriction and ligation of Ler DNA
The genomic DNA from Ler was cleaved to completion with the restriction endonucleases PstI, EcoRI, and TaqI. To the PstI ends of the restriction fragments a standard adapter was ligated, but to the EcoRI ends a long asymmetric adapter with a 3′-amino (NH2) group on the short strand was ligated (Table 1). The amino group, combined with PCR suppression (Siebert et al. 1995; Broude et al. 2000; Van der Linden et al. 2004), should prevent amplification of EcoRI- EcoRI fragments. Only PstI-EcoRI, and PstI- PstI fragments will be amplified. We used the modified EcoRI-adapter to reduce the complexity of the genomic representation, i.e. the number of unique amplified restriction fragments in the PCR mixture. To further reduce the complexity of the genomic representation, a third endonuclease was used, i.e. TaqI. No adapters were ligated to the TaqI sites. Consequently, all fragments that were digested by TaqI could not be amplified.
Restriction and ligation were performed simultaneously to prevent fragment-to-fragment ligation. To about 500 ng of genomic DNA, 50 μl of restriction-ligation mixture was added and the solution was incubated for 2 h at 37°C and 2 h at 65°C. The restriction-ligation mixture contained 5 U each of PstI, EcoRI, and TaqI (New England Biolabs, Beverly, MA), 10 μl of 5 times restriction-ligation buffer (10 mM Tris-acetate 10 mM magnesium acetate, 50 mM potassium acetate, 5 mM DTT, 50 ng/μl BSA, pH 7.5), 10 mM ATP, 2 U of T4 DNA ligase (Invitrogen, UK), and 5 pmol of each adapter (Table 1).
Amplification of the restriction-ligation mixture
The restriction-ligation mixture was diluted 20-fold with distilled water. From this diluted mix 12.5 μl was used as a template in a 50-μl PCR with 1.5 U of Taq polymerase (Promega, Madison, WI, USA) and 15 pmol each of the two cloning primers listed in Table 1. The PCR was carried out on a thermal cycler (PTC-200, MJ Research) using a touchdown PCR program. The amplification mixture was incubated for 2 min at 94°C, followed by eight cycles of 94°C for 10 s, 65°C (−1°C each cycle) for 30 s, and 72°C for 2 min, and 29 cycles of 94°C for 10 s, 56°C annealing temperature for 30 s, and 72°C for 2 min. The final extension was at 60°C for 30 min. The PCR mixture was purified, using the QIAquick PCR purification kit (Qiagen, Crawley, UK).
Cloning of restriction fragments from Ler and colony PCR
In order to individualize amplified restriction fragments from Ler, amplicons were ligated into a T-vector using the pGEM-T Easy Vector System (Promega, Madison, WI, USA). The vectors were transformed into competent XL-Blue (Stratagene, La Jolla, CA) bacteria, and the cells were plated on LB plates (Sambrook et al. 1989) containing ampicillin (100 μg/ml), IPTG (0.5 mM) and X-gal (80 μg/ml) for blue–white color screening. After overnight growth at 37°C, 2592 white colonies were picked, and individually grown in 70-μl aliquots of “LB-freeze” medium [LB medium supplemented with 100 μg/ml ampicillin, 36 mM K2HPO4, 13.2 nM KH2PO4, 1.7 mM trisodium citrate, 0.4 mM MgSO4. 7H2O, 6.8 mM (NH4)2SO4, and 4.4% (v/v) glycerol]. A 2-μl aliquot of each individual culture was then subjected to PCR using the M13 Forward and Reverse universal primers. Amplification was performed under the following conditions: initial denaturation at 94°C for 6 min, followed by 34 cycles of denaturation (94°C for 30 s), annealing (55°C for 1 min), and extension (72°C for 2 min). A final extension at 72°C for 3 min was included. This yielded a library of 2592 amplified restriction fragments from Ler. Each restriction fragment was flanked by adapter sequences and small portions of the pGEM-T Easy Vector.
Printing and processing of microarrays
The unpurified PCRs were dried, dissolved in 35 μl of VSMR print buffer A (Vanderbilt University, South Nashville, Tenn.) and spotted in triplicate on SuperChip poly-L-lysine slides (Erie Microarray) using a MicroGrid II arrayer (Biorobotics, UK). After printing, the slides were processed according to the Erie protocol for poly-L-lysine slides (http://www.eriemicroarray.com/support/tech.aspx).
Genotyping using the microarrays
Plant material and DNA isolation. The homozygous A. thaliana ecotypes Ler and Col were crossed, and the F1 progeny were self-fertilized. This resulted in a segregating F2 population, of which 80 individuals were used for the construction of a genetic linkage map. The DNA from Col and the segregating F2 population were isolated using the method described for isolation of Ler genomic DNA.
Generation of genomic representations
From the DNA samples obtained from the parents and the 80 F2 plants genomic representations were made by means of restriction enzyme digestion, adapter ligation and amplification. These genomic representations were labeled with Cy5 and hybridized to the microarrays. The images were analyzed for detection of clones that revealed polymorphism between the parents and segregation in the progeny. Details are given below.
For Ler and Col two different DNA samples per genotype were used to prepare the genomic representations. This DNA was used to prepare the labeled genomic representations that were hybridized in duplicate to the microarrays. From the 80 F2 plants, 40 genomic representations were hybridized once and the remaining 40 in duplicate. The preparation of genomic representations from a genotype started from a single batch of DNA, but from the endonuclease digestion onward the replicates were treated separately.
For the generation of the genomic representations for hybridization, the same complexity reduction method as described above (see ‘Restriction and ligation of Ler DNA’) was used. However, the adapter and primer sequences of the genomic representation deviated from the adapter sequences used for construction of the genomic representation printed on the slides (Table 1). This was done to prevent hybridization of the adapter sequences from the genomic representation used for hybridization to the adapter sequences of the printed fragments. From earlier experiments (data not shown) we concluded that application of different adapter sequences increased the signal to noise ratio considerably.
As a reference signal for all spots, we used a Cy3 labeled vector sequence that flanked all printed restriction fragments of Ler. This 275-bp vector sequence was obtained by amplification of the poly-linker sequence of the pGEM-T Easy Vector using M13 Forward and Reverse amplification primers with the empty vector as template.
Fluorescent labeling of the genomic representations
The genomic representations and reference DNA were purified using the QIAquick PCR purification kit (Qiagen, Crawley, UK). For each sample, approximately 750 ng of DNA was labeled with Cy5-dUTP and Cy3-dUTP (Amersham, NJ, USA), respectively, using the Fermentas DecaLabel DNA labeling kit. The genomic representations were labeled according to the manufacturer’s instructions, except that the reaction volume was reduced to 5 μl, the incubation time increased to 2 h, and 0.3 μl of 1 mM Cy5-dUTP or Cy3-dUTP was used instead of [32 P]dATP. The labeled genomic representations, referred to as targets, were not purified prior to hybridization.
Hybridization, washing and scanning
To each target, 5 μl of a 20-fold dilution of Cy3-labeled reference DNA was added and mixed with 50 μl of a 40:5 mixture of ExpressHyb buffer (Clontech, Heidelberg, Germany) and herring sperm DNA (10 g/l). After denaturating for 2 min at 95°C, these hybridization mixtures were hybridized to the microarrays overnight at 65°C. The slides were then washed according to Jaccoud et al. (2001) and scanned with an Affymetrix 428 scanner.
Image analysis and scoring for polymorphism
The software package DARTSOFT, Version 7.2.9 (Cayla et al., in preparation) was used to analyze each batch of scanned microarray images automatically. We analyzed three batches of images, i.e. from the parents (2×4), from 40 F2 plants hybridized once and from 40 F2 plants hybridized in duplicate. DARTSOFT localized the spots, rejected those with weak reference signals, computed and normalized the relative hybridization intensities, and identified clones that revealed genetic differences according to dominant scoring. These polymorphic clones were detected by means of a combination of ANOVA and fuzzy C-means clustering (Wenzl et al. 2004). A clone was considered to be polymorphic if the normalized hybridization signals from the different plants could be grouped into two distinct clusters, i.e. a cluster of low hybridization signals and a cluster of high signals, for which the between-cluster variance was at least 80% of the total variance. A polymorphic clone was incorporated into a 0/1 dominant scoring table if the probability of belonging to one of the two classes was above 0.95 averaged across all slides of the batch of images, and if it was scored with P > 0.95 in at least 90% of all these slides. Markers that showed conflicting scores between the replicates or could not be scored in either of the replicates were scored as unknown.
Construction of genetic linkage map
The automatically generated scoring table from DARTSOFT was converted into a format suitable for the software package JoinMap Version 3.0 (http://www.kyazma.nl) for construction of a genetic linkage map. Markers that showed an identical scoring pattern were removed from the scoring table, leaving one marker per unique segregation pattern. For the markers that showed an identical scoring pattern, we did not know whether the underlying clones had identical sequences or co-segregated because of close linkage. In the first step, the markers were assigned to linkage groups, based on the logarithm of the odds (LOD) ratio for each possible marker pair. The LOD value indicates the likelihood of linkage by comparing the probabilities of random association of markers in the progeny to association caused by linkage. We used LOD values in the range of 3.0–5.0, whereas the final assembly of linkage groups was completed using a LOD value of 3.0. In the second step, the calculation of a linear order of markers within a linkage group was calculated in JoinMap 3.0 using the default settings of the mapping parameters. In addition, we varied the settings of the most critical mapping parameters to validate the robustness of the map. We changed the threshold of linkage for marker pairs that should be included to calculate the map from a LOD value of 1 (default) to LOD values of 0.05 and 2. We also changed the χ2 threshold for the removal of loci with respect to jumps in the goodness-of-fit from 5 (default) to 0 (minimum). The genetic linkage maps generated with all these different mapping parameter settings were then compared.
Sequence analysis of the polymorphic clones from Ler
Sequencing procedure
The polymorphic Ler clones were sequenced using the ABI PRISM BigDye Terminator Cycle Sequencing Ready reaction kit (Perkin-Elmer, Wellesley, MA) and analyzed on an ABI 3700 sequencer. For the majority of the clones a full-length sequence could be obtained using the M13 Forward primer. For a few large fragments it was necessary to use the M13 Reverse sequencing primer to obtain the full-length sequence of the fragment.
Analysis of the sequences
The sequences were analyzed using the Lasergene software program Seqman (DNAstar Inc., Madison, WI). The vector and adapter sequences were removed and the original restriction sites restored.
Sequence alignments to the Col genome (Version 5.0, 2004) were performed using the Basic Local Alignment Search Tool (BLAST, Version 2.2.8) at the NCBI database (Altschul et al. 1997). The BAC clone with the highest sequence similarity was selected and used to obtain the position (in bp) on one of the five chromosomes. This was performed with the SeqViewer program (Version Feb 2004; Rhee et al. 2003) from The Arabidopsis Information Resource (TAIR; http://www.arabidopsis.org/servlets/sv). To scale the sequence map positions from Col (kb) to the genetic map positions (cM) of Ler, the two most distal markers on each chromosome were fixed. The program MapChart Version 2.11 (Voorrips 2002) was used to display the positions of the DArT markers on both the genetic linkage map of Ler and on the sequence map of Col.
Furthermore, the homologous Col sequences were checked for the presence of intact PstI or EcoRI restriction recognition sites at the same locations as in the Ler sequences. We also checked whether a TaqI site was present in the homologous Col sequence. The purpose of this analysis was to verify that the segregation of the DArT markers could be explained on the basis of SNPs or InDels in the sites recognized by the restriction enzymes used. This was an additional check on the quality of the DArT markers.
Sequences that showed no or very low similarity to the Col genome were aligned to the Ler sequence data from the Cereon Arabidopsis Landsberg Sequence Database (http://www.arabidopsis.org/Cereon/index.jsp) and the random Ler sequence database from The Institute for Genomic Research (http://www.tigr.org/tdb/e2k1/ath1/atgenome/Ler.shtml).
Results and discussion
The high-throughput fingerprinting technique DArT can detect DNA polymorphisms by scoring the presence or absence of specific DNA sequences in a defined genomic representation (i.e., a representative subset of genomic fragments) through hybridization to microarrays (Jaccoud et al. 2001; Wenzl et al. 2004). Although DArT has been applied in rice to detect polymorphisms, its usefulness for constructing genetic linkage maps has only recently been demonstrated (Wenzl et al. 2004) in barley. By taking advantage of the availability of the whole-genome sequence of A. thaliana, we were able to validate the quality and suitability of DArT for mapping. The quality of the data obtained was evaluated on the basis of the following criteria.
Reproducibility
The DArT marker scores for 40 F2 genotypes were generated on two series of slides. The marker scores from the first series of slides were compared with the marker scores from the second series. Two images generated for one F2 genotype hybridized to the array are available as Electronic Supplementary Material (Fig. S1). The two replicates started from the same DNA samples of the 40 F2 genotypes and one restriction-ligation event per genotype, but were based on separate amplifications and subsequent steps. The data were evaluated for the 190 clones that revealed segregation in the F2 population, which resulted in 7600 pairs of scores (40 × 190). In nine out of these 7600 scoring pairs conflicting scores were found. This corresponds to a reproducibility of 99.88%, similar to the value reported for DArT in barley (Wenzl et al. 2004).
Consistency of genetic differences between the parents, and Mendelian segregation in the F2 population
The Cy5-labeled genomic representations from Ler and Col were hybridized to the microarrays in four replicates, starting from two different DNA isolations per parent, as described in Materials and methods. The Cy5 hybridization signals were corrected for the amount of DNA spotted by calculating the Cy5/Cy3 ratios per spot, using the software DARTSOFT. The Cy5/Cy3 ratios per spot were averaged per clone among the three spots per slide, normalized per slide, and averaged among the four slides that were hybridized with a labeled PCR mixture from Col.
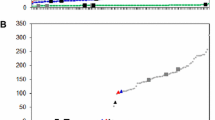
For the majority of the printed Ler clones, the normalized hybridization signal from Ler was approximately as high as the normalized signal from Col. These clones are located around the diagonal in Fig. 1, and do not reveal genetic differences between Ler and Col.

Hybridization signals from a Cy5 labeled Ler representation and from a Cy5 labeled Col representation. Cloned fragments from Ler were printed on the slides. Clones that did not show significant segregation in the F2 population (blue triangles) generally appeared to have a similar Col and Ler signal and therefore fall on the diagonal. For clones that showed Mendelian segregation in the F2 population (red triangles), the Ler signal was clearly higher than the Col signal, and these can be regarded as Ler specific
Clones located below the diagonal hybridized with fragments that were amplified in Ler, but not in Col and therefore can be regarded as genetic markers. Apart from clones that yielded low-intensity signals, all of these markers could be scored as reliable segregating markers in the progeny. We found no segregating markers that showed a higher Col signal than Ler signal. The reason for this is that only clones from Ler were printed.
Segregating markers were not always characterized by a strict presence versus absence pattern. Consistent differences in the intensity of the hybridization signal were sufficient to identify restriction fragments that segregated as reliable markers.
Robustness of the genetic linkage map
Based on the hybridization images for the 80 F2 plants, we generated a dominant scoring table for 190 segregating clones, using DARTSOFT. This F2 scoring table was formatted as an input file for JoinMap. The 190 markers showed 74 (~40%) unique segregation patterns. Using a LOD threshold value of 5.0, all 74 markers showed linkage to at least three other markers, allowing reliable mapping by three-point analysis. This LOD value resulted in six linkage groups with 18, 16, 14, 13, 9, and 4 markers, respectively. As A. thaliana has five chromosomes we lowered the LOD value to 3.0; at this value five linkage groups were obtained. Lowering the LOD value did not change the assignment of the remaining markers to the linkage groups, nor did it affect the order of the markers. The two joined groups indeed belonged to one chromosome, as was confirmed by alignment of the genetic and sequence maps (see below).
For most datasets JoinMap needs three rounds to determine the linear order of the markers (J. W. van Ooijen, personal communication). In the first round all markers are included that can be positioned well within a linkage group according to the χ2 tests. In the second and third rounds the markers are included that cause tension in the map (above the χ2 threshold). The amount of tension in the map is indicative for the quality of the data set. Initially we used a χ2 threshold of five (standard), beyond which markers were not positioned on the map. With this setting all markers could be positioned in a single round. However, the χ2 threshold could easily be lowered to zero (minimum), and all markers were still included in the first round, making a second and third round unnecessary. Although the number of markers is relatively low, inclusion of all markers during the first round is exceptional and indicates a high quality of the marker data. Overall the map was extremely robust, since all parameter settings used resulted in the same genetic linkage map.
As only clones derived from Ler were printed on the slides, the genetic linkage map obtained is a map for Ler (Fig. 2). The average spacing between the 74 loci is 5.4 cM, and there is no obvious clustering of the markers.

Colinearity between DArT markers on the genetic linkage map of Ler (left) and the same markers on the Col sequence map (right). The genetic linkage map of Ler is based on 190 DArT markers that segregated in an F2 population of 80 individuals obtained from a Col × Ler cross. For clarity only non-redundant markers are displayed. Approximate centromere locations are shown as ellipses. Ler markers for which no homologous sequence could be found in the Col genome are highlighted in green. Markers that showed high homology to (retro-) transposons are highlighted in red. For marker 4D04 (bold) on chromosome 1, the position on the Ler genetic map was not in agreement with the position on the Col sequence map
Microsoft Excel was used to display the DArT scores according to the order of the markers on the linkage groups (Supplementary Table S1) and to visualize the recombination events in all progeny. This allowed us to detect singletons. Singletons are often the result of scoring errors, and thus can be regarded as indicators for data quality. Errors may result in extra, apparent but nonexistent, crossovers, altering marker order, map length or both (Buetow 1991; Lincoln and Lander 1992). Some scoring errors can be resolved by thorough checking of data, but apparent “false errors” arising from, for example, double crossovers (DCOs) or DNA methylation events are not data-scoring errors (Knox and Ellis 2001).
In our dataset we identified eight singletons in the 74 unique segregation patterns. Six out of these eight were assayed more than once because of redundancy of printed clones. In all cases these redundant clones had identical scores, which indicates that these singletons were not a result of scoring errors. In two other cases replicates were not available. Most markers responsible for a singleton mapped far (>5.4 cM) from their flanking markers, making it more likely that a true double recombinant event had occurred.
Colinearity of the genetic linkage map and the sequence map
The 190 Ler clones used for the construction of the genetic linkage map were sequenced and compared to the whole-genome sequence of Col. This enabled us to position the DArT markers on the Col sequence map and evaluate the colinearity of markers on the two maps (Fig. 2) The sequence analysis resulted in 107 unique sequences, of which 97 (90.7%) could be located on the sequence map of Col. The order of all, except one (discussed later), of these markers on the genetic map coincided with the order on the sequence map.
The remaining 10 (9.3%) Ler sequences could not be positioned on the sequence map of Col. For seven out of these 10 sequences little or no homology to the Col genome sequence could be found. These sequences were aligned to Ler sequences in the NCBI Arabidopsis database, the Cereon Ler database, and the TIGR Ler database. All seven sequences matched sequences in one of the Ler databases. Although there is a possibility that some of these markers do not match the Col genome sequence because these regions were missed in the BAC-based sequencing approach, they are more likely to be part of large insertions in the Ler genome or deletions in the Col genome. Two of these sequences showed high homology to previously identified transposons unique for the Ler accession. One of these was the Tag1 element (5N02) and the other was Ta1-2 (5L08) (Bhatt et al. 1998; Voytas et al. 1990). According to our results the Tag1 element maps on the lower-arm of chromosome 1 (see clone 5N02 in Fig. 2). This is in agreement with the position shown by Bhatt et al. (1998).
Three sequences (4C21, 2N13, and 3M21) showed high homology to retrotransposons. As copies of these retrotransposons are present at multiple positions in the genome, it was not possible to determine a single location on the Col genome. Because multiple copies were present in the Col genome, we were surprised that segregation of these sequences in the F2 population could be followed reliably. However, we then found that all copies that showed high homology to these elements had a polymorphism in one of the restriction sites in Col, preventing amplification of any of these fragments from Col. In addition only a single copy, containing the restriction sites necessary for amplification, could be found in the Ler sequence databases.
The position of clone 4D04 on the sequence map did not correspond to that on the genetic map (Fig. 2). On the sequence map of Col this marker was positioned more than half a chromosome away from the position on the genetic map of Ler. A small distance would be consistent with some scoring errors. However, the large distance actually observed may point to a translocation of a part of the Ler genome relative to the Col genome.
The colinearity of the markers on the genetic map of Ler with these markers on the sequence map of Col is an additional proof of the high quality of the genetic map, and therefore of the high quality of the DArT markers.
Molecular basis of DArT polymorphisms
The inserts were generated using the restriction enzymes PstI, EcoRI, and TaqI. Both PstI and EcoRI recognize a 6-bp sequence, whereas TaqI recognizes a 4-bp motif. A standard adapter was ligated to the PstI sites, and a long asymmetric adapter with a 3′ amino group was ligated to the EcoRI sites (Table 1). This should result in the amplification of PstI-EcoRI fragments and some PstI-PstI fragments. Among the 107 clones sequenced, we found 106 PstI-EcoRI fragments and one PstI-PstI fragment. The efficiency of the modified EcoRI adapter is illustrated by the fact that no EcoRI-EcoRI fragments were found.
Comparison of the 107 sequences of the Ler clones with the Col genome sequence allowed us to analyze the molecular basis for the polymorphic behavior of the DArT markers (Fig. 3). The segregation of 33 markers (30.8%) could be explained on the basis of the presence of an EcoRI site in Ler that is absent in the homologous Col sequence. Sequence differences in PstI sites could explain 24 markers (22.4%). The co-digesting enzyme TaqI explained 28 markers (26.2%). A total of six markers (5.6%) revealed polymorphisms in more than one restriction site. For seven markers (6.5%) no homologous Col sequence could be found. By alignments to the available Ler sequences, we were able to confirm that these sequences were based on Ler specific insertions ranging in size from 200 to 680 bp. For the remaining nine markers (8.4%) we could not explain the segregation by sequence differences between Ler and Col. As PstI is a methylation sensitive enzyme, a possible explanation for the segregation of these markers is that the PstI sites were not methylated in Ler, but may have been methylated in Col. Mendelian inheritance of such methylation-based polymorphisms has been shown in a number of organisms (Messeguer et al. 1991; Cervera et al. 2002).

The molecular basis of DArT markers was generally polymorphism between Ler and Col in the restriction sites of the endonucleases used. In cases where no SNP or InDel could be detected in any of the restriction sites, the differential behavior was probably based on the presence of methylation in the PstI site in Col, but not in Ler
The above findings clearly show that DArT can effectively detect SNPs and InDels in restriction sites. An additional advantage is that DArT clones can readily be sequenced and thus provide information for conversion into PCR-based markers.
Degree of polymorphism and redundancy
Out of 2592 clones picked, 190 (7.3%) appeared to reveal polymorphism between Col and Ler. Based on high-quality DNA sequence data, Schmid et al. (2003) found 1241 SNPs and InDels between Col and Ler in 216,760 bp, equivalent to a polymorphism score of 0.57%. For the creation of the genomic representations we used three enzymes that recognized 6+6+4 = 16 bp. A rough estimate of the frequency of polymorphic DArT markers based on the sequence data from Schmid et al. (2003) would then be 16 × 0.57% = 9.1%. The degree of polymorphism we found (7.3%) is in reasonable agreement with this calculation.
From the 190 segregating clones 107 appeared to be unique. This implies a degree of redundancy of ~1.78 fold. The whole-genome sequence of Col allowed us to estimate the number of PstI-EcoRI and PstI-PstI fragments without TaqI sites in between. If methylation of PstI sites is ignored the number of fragments in the size range 50–1000 bp is 5237. We picked 2592 clones. If each of the 5237 fragments had an equal probability of being cloned and picked, the expected number of cloned fragments is 2045, resulting in an expected redundancy of 1.27. However, the observed redundancy is higher (1.78). We presume that this is caused by three phenomena. (1) Methylation will decrease the number of accessible PstI sites, and consequently the number of amplicons. (2) Differences in the efficiency of PCR among fragments may have resulted in unequal amplification of fragments. (3) Differences in cloning efficiency among amplicons may also have resulted in some sampling bias.
The redundancy lowered the number of markers, but it also provided us with the opportunity to check whether redundant clones showed identical segregation in the F2. Redundant clones appeared to be positioned always at exactly the same position on the genetic map of Ler. This again underlines the reproducibility of the DArT scores.
Although the total number of markers generated in this study is still limited, we could have doubled the number of markers if, in addition to the Ler fragments, fragments from Col had also been spotted. Furthermore, an enzyme combination that resulted in a more complex genomic representation would also have allowed us to generate more markers and reduce the redundancy.
Conclusion
We have successfully applied DArT with several modifications to the model plant A. thaliana. Using the available genome sequence, we were able to validate the quality of DArT and the molecular basis for polymorphism of the DArT markers. These results indicate that DArT provides high quality markers that can be used to construct medium-density genetic linkage maps. DArT can be fine-tuned to detect polymorphism in species with various genome sizes. This fine-tuning can be achieved by using the appropriate complexity reduction method or by making use of enrichment techniques prior to cloning. Therefore we argue that DArT is a good alternative to currently used techniques for whole-genome fingerprinting. Among the advantages of DArT are: (1) it provides high-quality markers, (2) it is not reliant on DNA sequence information, (3) it is non-gel based, (4) it is amenable to full automation, (5) is cost-effective, especially for highly multiplexed assays (>100 markers); and (6) integration of DArT maps is straightforward.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bhatt AM, Lister C, Crawford N, Dean C (1998) The transposition frequency of Tag1 elements is increased in transgenic Arabidopsis lines. Plant Cell 10:427–434
Borevitz JO, Liang D, Plouffe D, Chang H-S, Zhu T, Weigel D, Berry CC, Winzeler E, Chory J (2003) Large-scale identification of single-feature polymorphisms in complex genomes. Genome Res 13:513–523
Broude NE, Zhang L, Woodward K, Englert D, Cantor CR (2000) Multiplex allele-specific target amplification based on PCR suppression. Proc Natl Acad Sci USA 98:206–211
Buetow KH (1991) Influence of aberrant observations on high-resolution linkage analysis outcomes. Am J Hum Genet 5:985–994
Cervera MT, Ruiz-Garcia L, Martinez-Zarpater JM (2002) Analysis of DNA methylation in Arabidopsis thaliana based on methylation-sensitive AFLP markers. Mol Gen Genomics 268:543–552
Chee M, Yang R, Hubbell E, Berno A, Huang XC, Stern D, Winkler J, Lockhart DJ, Morris MS, Fodor SPA (1996) Accessing genetic information with high-density DNA arrays. Science 274:610–614
Cutler DJ, Zwick ME, Carrasquillo MM, Yohn CT, Tobin KP, Kashuk C, Mathews DJ, Shah NA, Eichler EE, Warrington JA, Chakravarti A (2001) High-throughput variation detection and genotyping using microarrays. Genome Res 11:1913–1925
Flavell AJ, Bolshakov VN, Booth A, Jing R, Russell J, Ellis TH, Isaac P (2003) A microarray-based high throughput molecular marker genotyping method: the tagged microarray marker (TAM) approach. Nucleic Acids Res 31:e115
Jaccoud D, Peng K, Feinstein D, Kilian A (2001) Diversity Arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res 29:e25
Jander G, Norris SR, Rounsley SD, Bush DF, Levin IM, Last RL (2002) Arabidopsis map-based cloning in the post-genome era. Plant Physiol 129:440–450
Jenkins S, Gibson N (2002) High-throughput SNP genotyping. Comp Funct Genom 3:57–66
Ji M, Hou P, Li S, He N, Lu Z (2004) Microarray-based method for genotyping of functional single nucleotide polymorphisms using dual-color fluorescence hybridization. Mutat Res 548:97–105
Knox MR, Ellis TH (2001) Stability and inheritance of methylation states at PstI sites in Pisum. Mol Gen Genom 265:497–507
Kwok PY (2000) High-throughput genotyping assay approaches. Pharmacogenomics 1:95–100
Li TX, Wang J, Bai Y, Sun X, Lu Z (2004) A novel method for screening species-specific gDNA probes for species identification. Nucleic Acids Res 32:e45
Lincoln SE, Lander ES (1992) Systematic detection of errors in genetic linkage data. Genomics 3:604–610
Luikart G, England PR, Tallmon D, Jordan S, Taberlet P (2003) The power and promise of population genomics: from genotyping to genome typing. Nature Genet 4:981–994
Lukowitz W, Gillmor CS, Scheible WR (2000) Positional cloning in Arabidopsis why it feels good to have a genome initiative working for you. Plant Physiol 132:795–805
Meinke DW, Cherry JM, Dean C, Rounsley SD, Koorneef M (1998) Arabidopsis thaliana: a model plant for genome analysis. Science 282:662–681
Messeguer R, Ganal MW, Steffens JC, Tanksley SD (1991) Characterization of the level, target sites and inheritance of cytosine methylation in tomato nuclear DNA. Plant Mol Biol 16:753–770
Pastinen T, Raitio M, Lindroos K, Tainola P, Peltonen L, Syvanen AC (2000) A system for specific, high throughput genotyping by allele-specific primer extension on microarrays. Genome Res 10:1031–1042
Pereira A, Aarts MGM (1998) Transposon tagging with the En-I system. In: Martinez-Zapater J, Salinas J (eds) Arabidopsis protocols. Humana Press, Totowa, NJ, pp 329–338
Peters JL, Cnudde F, Gerats T (2003) Forward genetics and map-based cloning approaches. Trends Plant Sci 8:484–491
Rhee SY et al (2003) The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res 31:224–228
Sachidanandam R et al (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NewYork
Schmid KJ, Rosleff Sörensen T, Stracke R, Törjék O, Altmann T, Mitchell-Olds T, Weisshaar B (2003) Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res 13:1250–1257
Siebert PD, Chenchik A, Kellogg DE, Lukyanov KA, Lukyanov SA (1995) An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res 23:1087–1088
Syvanen AC (1999) From gels to chips: ‘minisequencing’ primer extension for analysis of point mutations and single nucleotide polymorphisms. Hum Mutat 13:1–10
The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815
Törjék O, Berger D, Meyer RC, Müssig C, Schmid KJ, Sörensen TR, Weisshaar B, Mitchell-Olds T, Altman T (2003) Establishment of a high-efficiency SNP-based framework marker set for Arabidopsis. Plant J 36:122–140
Van Eijk MJT, Broekhof JLN, van der Poel HJA, Hogers RCJ, Schneiders H, Kamerbeek J, Verstege E, van Aart JW, Geerlings H, Buntjer JB, van Oeveren AJ, Vos P (2003) SNPWave: a flexible multiplexed SNP genotyping technology. Nucleic Acids Res 32:e47
Van der Linden CG, Wouters DCAE, Mihalka V, Kochieva EZ, Smulders MJM, Vosman B (2004) Efficient targeting of plant disease resistance loci using NBS profiling. Theor Appl Genet 109:384–393
Van der Wurff AWG, Chan YL, van Straalen NM, Schouten J (2000) TE-AFLP: combining rapidity and robustness in DNA fingerprinting. Nucleic Acids Res 28:e105
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuipers M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Voytas DF, Konieczny A, Cummings MP, Ausubel FM (1990) The structure, distribution and evolution of the Ta1 retrotransposable element family of Arabidopsis thaliana. Genetics 126:713–721
Wang DG et al (1998) Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 280:1077–1082
Wenzl P, Carling J, Kudrna D, Jaccoud D, Huttner E, Kleinhofs A, Kilian A (2004) Diversity Arrays Technology (DArT) for whole-genome profiling of barley. Proc Natl Acad Sci USA 101:9915–9920
Winzeler EA, Castillio-Davis CI, Oshiro G, Liang D, Richards DR, Zhou Y, Hartl DL (2003) Genetic diversity in yeast assessed with whole-genome oligonucleotide arrays. Genetics 163:79–89
Acknowledgements
We thank Andy Pereira for providing seeds from the A. thaliana ecotypes Ler, Col and from the F2 population used for mapping. We also thank our colleagues Gerard van der Linden, Mark Kemper, Mark Fiers and Hong Tran (Plant Research International) and Eric Huttner (DArT Pty./Ltd.) for constructive discussions during the course of this work and of manuscript preparation. This project was supported by a travel grant from the Netherlands Organization for Scientific Research (NWO)
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Wittenberg, A.H.J., Lee, T.v.d., Cayla, C. et al. Validation of the high-throughput marker technology DArT using the model plant Arabidopsis thaliana. Mol Genet Genomics 274, 30–39 (2005). https://doi.org/10.1007/s00438-005-1145-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-005-1145-6